Introduction
Progressive joint destruction, which is a
pathognomonic characteristic of rheumatoid arthritis (RA), is
caused by chronic inflammation and excessive osteoclastogenesis
(1,2). Under physiological conditions, a
balance between bone formation and resorption is maintained for
skeletal homeostasis. In pathological states, such as RA, this
balance is disrupted in favor of osteoclast-mediated bone
resorption. Excessive osteoclastogenesis occurring at the frontline
of the synovium-bone interface induces bone resorption (3). Metabolic activation of osteoclasts
for potentiating the bone-resorbing ability requires ‘biochemical
fueling’ from complex cellular interactions between cells of the
osteoclast lineage and mesenchymal cells and lymphocytes (4–6).
These interactions are controlled by various
cytokines and by the receptor activator of nuclear factor-κB
(NF-κB) ligand (RANKL). A large number of cytokines have been shown
to regulate osteoclast formation and function. In addition, a
number of cytokines were recently shown to have a major influence
on the ability of osteoclasts to resorb bone. RANKL is regarded as
an essential cytokine that involves the interaction between the
immune and bone systems. RANKL is necessary for the differentiation
of osteoclast precursors into mature osteoclasts, and its
deficiency is sufficient to block osteoclast formation completely
(7,8). As a member of the tumor necrosis
factor (TNF) superfamily, RANKL is expressed mainly by osteoblasts.
In pathological conditions, including RA, this role is deferred to
active fibroblast-like synoviocytes and T cells residing in the
inflamed joint (9,10). RANKL expression is upregulated in,
and constitutes an important prerequisite for, osteoclast
differentiation in animal models of RA and in rheumatoid synovial
tissue (11,12). In addition, RANKL expression is
upregulated by inflammatory cytokines, such as TNF-α,
interleukin-1β (IL-1β), IL-6, IL-17 and IL-23 (13–17).
Cytokines enhance osteoclastogenesis indirectly by
upregulating RANKL expression. However, the theories on
RANKL-dependent osteoclastogenesis are controversial. Certain
authors demonstrated that TNF-α, IL-1β and IL-6 induced osteoclast
differentiation directly, in a RANKL-independent manner (18–20), whereas others showed that a
permissive level of RANKL is necessary for TNF-α-induced
osteoclastogenesis, thus concluding that TNF-α alone cannot induce
osteoclast formation (21).
We considered that this controversy regarding
cytokine-induced, RANKL-independent osteoclastogenesis stems from
the fact that these previous experiments were performed under
different conditions. To minimize the confounding factors that may
arise from the experimental system of osteoclastogenesis, we
adopted a monocellular culture system instead of a coculture
system, consisting of osteoblasts and bone marrow cells. Mouse bone
marrow-derived macrophages (BMMs) were stimulated with the
macrophage colony-stimulating factor (M-CSF) and various
concentrations of RANKL. Inflammatory cytokines (TNF-α, IL-1β,
IL-6, IL-17 and IL-23) were added to the osteoclastogenesis culture
system at two different time-points: at 0 h after the stimulation
with RANKL (early phase), when undifferentiated osteoclast
precursors were predominant, and at 48 h after the stimulation with
RANKL (delayed phase), when half-matured osteoclasts appeared. In
the present study, we identified the direct influence of each
inflammatory cytokine on RANKL-induced osteoclast differentiation
and its effect on the bone-absorbing ability of osteoclasts using a
monocellular culture system.
Materials and methods
Reagents and proteins
Recombinant mouse IL-1β, IL-6, IL-17, IL-23 and
TNF-α were purchased from R&D Systems (Minneapolis, MN, USA).
Soluble RANKL was purchased from PeproTech (London, UK). The
tartrate-resistant acid phosphatase (TRAP) kit was purchased from
Sigma-Aldrich (St. Louis, MO, USA) and a Cell Counting kit (CCK)-8
was purchased from Dojindo Laboratories (Kumamoto, Japan). The
pNF-κB-Luc, pAP-1-Luc, CREB-Luc and pNFAT-Luc plasmids were
purchased from Stratagene (La Jolla, CA, USA).
Animals
Six-week-old male DBA/1J mice were purchased from
SLC, Inc., (Shizuoka, Japan). Animals were maintained under
specific pathogen-free conditions at the Institute of Medical
Science, the Catholic University of Korea, and were fed standard
mouse chow (Ralston Purina) and water ad libitum. All
experimental procedures were examined and approved by the Animal
Research Ethics Committee of the Catholic University of Korea,
which conforms to all USA National Institutes of Health guidelines
(approval ID 2011-0062-02).
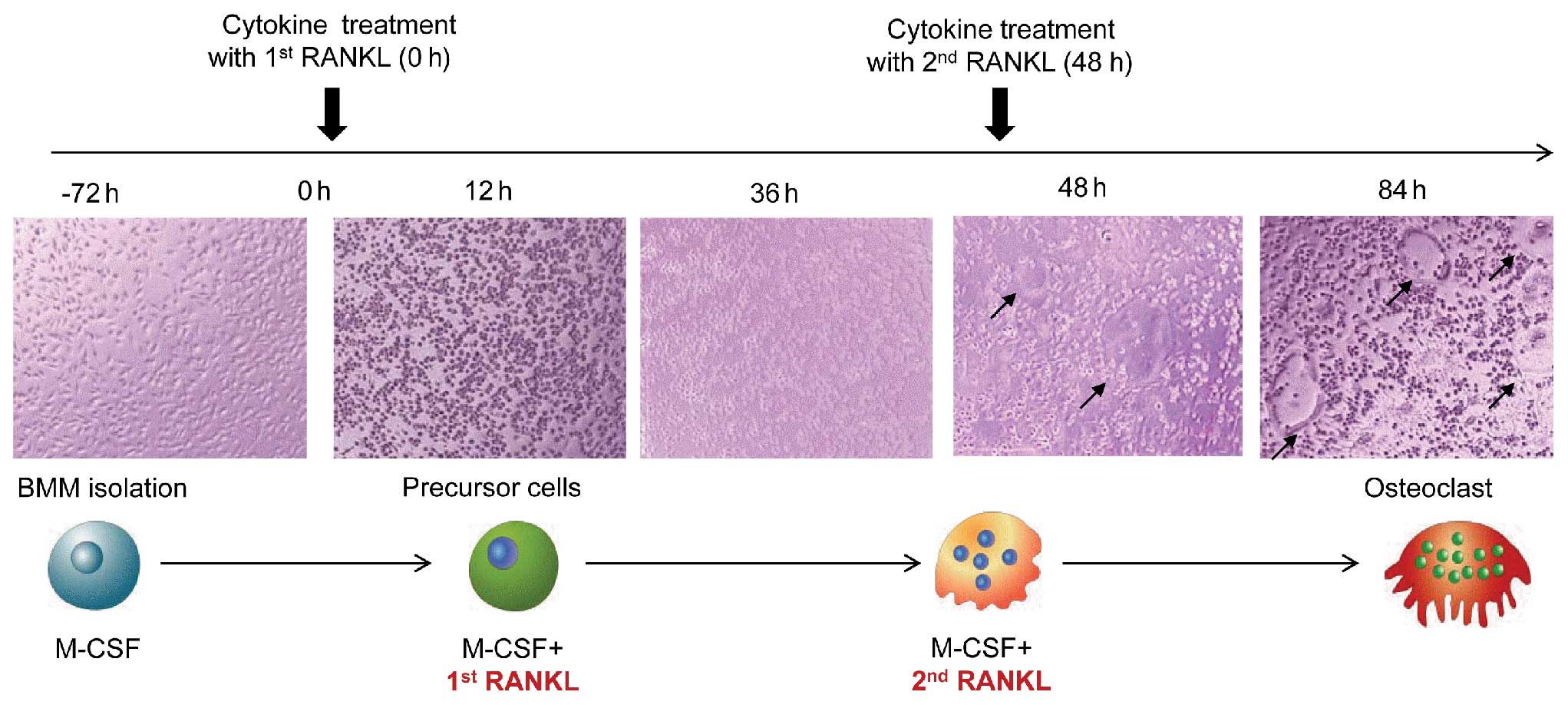
In vitro osteoclastogenesis
BMMs were isolated from the tibia and femur of
6-week-old DBA/1J male mice by flushing the bone marrow cavity with
minimum essential medium-α (α-MEM; Invitrogen, Carlsbad, CA, USA).
The cells were then centrifuged, exposed to hypotonic ACK buffer
(0.15 mM NH4Cl, 1 mM KCO3 and 0.1 mM EDTA, pH
7.4) at room temperature for 30 sec to remove red blood cells, and
incubated with α-MEM containing penicillin/streptomycin and 10%
heat-inactivated fetal bovine serum for 12 h to separate the
floating and adherent cells. Floating cells were collected,
suspended in α-MEM, counted, seeded on 24-well plates (Nalge Nunc
International, Naperville, IL, USA) at 2×105 cells/well,
and cultured in α-MEM in the presence of 30 ng/ml M-CSF for 3 days
to form macrophage-like osteoclast precursor cells. After 3 days,
adherent cells were used as BMMs after washing out the nonadherent
cells, including lymphocytes. These osteoclast precursor cells were
further cultured in the presence of 30 ng/ml M-CSF, various
concentrations of RANKL and cytokines to generate osteoclasts. The
RANKL concentrations used in the present study ranged from 1 to 100
ng/ml. Various concentrations of cytokines and RANKL were added
into the osteoclastogenesis culture system at two different
time-points; early cytokine treatment was performed with the first
RANKL administration, and late cytokine treatment was performed on
Day 2, when the second RANKL treatment was administered to the
osteoclast culture media. On Day 2, the media were replaced with
fresh medium containing M-CSF, RANKL, and cytokines. Precursor
cells began to fuse between 36 and 48 h and mature osteoclasts were
observed at 60 h after RANKL stimulation (Fig. 1).
TRAP staining
A commercial kit (catalog no. 387A; Sigma-Aldrich)
was used according to the manufacturer’s instructions, omitting the
counterstaining with hematoxylin. TRAP-positive cells containing
three or more nuclei were counted as osteoclasts. TRAP-positive
cells were counted three times without knowledge of the previous
counts of osteoclasts.
Bone resorption assay
To assess the effect of each cytokine on
RANKL-induced bone resorption, an osteoclast bone resorption assay
was performed using a commercially available bone resorption assay
kit (Cosmo Bio Co., Ltd., Tokyo, Japan). BMMs were cultured on
CaP-coated plates in the presence of M-CSF (30 ng/ml) and RANKL (50
and 100 ng/ml) in the presence or absence of various concentrations
of cytokines for 7 days. Cytokine treatment was performed at
different time-points: at 0 h from the first RANKL treatment and at
48 h after the first RANKL treatment, in accordance with previous
TRAP staining experiments. The cells were removed from the plates
(by wiping their surface), followed by staining of the slides with
toluidine blue (1 μg/ml). The resorbed areas on the plate were
visualized using light microscopy.
Cell proliferation assays
Cytotoxicity assays were conducted using a CCK-8
(Dojindo Laboratories, Kumamoto, Japan), which produces a highly
water-soluble formazan dye. Cells were seeded in 96-well plates
(5×104 cells/well, 100 μl/well) and cultured in the
presence of M-CSF, RANKL, and the indicated concentrations of
various cytokines. At the end of the culture period, 10 μl of the
CCK-8 reagent was added to each well. After 1 h of incubation at
37°C, the absorbance was determined at 450 nm using a microplate
reader (Vmax; Molecular Devices, Palo Alto, CA, USA).
Transfection and luciferase reporter gene
activity assay
For transfection, RAW264.7 cells were seeded in
6-well plates (1×106 cells/well) and transfection with
pNF-κB-Luc, pAP-1-Luc, CREB-Luc and pNFAT-Luc plasmids was
performed using FuGene® HD according to the
manufacturer’s instructions (Roche Diagnostics, Mannheim, Germany).
After 48 h, the medium was replaced with fresh medium and cells
were stimulated with various cytokines. The cells were incubated
for an additional 24 h and luciferase activity was determined using
a luciferase assay kit (Promega, Madison, WI, USA). Luciferase
activities were normalized to the corresponding β-galactosidase
levels (Promega).
Statistical analysis
Data were analyzed using the SPSS software version
16.0 (SPSS Inc., Chicago, IL, USA). Data are presented as the means
± standard deviation (SD). Results were analyzed using the
Kruskal-Wallis test, followed by the Mann-Whitney U-test. P-values
<0.05 were considered to indicate statistically significant
differences.
Results
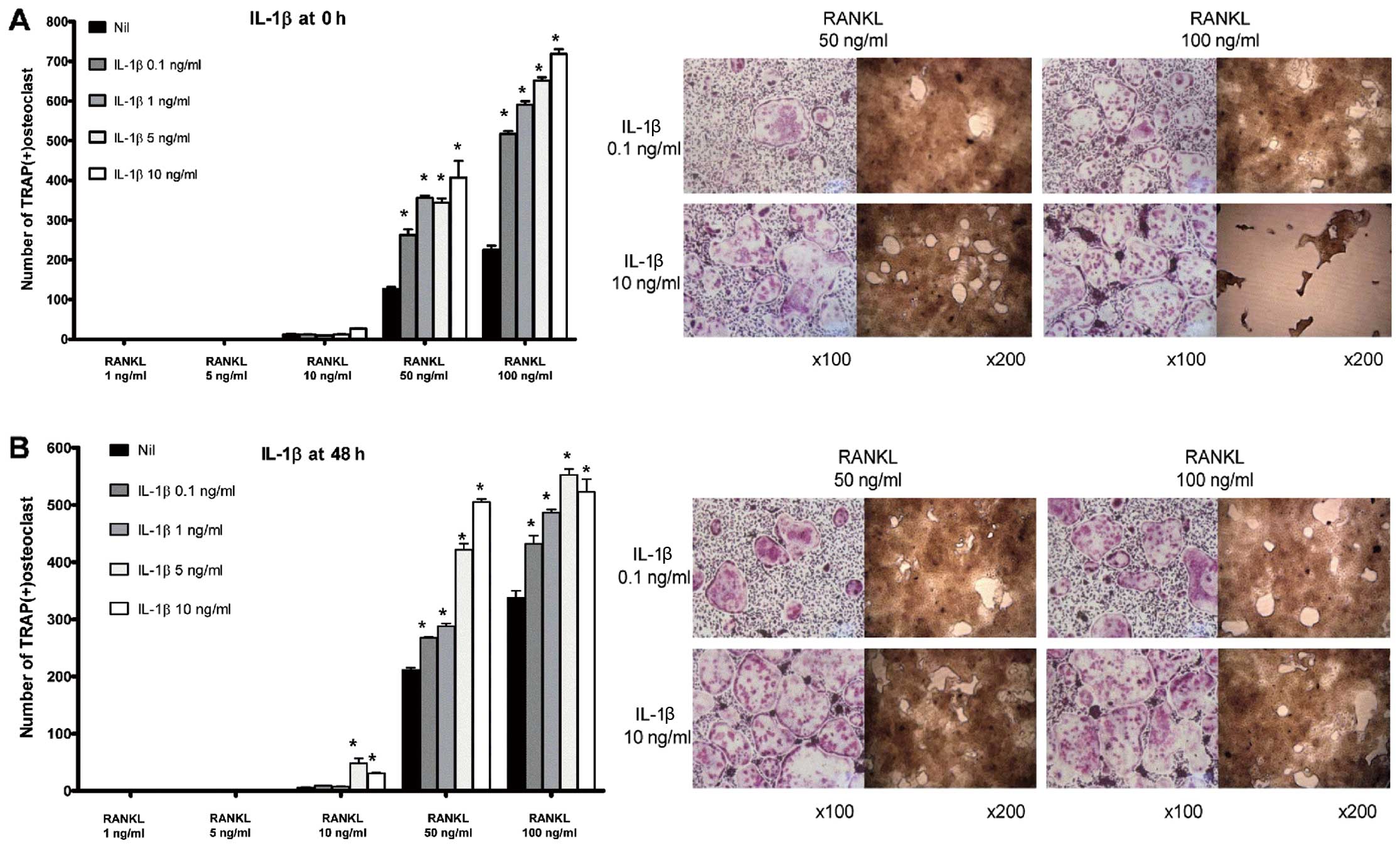
IL-1β induces osteoclastogenesis in BMMs
in a dose-dependent manner
Osteoclastogenesis was not induced either in the
absence of RANKL or in the presence of RANKL at a concentration
<50 ng/ml. Thus, a permissive level of RANKL ( ≥50 ng/ml) was
required for osteoclastogenesis. Both in the ‘early’ (Fig. 2A) and ‘late’ phases (Fig. 2B) of osteoclastogenesis, IL-1β
increased the number of osteoclasts and their bone-absorbing
ability. At all given concentrations of IL-1β, the number of
TRAP-positive multinucleated cells was increased in its presence
compared with the control group. In addition, the
pro-osteoclastogenic effect of IL-1β appeared to be dose-dependent
in both phases of osteoclastogenesis.
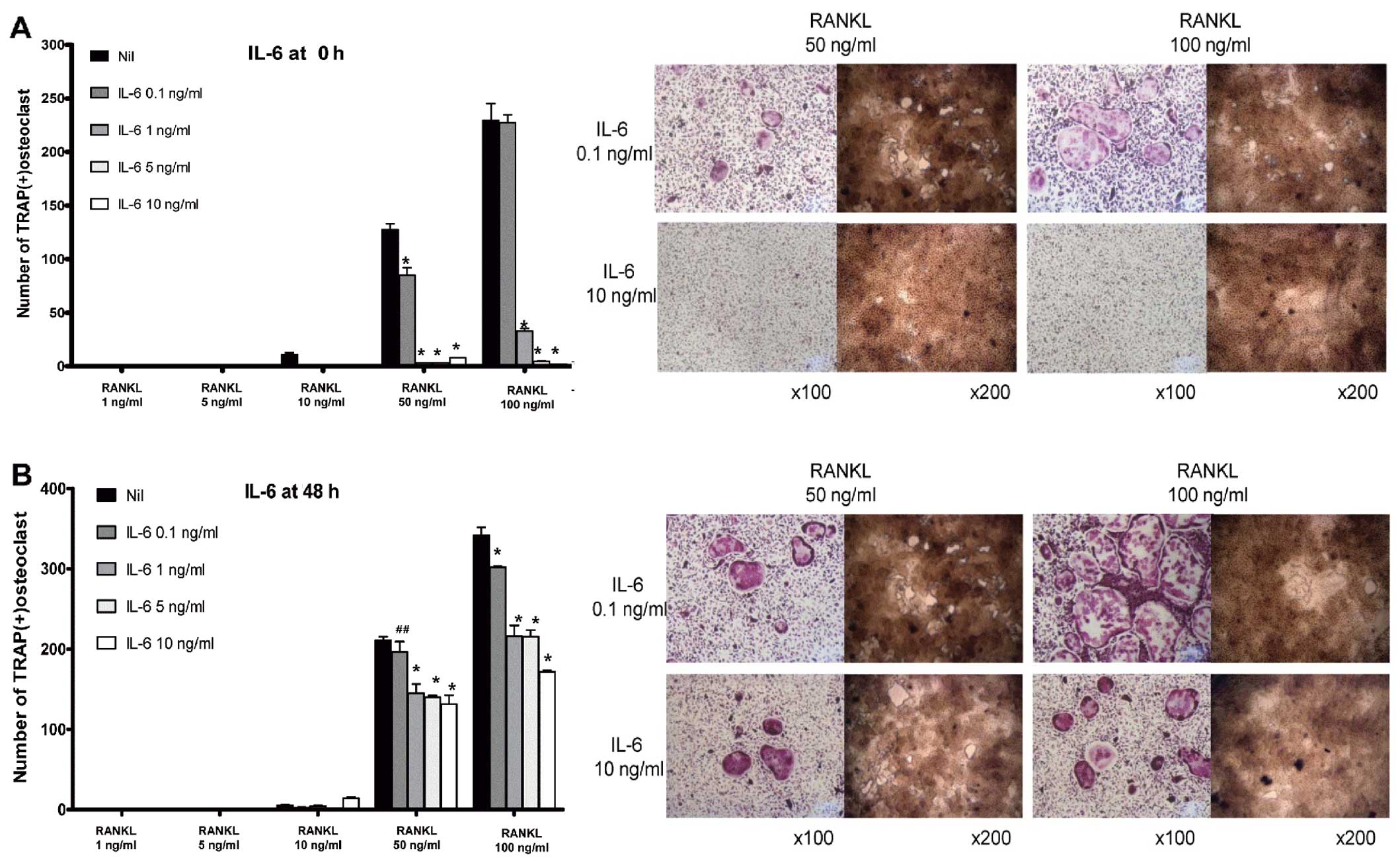
IL-6 decreases osteoclastogenesis in BMMs
in a dose-dependent manner
In contrast to IL-1β, IL-6 decreased osteoclast
formation and bone-absorption ability, both in the ‘early’ and
‘late’ phases of osteoclastogenesis. In the early phase of
osteoclastogenesis, IL-6 decreased the number of multinucleated
osteoclasts markedly from a dose of 1 ng/ml (Fig. 3A). In addition, in the late phase
of osteoclastogenesis, IL-6 reduced the number of TRAP-positive
multinucleated cells at all the given concentrations of IL-6 in a
dose-dependent manner (from 0.1 to 10 ng/ml) (Fig. 3B).
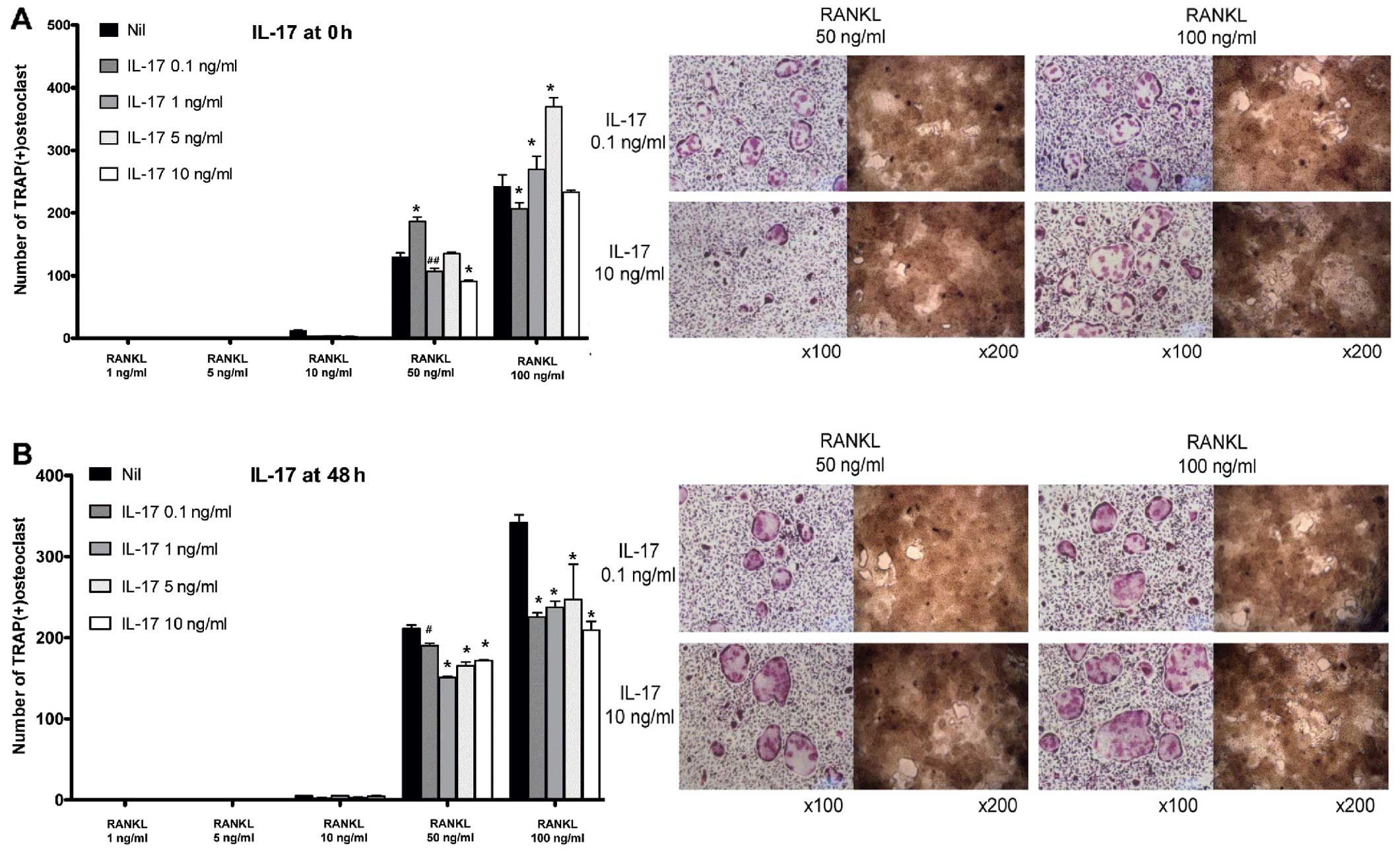
Suppressive effects of IL-17 on the late
phase of osteoclastogenesis
In BMM-induced osteoclastogenesis, IL-17 acted
differently on the time to challenge. In the early phase, IL-17 did
not yield coherent results (Fig.
4A). In certain conditions, IL-17 appeared to act as an
anti-osteoclastogenic factor, while in other conditions, IL-17
increased osteoclastogenesis. Conversely, challenge with IL-17 in
the late phase of osteoclastogenesis decreased the number of
osteoclasts at all given concentrations (from 0.1 to 10 ng/ml),
although the pattern was not dose-dependent (Fig. 4B).
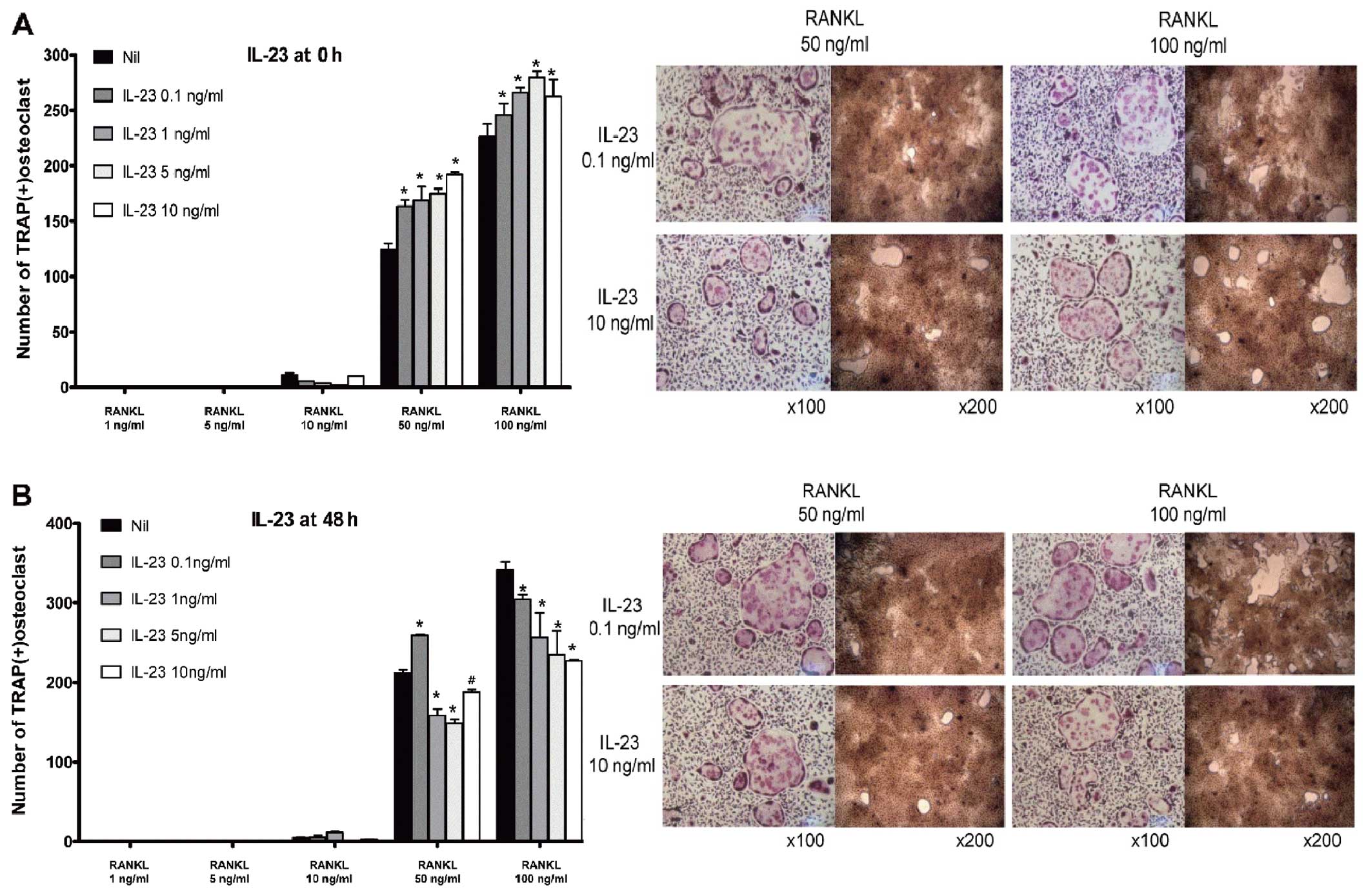
Contrary effect of IL-23 according to the
phase of osteoclastogenesis
In the early phase of osteoclastogenesis, IL-23
enhanced osteoclast formation and its bone-absorption ability in a
dose-dependent manner (Fig. 5A).
In the late phase, however, IL-23 suppressed osteoclast formation.
IL-23 exhibited a pattern that was similar to that of IL-1β when it
was applied in the late phase of osteoclastogenesis (Fig. 5B), although the osteoclastogenic
potency of IL-23 was significantly lower than that of IL-1β.
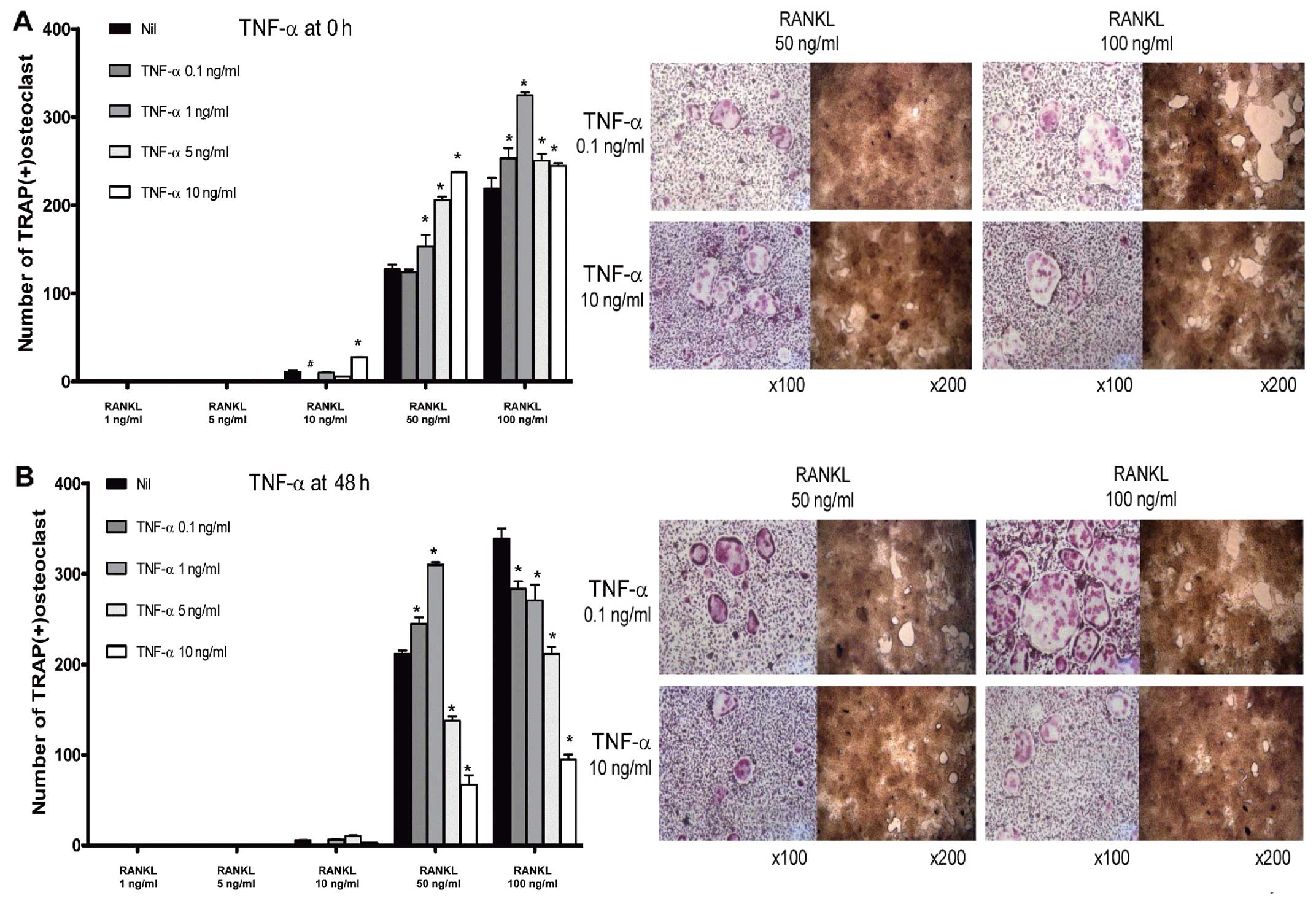
Pro-osteoclastogenic effects of TNF-α in
the early phase of osteoclastogenesis
Osteoclast precursor cells exhibited complex
responses to treatment with TNF-α. Stimulation of BMMs with a lower
concentration of RANKL (50 ng/ml) in the early phase of
osteoclastogenesis resulted in enhanced osteoclastogenesis
following the challenge with TNF-α, in a dose-dependent manner. At
a higher concentration of RANKL (100 ng/ml), TNF-α enhanced
osteoclast formation, although dose-dependency was not observed
(Fig. 6A). In the late phase,
TNF-α seemed to act as an anti-osteoclastogenic factor at a higher
concentration of RANKL (100 ng/ml). At a lower concentration of
RANKL (50 ng/ml), BMMs responded to TNF-α in a different way. At
lower concentrations of TNF-α (0.1 and 1 ng/ml), osteoclast
formation was increased. However, the number of osteoclasts was
decreased at higher doses of TNF-α (5 and 10 ng/ml) (Fig. 6B).
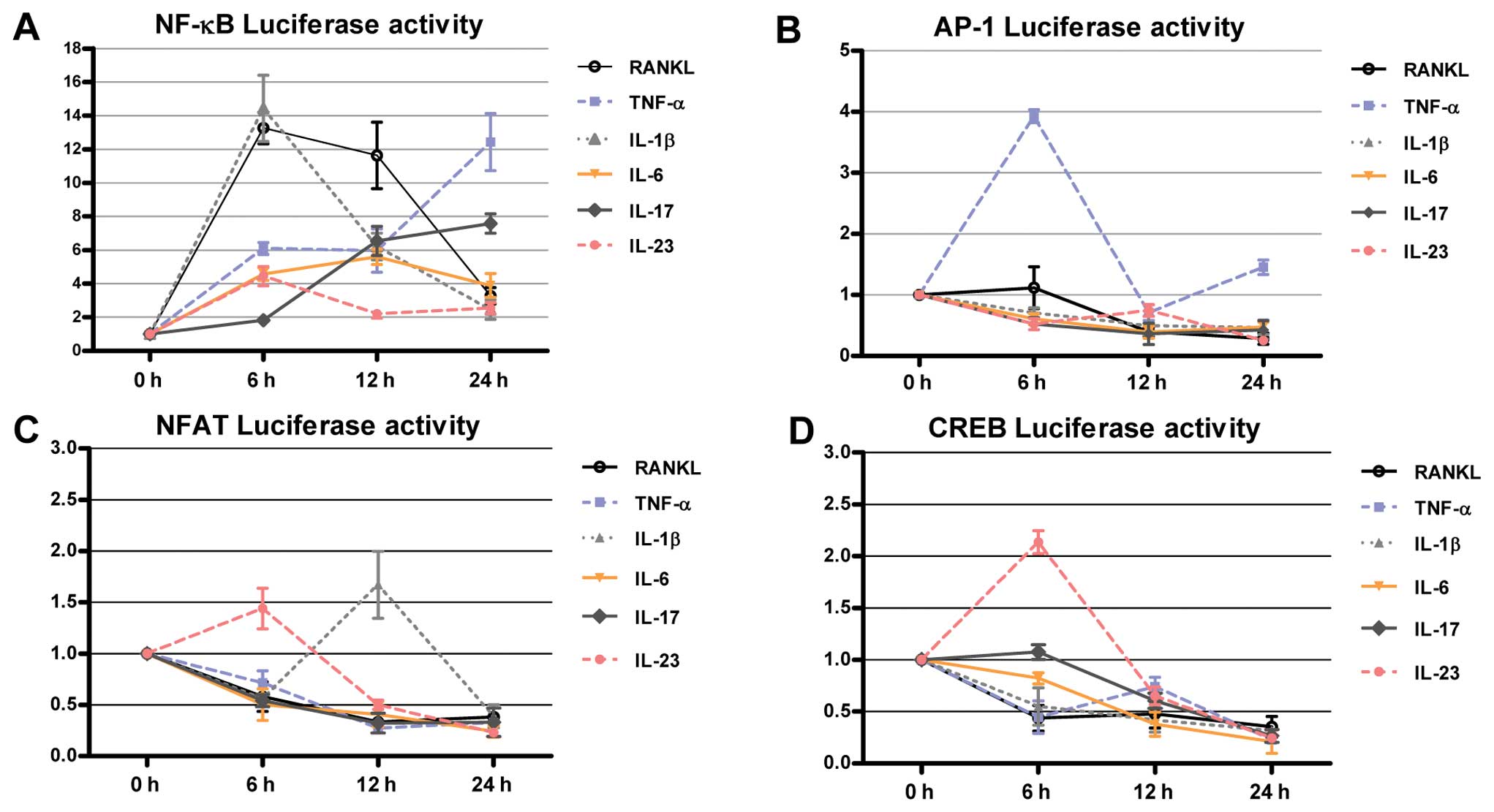
IL-1β and RANKL activate NF-κB at an
early phase, with a similar pattern
To assess the changes in NF-κB, AP-1, NFAT and CREB
activity following treatment with the cytokines mentioned above,
transient transfection was performed using luciferase constructs.
RAW264.7 cells were incubated in the presence of RANKL, TNF-α,
IL-1β, IL-6, IL-17 and IL-23. A luciferase assay was performed
serially from 6 to 24 h after stimulation with each cytokine. IL-1β
yielded an early increase of NF-κB, mirroring the RANKL-induced
signaling pathway (Fig. 7A).
Treatment with other cytokines also increased NF-κB activity,
although the degree of this increase and the peak time-points
varied. Other cytokines exhibited specific patterns of activation
of the signaling pathway (Fig.
7B-D).
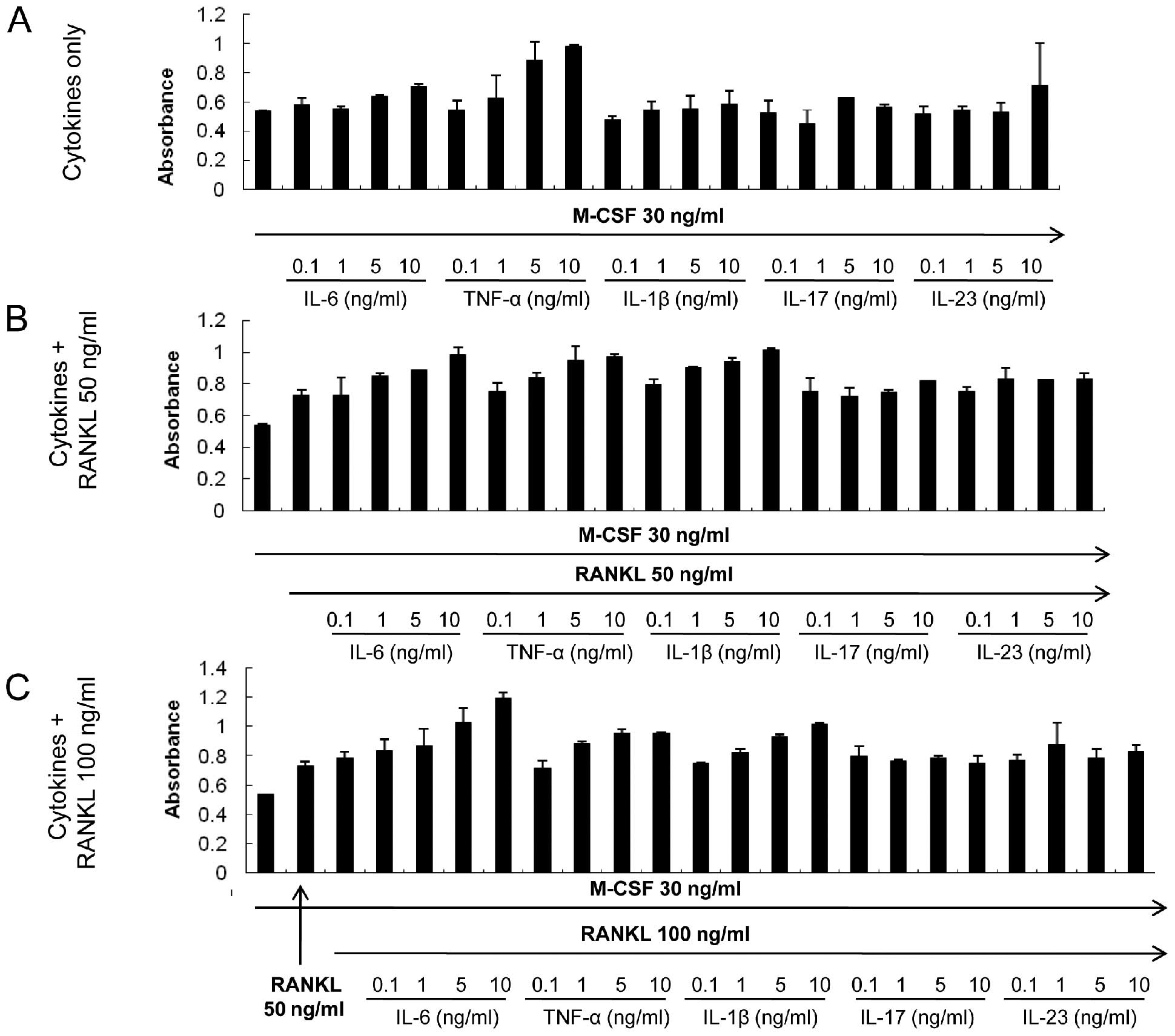
Treatment with the cytokines does not
have cytotoxic effects
A suppressive effect on osteoclastogenesis was
observed for some cytokines: IL-6, IL-17, IL-23 and TNF-α. CCK-8
assay was performed to ensure that the inhibitory effect of those
cytokines was not due to enhanced apoptosis or cytotoxicity. None
of the cytokines resulted in apoptotic or toxic effects in BMMs at
the concentrations used in the present experiments (Fig. 8). Moreover, IL-6 stimulated the
proliferation of BMMs in a dose-dependent manner, a tendency that
was augmented with increasing RANKL concentrations (Fig. 8B and C).
Discussion
In the present study, we investigated the effect of
several cytokines on osteoclastogenesis using a monocellular
culture system, and clarified whether the proinflammatory cytokines
IL-1β, IL-6, IL-17, IL-23 and TNF-α have a similar effect on
osteoclastogenesis, as they are known to be similar types of
proinflammatory cytokines. However, our findings demonstrated that
IL-1β increased the number of osteoclasts and their bone-absorption
ability, whereas IL-6 yielded the opposite effect.
One of the challenges of comparing certain
characteristics of a molecule with accuracy, is that a strict
control of confounding factors is required. Even if the features of
the molecules are successfully compared, applying this approach to
an in vivo system and in physiological conditions is
difficult. To minimize the confounding factors and to determine the
pure effect of cytokines on osteoclastogenesis, we adopted a
monocellular culture system using only osteoclast precursor cells
derived from mouse bone marrow. A coculture system is composed of
osteoblasts and bone marrow cells stimulated with vitamin D3 and
prostaglandin E2 (22). The
existence of various cell components other than precursor cells
produces several confusing variables that do not reflect the pure
effect of cytokines.
The cytokine milieu determines the effects of
inflammation on bone. There is extensive research on the role of
proinflammatory cytokines in bone homeostasis (23–25). Blockade of the inflammatory
cytokines results in the control of chronic inflammation as well as
in protective effects on bone. In general, TNF-α, IL-1β and IL-6
promote bone resorption via either the direct or indirect promotion
of osteoclastogenesis. TNF-α inhibition has been successful in the
therapy of patients with RA, yielding control of inflammation as
well as retardation of structural changes in the joints involved
(26). Moreover, suppression of
IL-1 and IL-6 resulted in the marked suppression of inflammation
and joint destruction in patients with RA (27–29). The IL-23/IL-17 axis was previously
demonstrated to be a potential linker between inflammation and bone
loss. Inhibition of IL-23 and IL-17 attenuated bone erosion in an
animal model of arthritis (30,31). Based on the beneficial effects of
anticytokine treatments observed in human and animal trials, it
could be assumed that the above-mentioned cytokines have
osteoclastogenic properties. However, the role of proinflammatory
cytokines on osteoclast precursor cells remains unclear.
Therefore, we determined whether the various
cytokines have different effects on osteoclast precursor cells in
the context of their maturation stages. Two different time-points
were selected for cytokine stimulation in osteoclast precursor
cells; the first was set at an early time after RANKL stimulation,
when precursor cells had not yet initiated the differentiation
process, and the other was set at a later time (48 h after the
initial stimulation with RANKL), when osteoclast precursor cells
had initiated the differentiation process, i.e., ‘committed
osteoclasts’. IL-1β and IL-6 had a consistent effect on precursor
cells, regardless of maturation status. IL-23 and TNF-α yielded
different responses depending on maturation status. IL-23 and TNF-α
may be helpful for osteoclastogenesis when the fate of precursor
cells is not committed.
We postulated that a decreasing number of mouse BMMs
may reflect the suppressive effect of some cytokines, such as IL-6
and TNF-α, due to cytotoxic effects of cytokines and apoptosis. A
CCK-8 assay demonstrated that cytokines did not induce apoptosis or
cellular toxicity at the given concentrations. The suppressive
tendency of some cytokines, such as IL-17, IL-23 and TNF-α, may
have also resulted from a disruption in the balance of BMMs, with a
shift towards activated macrophages. If a larger proportion of BMMs
is differentiated into activated macrophages in response to
cytokine stimulation, a smaller proportion of candidate cells can
fuse and form mature osteoclasts, leading to a decreased number of
osteoclasts.
The mechanism underlying the pro-osteoclastogenic
potential of IL-1β could be partly explained by the sharing of the
early NF-κB signaling pathway with RANKL. RANKL and IL-1β led to
the early upregulation of NF-κB in RAW264.7 cells. NFAT is a
candidate molecule that could be activated by RANKL stimulation.
However, stimulation by RANKL did not lead to the activation of the
signaling NFAT. Therefore, we analyzed NFAT signaling again by
dissecting it into NFATc1 and NFATc2. Notably, NFATc1 and NFATc2
exhibited reciprocally activating patterns (data not shown). At the
early time-point of 6 h, the NFATc1/NTATc2 ratio increased
following RANKL stimulation. At the later time-point of 24 h, the
ratio was reversed. RANKL also decreased the temporal ratio of
NFATc1 (24/6 h) and increased the ratio of NFATc2 (24/6 h) in a
dose-dependent manner. These results indicate that NFATc1 plays an
important role during early RANKL-induced osteoclastogenesis.
In the present study, we demonstrated that cytokines
have specific characteristic osteoclastogenic properties. Cytokines
exhibited their osteoclastogenesis-related activity only when a
permissive level of RANKL existed. IL-1β enhanced
osteoclastogenesis, regardless of RANKL concentration or the
maturation status of precursor cells. IL-6 interrupted the process
of osteoclastogenesis in osteoclast precursor cells under all the
given conditions. IL-17 slightly favored a suppressive effect in
the late stage of osteoclastogenesis. IL-23 and TNF-α appeared to
have osteoclastogenic properties in the early stage of
osteoclastogenesis, when BMMs are ‘just’ osteoclast precursor
cells.
In conclusion, IL-1β was the most potent
osteoclastogenic cytokine among the proinflammatory molecules
studied, and IL-6 disturbed osteoclast formation, as well as the
bone-absorption ability of these cells. We consider this a notable
phenomenon, as shown in in vitro experiments. Nevertheless,
our in vitro results should not be extended for more complex
in vivo biological systems.
Acknowledgements
This study was supported by the Basic Science
Research Program through the National Research Foundation of Korea
(NRF) funded by the Ministry of Education, Science and Technology
(331-2008-1-E00144 and 2009-0074198) and a grant from the National
Project for Personalized Genomic Medicine, Ministry for Health
& Welfare, Republic of Korea (A111218-PG01).
References
|
1
|
Sato K and Takayanagi H: Osteoclasts,
rheumatoid arthritis, and osteoimmunology. Curr Opin Rheumatol.
18:419–426. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Schett G: Osteoimmunology in rheumatic
diseases. Arthritis Res Ther. 11:2102009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gravallese EM, Manning C, Tsay A, Naito A,
Pan C, Amento E and Goldring SR: Synovial tissue in rheumatoid
arthritis is a source of osteoclast differentiation factor.
Arthritis Rheum. 43:250–258. 2000.PubMed/NCBI
|
|
4
|
Martin TJ and Ng KW: Mechanisms by which
cells of the osteoblast lineage control osteoclast formation and
activity. J Cell Biochem. 56:357–366. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Alnaeeli M, Penninger JM and Teng YT:
Immune interactions with CD4+ T cells promote the
development of functional osteoclasts from murine CD11c+
dendritic cells. J Immunol. 177:3314–3326. 2006.PubMed/NCBI
|
|
6
|
Choi Y, Woo KM, Ko SH, Lee YJ, Park SJ,
Kim HM and Kwon BS: Osteoclastogenesis is enhanced by activated B
cells but suppressed by activated CD8(+) T cells. Eur J Immunol.
31:2179–2188. 2001.
|
|
7
|
Xing L, Schwarz EM and Boyce BF:
Osteoclast precursors, RANKL/RANK, and immunology. Immunol Rev.
208:19–29. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cappellen D, Luong-Nguyen NH, Bongiovanni
S, Grenet O, Wanke C and Susa M: Transcriptional program of mouse
osteoclast differentiation governed by the macrophage
colony-stimulating factor and the ligand for the receptor activator
of NFkappa B. J Biol Chem. 277:21971–21982. 2002. View Article : Google Scholar
|
|
9
|
Kim KW, Cho ML, Lee SH, Oh HJ, Kang CM, Ju
JH, Min SY, Cho YG, Park SH and Kim HY: Human rheumatoid synovial
fibroblasts promote osteoclastogenic activity by activating RANKL
via TLR-2 and TLR-4 activation. Immunol Lett. 110:54–64. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Horwood NJ, Kartsogiannis V, Quinn JM,
Romas E, Martin TJ and Gillespie MT: Activated T lymphocytes
support osteoclast formation in vitro. Biochem Biophys Res Commun.
265:144–150. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pettit AR, Walsh NC, Manning C, Goldring
SR and Gravallese EM: RANKL protein is expressed at the pannus-bone
interface at sites of articular bone erosion in rheumatoid
arthritis. Rheumatology. 45:1068–1076. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Schett G, Hayer S, Zwerina J, Redlich K
and Smolen JS: Mechanisms of disease: the link between RANKL and
arthritic bone disease. Nat Clin Pract Rheumatol. 1:47–54. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang YH, Heulsmann A, Tondravi MM,
Mukherjee A and Abu-Amer Y: Tumor necrosis factor-alpha (TNF)
stimulates RANKL-induced osteoclastogenesis via coupling of TNF
type 1 receptor and RANK signaling pathways. J Biol Chem.
276:563–568. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
O’ Gradaigh D, Ireland D, Bord S and
Compston JE: Joint erosion in rheumatoid arthritis: interactions
between tumour necrosis factor alpha, interleukin 1, and receptor
activator of nuclear factor kappaB ligand (RANKL) regulate
osteoclasts. Ann Rheum Dis. 63:354–359. 2004.
|
|
15
|
Tamura T, Udagawa N, Takahashi N, Miyaura
C, Tanaka S, Yamada Y, Koishihara Y, Ohsugi Y, Kumaki K, Taga T, et
al: Soluble interleukin-6 receptor triggers osteoclast formation by
interleukin 6. Proc Natl Acad Sci USA. 90:11924–11928. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lubberts E, van den Bersselaar L,
Oppers-Walgreen B, Schwarzenberger P, Coenen-de Roo CJ, Kolls JK,
Joosten LA and van den Berg WB: IL-17 promotes bone erosion in
murine collagen-induced arthritis through loss of the receptor
activator of NF-kappa B ligand/osteoprotegerin balance. J Immunol.
170:2655–2662. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ju JH, Cho ML, Moon YM, Oh HJ, Park JS,
Jhun JY, Min SY, Cho YG, Park KS, Yoon CH, Min JK, Park SH, Sung YC
and Kim HY: IL-23 induces receptor activator of NF-kappaB ligand
expression on CD4+ T cells and promotes
osteoclastogenesis in an autoimmune arthritis model. J Immunol.
181:1507–1518. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kobayashi K, Takahashi N, Jimi E, Udagawa
N, Takami M, Kotake S, Nakagawa N, Kinosaki M, Yamaguchi K, Shima
N, Yasuda H, Morinaga T, Higashio K, Martin TJ and Suda T: Tumor
necrosis factor alpha stimulates osteoclast differentiation by a
mechanism independent of the ODF/RANKL-RANK interaction. J Exp Med.
191:275–286. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kim JH, Jin HM, Kim K, Song I, Youn BU,
Matsuo K and Kim N: The mechanism of osteoclast differentiation
induced by IL-1. J Immunol. 183:1862–1870. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kudo O, Sabokbar A, Pocock A, Itonaga I,
Fujikawa Y and Athanasou NA: Interleukin-6 and interleukin-11
support human osteoclast formation by a RANKL-independent
mechanism. Bone. 32:1–7. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lam J, Takeshita S, Barker JE, Kanagawa O,
Ross FP and Teitelbaum SL: TNF-alpha induces osteoclastogenesis by
direct stimulation of macrophages exposed to permissive levels of
RANK ligand. J Clin Invest. 106:1481–1488. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Akatsu T, Takahashi N, Debari K, Morita I,
Murota S, Nagata N, Takatani O and Suda T: Prostaglandins promote
osteoclastlike cell formation by a mechanism involving cyclic
adenosine 3′,5′-monophosphate in mouse bone marrow cell cultures. J
Bone Miner Res. 4:29–35. 1989.PubMed/NCBI
|
|
23
|
Baker-LePain JC, Nakamura MC and Lane NE:
Effects of inflammation on bone: an update. Curr Opin Rheumatol.
23:389–395. 2011. View Article : Google Scholar
|
|
24
|
Brennan FM and McInnes IB: Evidence that
cytokines play a role in rheumatoid arthritis. J Clin Invest.
118:3537–3545. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Walsh NC, Crotti TN, Goldring SR and
Gravallese EM: Rheumatic diseases: the effects of inflammation on
bone. Immunol Rev. 208:228–251. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tracey D, Klareskog L, Sasso EH, Salfeld
JG and Tak PP: Tumor necrosis factor antagonist mechanisms of
action: a comprehensive review. Pharmacol Ther. 117:244–279. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Furst DE: Anakinra: review of recombinant
human interleukin-I receptor antagonist in the treatment of
rheumatoid arthritis. Clin Ther. 26:1960–1975. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gabay C, Lamacchia C and Palmer G: IL-1
pathways in inflammation and human diseases. Nat Rev Rheumatol.
6:232–241. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Fonseca JE, Santos MJ, Canhao H and Choy
E: Interleukin-6 as a key player in systemic inflammation and joint
destruction. Autoimmun Rev. 8:538–542. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yago T, Nanke Y, Kawamoto M, Furuya T,
Kobashigawa T, Kamatani N and Kotake S: IL-23 induces human
osteoclastogenesis via IL-17 in vitro, and anti-IL-23 antibody
attenuates collagen-induced arthritis in rats. Arthritis Res Ther.
9:R962007. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Koenders MI, Lubberts E, Oppers-Walgreen
B, van den Bersselaar L, Helsen MM, Di Padova FE, Boots AM, Gram H,
Joosten LA and van den Berg WB: Blocking of interleukin-17 during
reactivation of experimental arthritis prevents joint inflammation
and bone erosion by decreasing RANKL and interleukin-1. Am J
Pathol. 167:141–149. 2005. View Article : Google Scholar : PubMed/NCBI
|