Introduction
Cirrhosis, which is induced by various factors such
as chronic viral hepatitis, autoimmune hepatitis, fatty liver
disease, or hereditary metabolic disorders, is a major cause of
morbidity and mortality worldwide (1–4).
Due to the stimulation of hepatocellular damage and inflammation,
cirrhosis is characterized by the remodeling of liver tissues with
excess deposition of extracellular matrix (ECM) components such as
collagens. Liver fibrosis is the intermediate link in the
progression from chronic liver disease to cirrhosis. Therefore,
early prevention or reversion of liver fibrosis is key in improving
the prognosis of various chronic liver diseases. In the progression
of liver fibrosis, hepatic stellate cells (HSCs), which may be the
most important cell type for the production of collagens, are
considered to be responsible for the accumulation of ECM proteins
(4). HSCs become activated in
response to inflammatory stimuli and undergo myofibroblastic
transdifferentiation. Therefore, a possible therapeutic strategy is
to treat liver fibrosis by suppressing the activation of HSCs.
microRNAs (miRNAs) are a class of small,
evolutionarily conserved, non-coding RNAs, which suppress the
expressions of protein coding genes by base pairing with the 3′
untranslated region (3′UTR) of their target messenger RNAs (mRNAs)
(5). miRNAs can repress gene
expression by either of two mechanisms: target mRNAs are degraded
by perfect or nearly perfect pairing, or translational repression
is caused by the imperfect pairing. To date, the number of known
miRNAs has grown exponentially, and more than 1,000 miRNAs are
known to be encoded by the human genome (6), half of which have been
experimentally validated (7).
miRNAs have been associated with numerous basic cellular processes
such as cell death, proliferation and differentiation (8–11).
Furthermore, the dysregulation of miRNAs has also been correlated
with a wide spectrum of human diseases such as cancer (12). For example, miR-17-5p is found to
be overexpressed in hepatocellular carcinoma (HCC) and increased
miR-17-5p expression levels are correlated with poor prognosis of
patients with HCC (13,14).
In addition, a number of recent reviews have shown
that miRNA dysregulation is involved in the activation of HSCs. The
reduced expressions of miRNA-150 and miRNA-194 are found in
activated HSCs, and their overexpressions inhibit the activation
and proliferation of HSCs (15).
On the contrary, reduced expressions of miR-27a and -27b allow
culture-activated rat HSCs to return to a quiescent phenotype with
abundant vitamin A storage and decreased cell proliferation
(16). Furthermore, miR-214-5p is
upregulated in human and mouse livers in a fibrosis
progression-dependent manner and the overexpression of miR-214-5p
in LX-2 cells increases the expressions of fibrosis-related genes,
such as matrix metal-loproteinase-2 (MMP-2), MMP-9, α-smooth muscle
actin (α-SMA), and transforming growth factor-β1 (TGF-β1) (17). However, little is known about the
potential molecular mechanism of these miRNAs in liver
fibrosis.
In the present study, we demonstrated that miR-150
was significantly downregulated in LX-2 cells treated with TGF-β1
when compared with the control. It was confirmed that the
overexpression of miR-150 inhibited the activation and
proliferation of HSCs with no effect on cell survival. In addition,
we found that miR-150 was a potential regulator of type IV collagen
protein expression via its interaction with Col4A4 3′UTR. Our data
also suggested that miR-150 may be a regulator of type I collagen
expression through the Sp1 signal pathway.
Materials and methods
Cell culture
The human LX-2 cell strain was obtained from JENNIO
Biological Technology, Guangdong, China. It was cultured in DMEM
containing 10% fetal bovine serum, 100 U/ml penicillin G sodium
salt and 100 U/ml streptomycin sulfate (Gibco, Carlsbad, CA, USA).
The cells were grown in a 37°C incubator with 5% CO2.
Exponentially growing cells were seeded in a 6-well plate at a
density of 1×106 cells/well and were then transfected
with the miR-150 mimics or a negative control (Shanghai GenePharma
Co., Ltd., Shanghai, China) for 24 h. Cells were also treated with
TGF-β1 (R&D Systems, Shanghai, China) for different experiment
purposes. Cells were harvested for RNA/miRNA isolation, and whole
cell extracts were subjected to western blot analysis.
Exiqon miRCURY LNA™ miRNA array
Total RNA of HSCs was extracted and labeled with a
miRCURY Hy3/Hy5 labeling kit (Exiqon, Vedbaek, Denmark). Then the
labeled samples were hybridized to the miRCURY LNA array version
8.0 (Exiqon). The hybridization was performed following the miRCURY
LNA array manual. Following hybridization, the slides were washed
by Wash buffer kit (Exiqon), dried and scanned on a GenePix 4000B
array scanner (Molecular Devices Co., Sunnyvale, CA, USA). The
results were carried out using unsupervised hierarchical clustering
(Cluster 3.0) and TreeView analysis (Stanford University, Stanford,
CA, USA).
Quantitative real-time PCR
Total RNA was extracted from LX-2 cells using the
miRNeasy Mini Kit (Qiagen, Valencia, CA, USA). Also, fifty
nanograms of total RNA were reverse-transcribed to cDNA using the
ReverTra Ace® qPCR RT Kit (Toyobo, Osaka, Japan) in
accordance with the manufacturer’s instructions. Gene expression
was measured by real-time PCR using cDNA, SYBR-Green real-time PCR
Master Mix (Toyobo), and a set of gene-specific oligonucleotide
primers; α-1 (I) collagen (Col1A1), forward,
5′-CCCGGGTTTCAGAGACAACTTC-3′ and reverse, 5′-TCCACATGCTTTATTCCAG
CAATC-3′; α-2 (I) collagen (Col1A2), forward,
5′-AAGGGTCCCTCTGGAGAACC-3′ and reverse, 5′-TCTAGAGCCAGGGAGACCCA-3′;
α-4 (IV) collagen (Col4A4), forward, 5′-TGAAGGGAAATCCCGGTGTG-3′ and
reverse, 5′-CAGGTGGCTCTACCAACAGG-3′; α-SMA, forward,
5′-TCGGATGAGCTACAGAGGCACAA-3′, and reverse,
5′-GTCACTCCTCATGAAGCGCTTAGG-3′; Sp1, forward,
5′-TCGGATGAGCTACAGAGGCACAA-3′ and reverse,
5′-GTCACTCCTCATGAAGCGCTTAGG-3′; GAPDH, forward,
5′-GCACCGTCAAGGCTGAGAAC-3′, and reverse, 5′-TGGTGAAGACGCCAGTGGA-3′;
U6, forward, 5′-CTCGCTTCGGCAGCACA-3′ and reverse,
5′-AACGCTTCACGAATTTGCGT-3′. To detect miR-150 expression, the RT
reaction was performed using the TaqMan microRNA Assay (Applied
Biosystems, Foster City, CA, USA) according to the manufacturer’s
instructions. The GAPDH and U6 snRNA (Applied Biosystems) levels
were measured and used to normalize the relative abundance of mRNAs
and miRNAs, respectively. The expression level (2−ΔΔCt)
of miR-150 was calculated as previously described (18).
Protein extraction and western blot
assay
Proteins were subjected to sodium dodecy1
sulfate-polyacrylamide gel electrophoresis and then transferred
onto Immobilon-P membranes. After blocking, the membranes were
incubated with primary antibodies [mouse monoclonal antibody
against α-SMA (ab5694) and rabbit polyclonal antibody against type
IV collagen (ab6586); rabbit polyclonal antibody against type I
collagen (ab34710) and GAPDH (ab9485); mouse monoclonal antibody
against Smad2 (ab119907) and rabbit polyclonal antibody against
phospho-Smad2 (p-Smad2, ab53100); rabbit polyclonal antibody
against Smad3 (ab40854) and p-Smad3 (ab52903); mouse monoclonal
antibody against Sp1 (ab77441) and goat polyclonal antibody against
C-myb (ab62824) (Abcam, Cambridge, MA, USA)] followed by
peroxidase-conjugated secondary antibodies (Fuzhou Maixin
Biological Technology Co., Ltd., Fujian, China). The
antigen-antibody complex was developed by enhanced
chemiluminescence, exposed in the dark room and analyzed for
integral absorbance (IA) of the protein bands using quantitative
software, Quantity One 4.4.
Proliferation and apoptosis assays
Cells were seeded in a 96-well plate at a density of
1×103 cells/well and the cells were then transfected
with the miR-150 mimics or a negative control for 24 h before the
assessment of the proliferation and apoptosis. Cell proliferation
was determined by MTT assay according to the instructions of an MTT
cell proliferation assay kit (Beyotime Institute of Biotechnology).
The optical density (OD) was measured at 570 nm on a 550 Microplate
Reader (Bio-Rad, USA). The DNA fragmentation for cell apoptosis was
measured using a DNA/histone-complex ELISA kit (Roche) as described
(19). The OD at 490 nm was
measured.
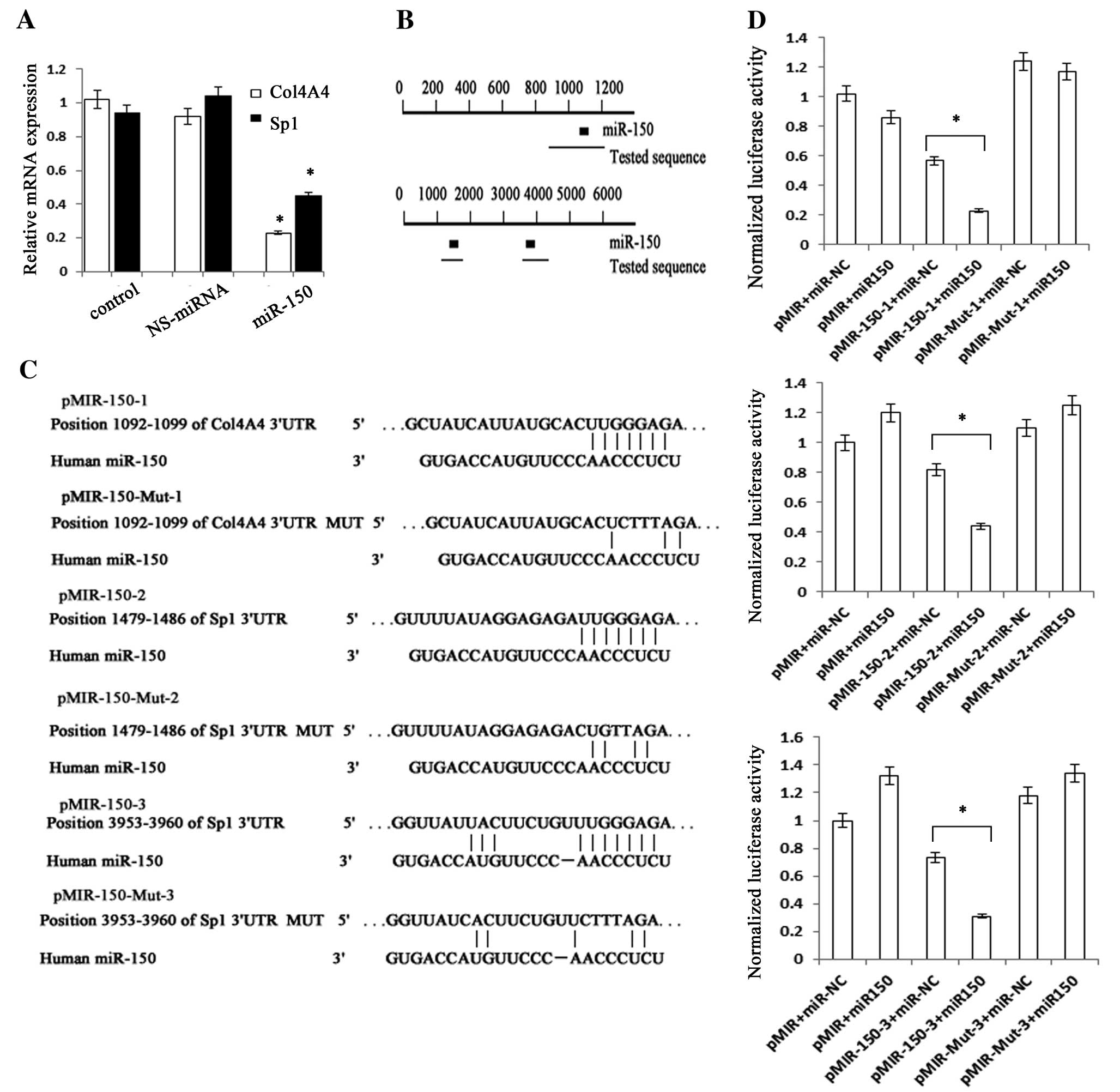
Luciferase activity assay
According to the targetscan analysis,
oligonucleotides, which contained human Col4A4 and Sp1 3′UTR target
sequence, were annealed and cloned into the pMIR-Report™ Luciferase
plasmid (Applied Biosystems) following the manufacturer’s protocol
to generate pMIR-150 vectors including pMIR-Col4A4-150 and
pMIR-Sp1-150. Col4A4-3′UTR for miR-150 (position of 1092–1099)
forward, 5′-GTGCGTGCTAATGGGACTGA-3′ and reverse,
5′-GGATGCTCCTGTAACAGCCA-3′. Sp1-3′UTR for miR-150 (position of
1479–1486) forward, 5′-TGATGTGTGGGCTTCTGAGT-3′ and reverse,
5′-ATGCTTTTATGGCTGGGCCT-3′. Sp1-3′UTR for miR-150 (position of
3953–3960) forward, 5′-AAGGTCGCAGCAGTAGCTTT-3′ and reverse,
5′-GAGACAAGGAAGACTGGGGC-3′. Empty vector pMIR without the inserts
was used as a negative control. pMIR-Report β-gal control plasmid
was used for transfection normalization. LX-2 cells were cultured
in 24-well plates and transfected with 800 ng of pMIR-150 or pMIR
together with 100 ng of pMIR-β-gal and 20 pmol of miR-150 precursor
or miRNA negative control (miR-NC) (Shanghai GenePharma Co., Ltd.,
Shanghai, China). Lipofectamine 2000 (Invitrogen, Carlsbad, CA,
USA) was used for transfection. Forty-eight hours after
transfection, luciferase and β-gal activity were measured using the
Dual-Light System (Applied Biosystems).
Statistical analysis
Data from at least three independent experiments
were expressed as the means ± SD. Statistical analysis was
performed using Student’s t-test and P<0.05 was considered to
indicate a statistically significant difference.
Results
TGF-β1 causes differential expression of
miRNAs in cultured LX-2 cells
TGF-β1 is considered to be the most important
mediator to induce HSC transformation into myofibroblasts and
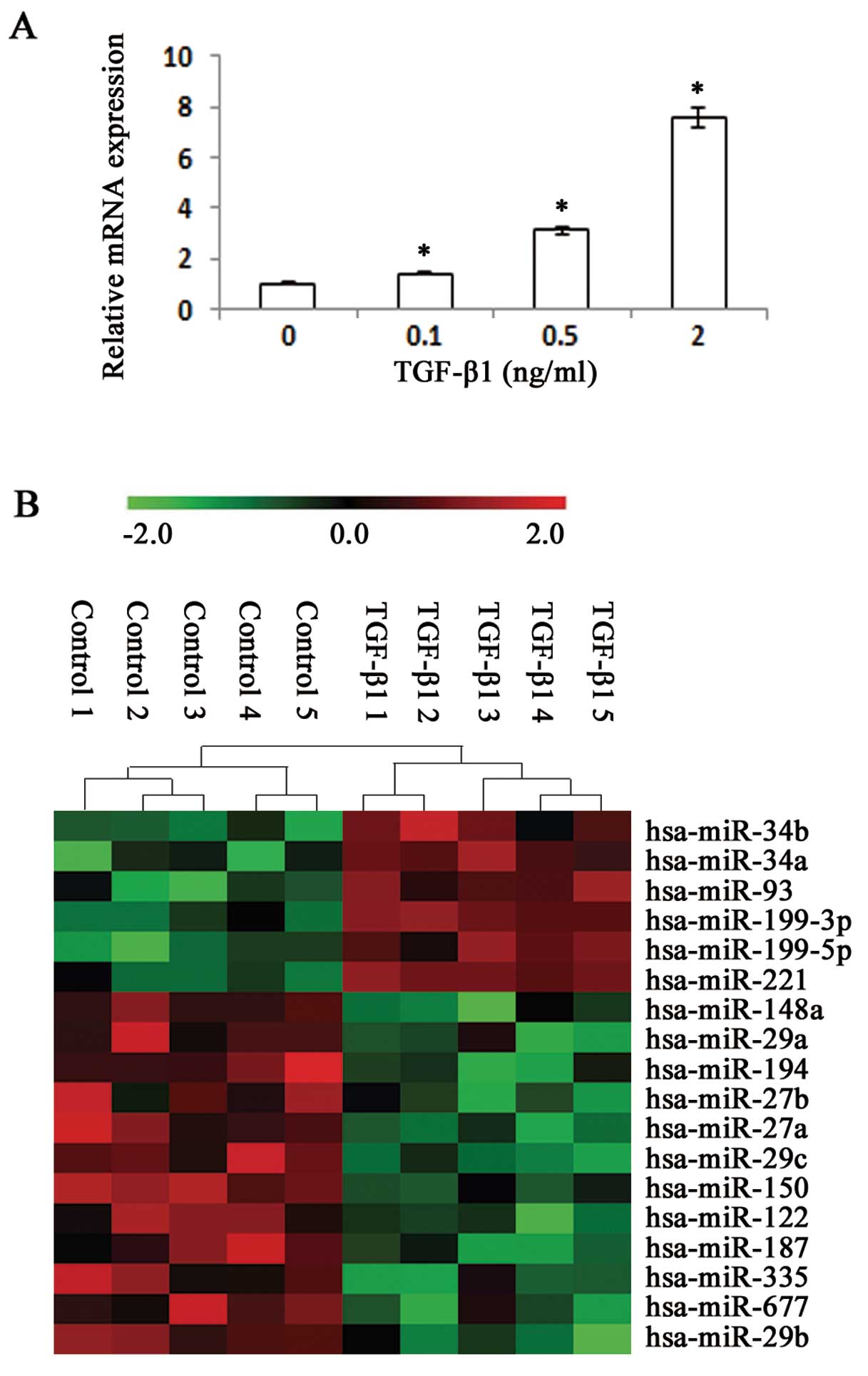
upregulate expression of collagen proteins (20). The expression of Col1A1 mRNA was
increased dose-dependently by TGF-β1 in cultured LX-2 cells
(Fig. 1A). To screen
differentially expressed miRNAs induced by TGF-β1, we performed
microarray analysis on RNA extracts from LX-2 cells with TGF-β1 and
the control. miRNAs were considered differentially expressed
according to the following standards: the differences of miRNA
expression levels showed significance both in unpaired Student’s
t-test (P<0.01) and significance analysis of microarray test
(q-value <5%). The expression of several miRNAs was noted to be
altered between cultured LX-2 cells with TGF-β1 and the control
(Fig. 1B and Table I). miRNA-150 was among the most
significantly altered miRNAs and its expression level was confirmed
by quantitative real-time PCR (Fig.
2). Additionally, although there are a few studies on the
involvement of miR-150 in liver fibrosis, the role of miR-150 in
liver fibrosis remains unclear. Hence, we regarded miR-150 as the
object of subsequent experiments.
 | Table I.Log2 (TGF-β1/control) ratio
of differentially expressed miRNAs that reached statistical
significance by t-test (P<0.01) and further confirmed by SAM
test (q<5%). |
Table I.
Log2 (TGF-β1/control) ratio
of differentially expressed miRNAs that reached statistical
significance by t-test (P<0.01) and further confirmed by SAM
test (q<5%).
| miRNA | Log2
(TGF-β1/control) ratio | P-value |
|---|
| Upregulated miRNAs
(n=6) |
| miR-221 | 1.61 | 0.0007 |
| miR-199a-3p | 1.53 | 0.0063 |
| miR-34a | 1.21 | 0.0024 |
| miR-199a-5p | 0.82 | 0.0029 |
| miR-34b | 0.63 | 0.0012 |
| miR-93 | 0.54 | 0.0041 |
| Downregulated
miRNAs (n=12) |
| miR-150 | −1.86 | 0.0001 |
| miR-29b | −1.56 | 0.0017 |
| miR-148a | −1.32 | 0.0028 |
| miR-122 | −1.15 | 0.0005 |
| miR-29c | −0.97 | 0.0029 |
| miR-677 | −0.75 | 0.0077 |
| miR-29a | −0.74 | 0.0089 |
| miR-187 | −0.58 | 0.0068 |
| miR-335 | −0.56 | 0.0066 |
| miR-194 | −0.48 | 0.0021 |
| miR-27a | −0.48 | 0.0003 |
| miR-27b | −0.48 | 0.0040 |
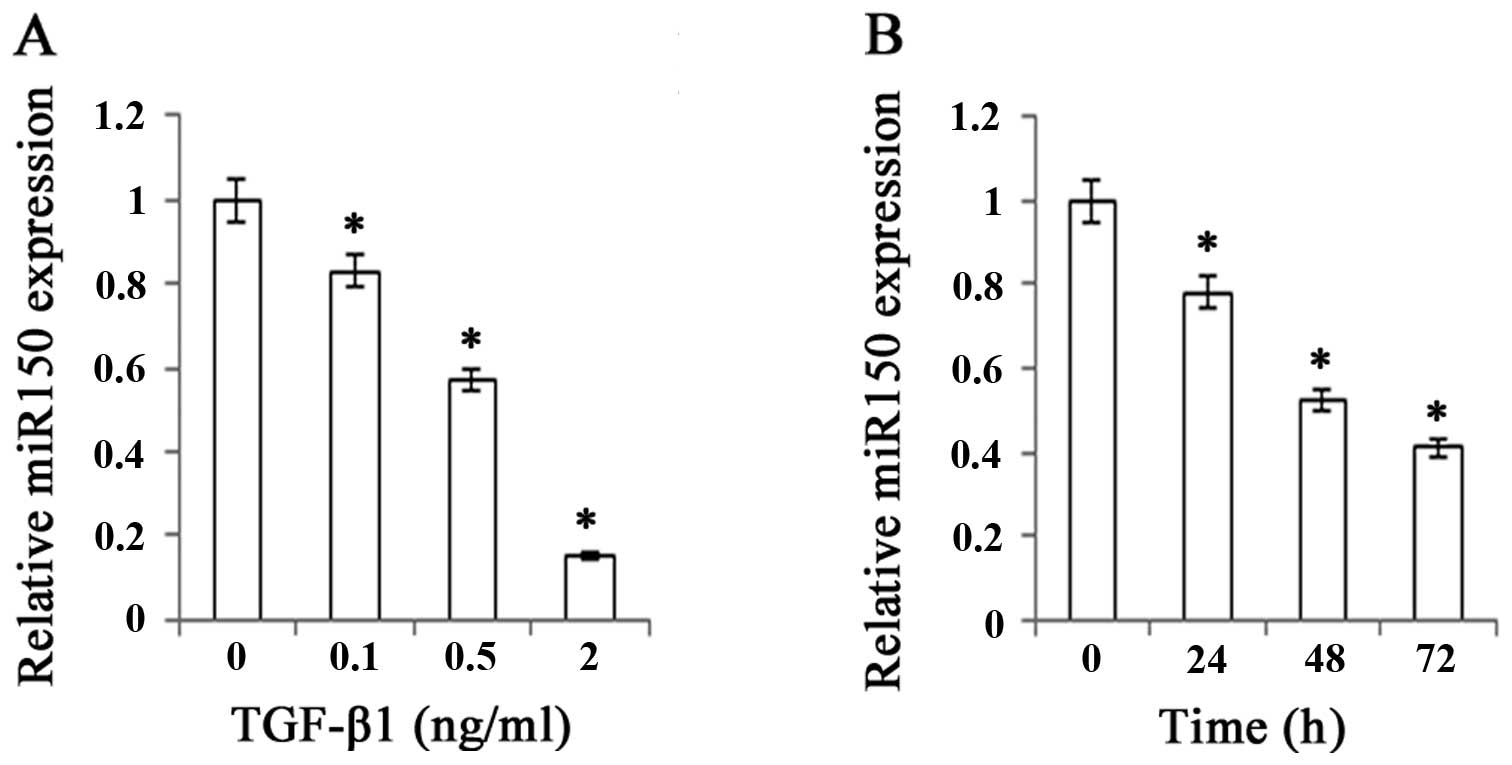
Dose- and time-dependent reduction of
miR-150 by TGF-β1
There was a significant decrease in miR-150
expression level as we increased the concentrations of TGF-β1 from
0.1 to 2 ng/ml, indicating that the inhibitory effect of TGF-β1 on
miR-150 was dose-dependent (Fig.
2A). Furthermore, miR-150 expression level was also examined at
0, 24, 48 and 72 h after the treatment of TGF-β1 (Fig. 2B). It was significantly decreased
with the prolongation of time, the lowest being at 72 h after the
treatment of TGF-β1. These findings raised the possibility that
miR-150 may play a pivotal role in the progression of liver
fibrosis.
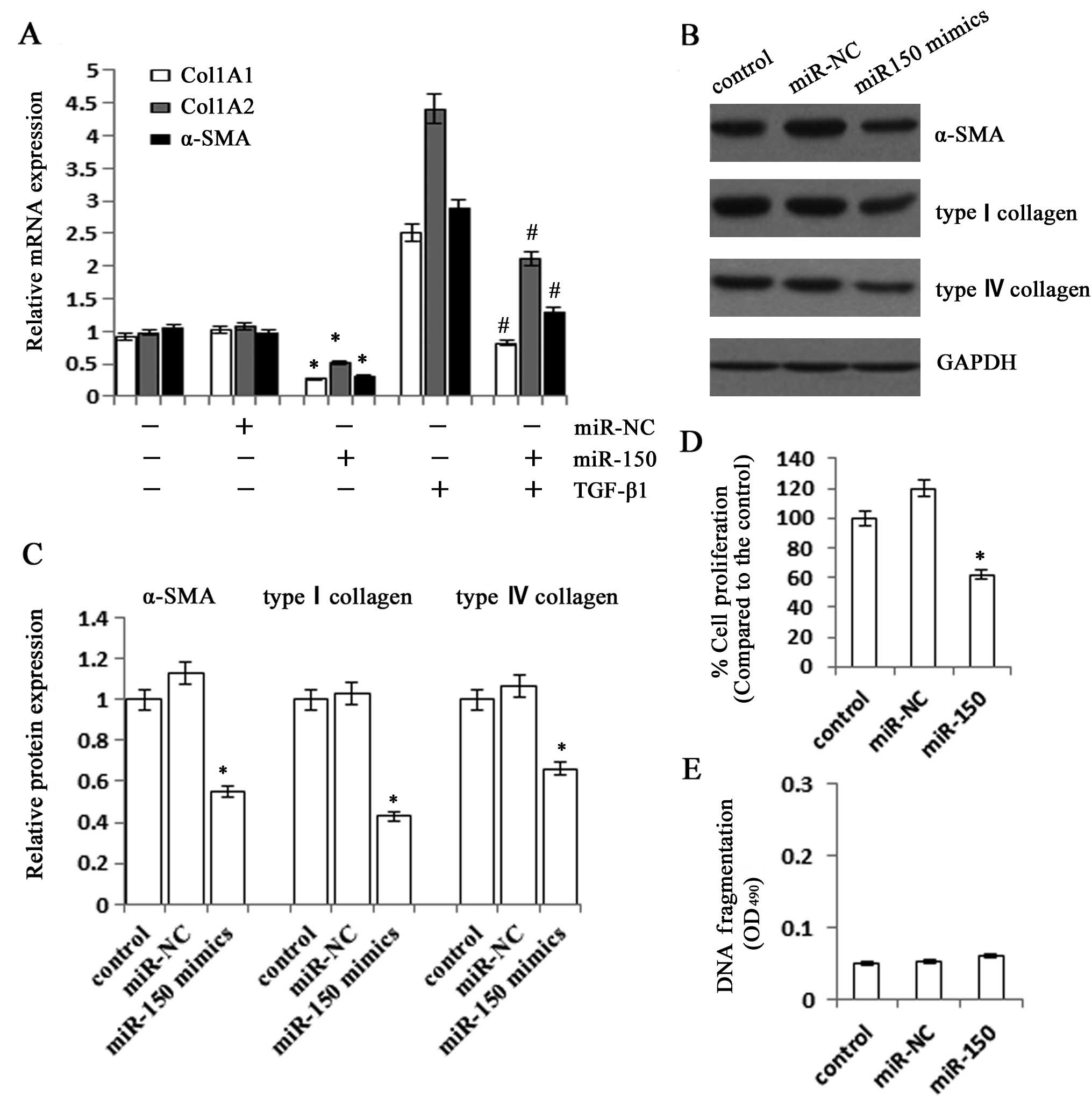
Effects of miR-150 overexpression on the
activation of LX-2 cells
Next, we investigated the effects of miR-150
overexpression on the activation of HSCs. When compared with the
control, the mRNA expression levels of Col1A1 and Col1A2 in cells
transfected with miR-150 mimics were markedly suppressed to 27.1
and 52.3%, respectively (Fig.
3A). The miR-150 mimics also significantly reduced the mRNA
expression level of α-SMA to 31.1%. It was further confirmed that
all the mRNA expression levels of Col1A1, Col1A2 and α-SMA were
inhibited in miR-150-overexpressing cells under the stimulation of
TGF-β1. Consistent with the results of the mRNA expression levels,
immunoblot analysis showed that the protein expression levels of
type I collagen and α-SMA were also suppressed by the
overexpression of miR-150 (Fig.
3B). Then, we examined the role of miR-150 in regulating the
proliferation and apoptosis of LX-2 cells. As shown by the MTT
assay, the cells transfected with miR-150 mimics had significantly
slower growth rates than the control, which was inhibited to 62.1%
(Fig. 3D). However, there was no
significant change in apoptosis rate in the cells transfected with
miR-150 mimics when compared with the control (Fig. 3E). These data suggested that the
expression of miR-150 reduced the production of ECM protein and
suppressed HSC activation, whereas it did not influence the
survival of LX-2 cells.
Col4A4 and Sp1 are the targets of
miR-150
To explore the molecular mechanism of miR-150 in the
collagen production of HSCs, we predicted the targets of miR-150
using bioinformatics analysis. We predicted that Col1A1 and Col1A2
were not targets of miR-150. Therefore, we conjectured that the
reduction of Col1A1 and Col1A2 may be controlled by other
mechanisms. It has been demonstrated that the expression of Col1A1
mRNA induced by TGF-β is through a pathway including Sp1 and
phosphorylated Smad2/3 (21). The
promoter activity of Col1A1 could be reduced by the knockdown of
Sp1 and Smad2. Since Sp1 is a transcriptional regulator of Col1A1
expression induced by TGF-β, we considered whether it was a
potential target gene for miR-150. We predicted that miR-150 could
interact with the 3′UTRs of human Col4A4 and Sp1 mRNA using
TargetScan Human Release 6.2 (http://www.targetscan.org/) (Fig. 4B and C). The sequence of each
target region including the 3′UTRs of Col4A4 and Sp1 mRNA was
cloned into pMIR-Report™ Luciferase plasmid. The construct was
cotransfected into LX-2 cells along with miR-150 precursor or
miR-NC. β-gal reporter control plasmid was cotransfected to monitor
transfection efficiency. miR-150 precursor significantly reduced
luciferase activities driven by the wild-type 3′UTRs of Col4A4 and
Sp1 when compared with their respective miR-NC. By contrast,
miR-150 precursor could not inhibit luciferase activities of
mutated type Col4A4 3′UTR, mutated type Sp1 3′UTR and empty vector
(Fig. 4D). To further verify the
above results, we measured the mRNA and protein expression levels
of Col4A4 and Sp1 in cells transfected with miR-150 mimics. As
expected, the overexpression of miR-150 not only decreased the mRNA
expressions of Col4A4 and Sp1, but also reduced the protein
expressions of type IV collagen and Sp1 (Figs. 3B, 4A and 5A). These results suggested that Col4A4
and Sp1 are the targets of miR-150.
Regulation of type I collagen expression
by miR-150
The degradation of type IV collagen could be
directly induced by miR-150, but the underlying molecular mechanism
of the reduction of type I collagen and α-SMA remains unclear.
Next, further study was performed to confirm whether the pathway
including Sp1 and Smad2/3 is responsible for the reduction of
Col1A1. There was a significant decrease in the protein expression
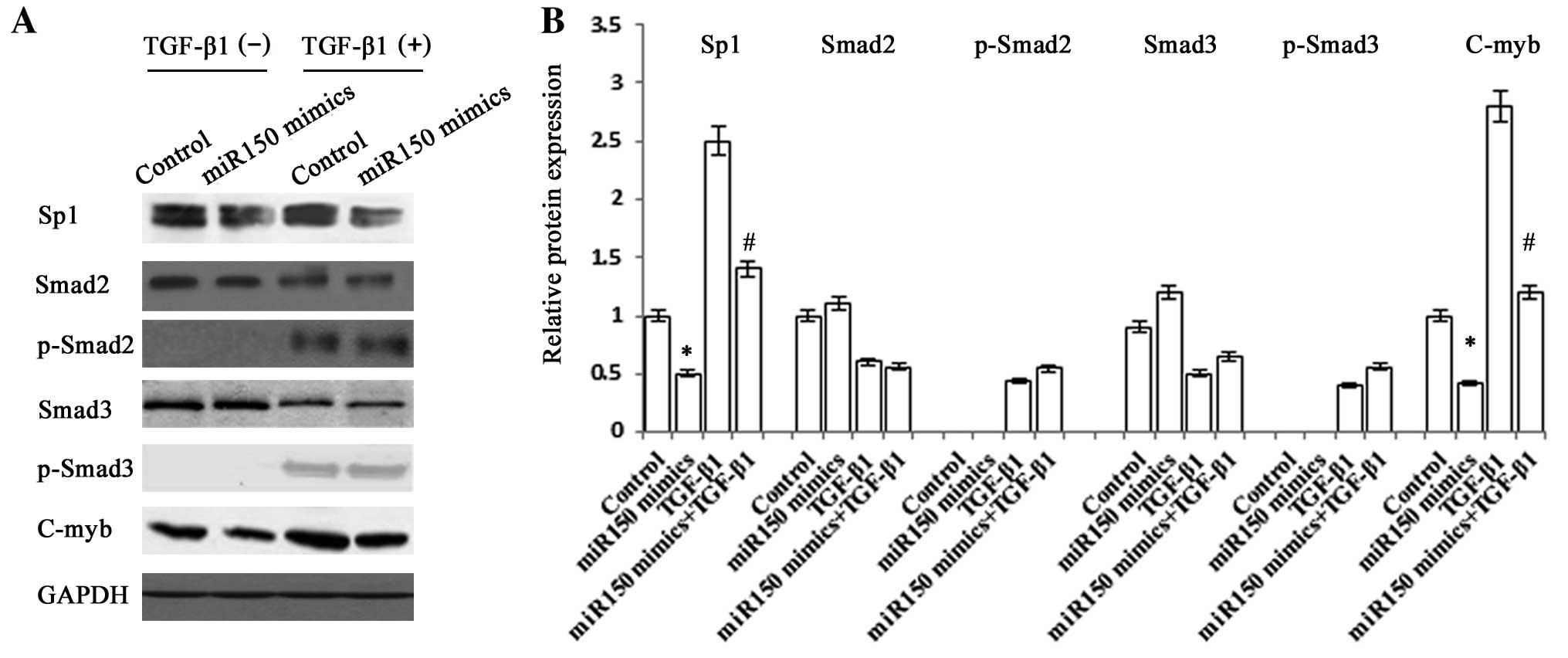
levels of Sp1 in miR-150-overexpressing cells (Fig. 5). However, these cells showed no
significant change in the protein expression levels of Smad2,
p-Smad2, Smad3 and p-Smad3. These data showed that the upstream of
TGF-β signaling pathway was not affected by the effect of miR-150.
We also found that C-myb, which is a target of miR-150 and plays a
role in regulating the expression of α-SMA and ECM proteins such as
type I collagen, was reduced in miR-150-overexpressing cells
(15). These results suggested
that miR-150 reduces the production of type I collagen via a
complex pathway including the regulation of C-myb and Sp1.
Discussion
HSCs play an important role in the progression of
hepatic fibrosis and are activated by liver inflammation factors or
injuries (22,23). The activated HSCs undergo
proliferation and secrete excessive ECM proteins such as type I and
IV collagen (24). The type I
collagen, which is encoded by Col1A1 and Col1A2, accounts for 36%
of the total collagens in ECM of healthy liver and deposits in the
perisinusoidal space during liver fibrogenesis (25). The most markedly upregulated
collagen is the type IV collagen in liver fibrosis, which
constitutes less than 10% of total collagen in the normal liver
(26-28). The objective of the present study
was to explore the antifibrotic effect of miR-150 as well as the
underlying molecular mechanism of suppression of HSC activation by
miR-150.
It is may be that different HSC behaviors are
controlled by different miRNAs, particularly in the synthesis of
the ECM protein. For example, miR-29b, which is a regulator of type
I collagen mRNA and protein, could suppress the activation of HSCs
(29,30). In this study, the expression of
miR-150 was found to be significantly reduced in the TGF-β1 group
in a dose- and time-dependent manner. Therefore, we speculated that
miR-150, which is inhibited in LX-2 cells treated with TGF-β1, may
play a role in the antifibrotic effect. As expected, following the
restoration of miR-150 in LX-2 cells, the mRNA and protein
expressions of type I collagen and α-SMA were markedly decreased
and the cell growth rate was significantly inhibited, which is
consistent with a previous study (15). Our data suggested that miR-150
could inhibit the production of ECM proteins and the activation of
HSCs. However, there was no change in the apoptotic rate of DNA
fragment in cells transfected with miR-150 as compared with the
control, indicating that miR-150 did not affect the cell
survival.
LX-2 cells, which are classified as an activated
phenotype that expresses high levels of α-SMA and collagens, are
selected for all the experiments (31). Using TargetScan Human Release 6.2,
we predicted that miR-150 may interact with the 3′UTR of Col4A4. It
was further verified by the dual luciferase reporter experiment.
Therefore, our data suggested that miR-150 was a potential
regulator of type IV collagen mRNA and protein expressions.
Furthermore, Sp1, which was a predicted target gene for miR-150,
had two target regions for miR-150. Each of the target regions was
cloned and inserted into the pMIR-Report™ luciferase plasmid. Our
data demonstrated that miR-150 targets both target regions of 3′UTR
of Sp1, which may be partly responsible for the reduction of Col1A1
mRNA and protein expressions in LX-2 cells transfected with miR-150
mimics. However, the Smad2 and phosphorylation of Smad2 expressions
under TGF-β1 stimulation, which are the downstream of the TGF-β1
pathway, showed no change in LX-2 cells overexpressing miR-150 as
compared with the control. The same was also seen in the expression
of the Smad3 and phosphorylation of Smad3. Therefore, it is
considered that miR-150 may affect the downstream of Smad2/3. In
our study, it is not evident how miR-150 regulates the expression
of Col1A2 and α-SMA. We hypothesize that the reduction of Col1A2
and α-SMA may be associated with C-myb, which has been proved to be
a target of miR-150 and to play a role in promoting the activation
of HSCs (15). It has been found
that C-myb expression is increased in activated HSCs and is
associated with the development of fibrosis in animal models
(32). Moreover, Lee et al
(33) found that the type I
collagen and α-SMA could be induced via C-myb under the stimulation
of oxidative stress. Our study confirmed that the overexpression of
miR-150 reduced the C-myb protein expression.
Collectively, our study illustrates that miR-150,
which is crucial in the development of liver fibrosis, could
inhibit type I and IV collagen in activated HSCs, at least in part,
via the reduction of Sp1 and Col4A4 expression.
Acknowledgements
The study was supported by the
Zhejiang Extremely Key Subject of Surgery, National Natural Science
Foundation of China (81000176/H0317 and 81100292/H0317), the
Zhejiang Provincial Natural Science Foundation of China (Y2090326
and Y2110634), the Wang Bao-En Liver Fibrosis Foundation (no.
20100002 and 20120127), the Wenzhou Municipal Science and
Technology Bureau (Y20110033 and Y20120127) and the key disciplines
in Colleges and Universities of Zhejiang Province.
References
|
1.
|
Schuppan D, Krebs A, Bauer M and Hahn EG:
Hepatitis C and liver fibrosis. Cell Death Differ. 10(Suppl 1):
S59–S67. 2003. View Article : Google Scholar
|
|
2.
|
Gressner OA, Rizk MS, Kovalenko E,
Weiskirchen R and Gressner AM: Changing the pathogenetic roadmap of
liver fibrosis? Where did it start; where will it go? J
Gastroenterol Hepatol. 23:1024–1035. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Kong X, Horiguchi N, Mori M and Gao B:
Cytokines and STATs in liver fibrosis. Front Physiol. 3:692012.
View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Friedman SL: Mechanisms of hepatic
fibrogenesis. Gastroenterology. 134:1655–1669. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Vettori S, Gay S and Distler O: Role of
microRNAs in fibrosis. Open Rheumatol J. 6:130–139. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Berezikov E, Guryev V, van de Belt J,
Wienholds E, Plasterk RH and Cuppen E: Phylogenetic shadowing and
computational identification of human microRNA genes. Cell.
120:21–24. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Kozomara A and Griffiths-Jones S: miRBase:
integrating microRNA annotation and deep-sequencing data. Nucleic
Acids Res. 39:D152–D157. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Brennecke J, Hipfner DR, Stark A, Russell
RB and Cohen SM: bantam encodes a developmentally regulated
microRNA that controls cell proliferation and regulates the
proapoptotic gene hid in Drosophila. Cell. 113:25–36. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Schratt GM, Tuebing F, Nigh EA, et al: A
brain-specific microRNA regulates dendritic spine development.
Nature. 439:283–289. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Chen CZ, Li L, Lodish HF and Bartel DP:
MicroRNAs modulate hematopoietic lineage differentiation. Science.
303:83–86. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Mott JL, Kobayashi S, Bronk SF and Gores
GJ: mir-29 regulates Mcl-1 protein expression and apoptosis.
Oncogene. 26:6133–6140. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Kloosterman WP and Plasterk RH: The
diverse functions of microRNAs in animal development and disease.
Dev Cell. 11:441–450. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Chen L, Jiang M, Yuan W and Tang H:
miR-17-5p as a novel prognostic marker for hepatocellular
carcinoma. J Invest Surg. 25:156–161. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Yang F, Yin Y, Wang F, et al: miR-17-5p
promotes migration of human hepatocellular carcinoma cells through
the p38 mitogen-activated protein kinase-heat shock protein 27
pathway. Hepatology. 51:1614–1623. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Venugopal SK, Jiang J, Kim TH, et al:
Liver fibrosis causes downregulation of miRNA-150 and miRNA-194 in
hepatic stellate cells, and their overexpression causes decreased
stellate cell activation. Am J Physiol Gastrointest Liver Physiol.
298:G101–G106. 2010. View Article : Google Scholar
|
|
16.
|
Ji J, Zhang J, Huang G, Qian J, Wang X and
Mei S: Over-expressed microRNA-27a and 27b influence fat
accumulation and cell proliferation during rat hepatic stellate
cell activation. FEBS Lett. 583:759–766. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Iizuka M, Ogawa T, Enomoto M, et al:
Induction of microRNA-214-5p in human and rodent liver fibrosis.
Fibrogenesis Tissue Repair. 5:122012. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Schmittgen TD and Livak KJ: Analyzing
real-time PCR data by the comparative C(T) method. Nat Protoc.
3:1101–1108. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Thompson WJ, Piazza GA, Li H, et al:
Exisulind induction of apoptosis involves guanosine 3′,5′-cyclic
monophosphate phosphodiesterase inhibition, protein kinase G
activation, and attenuated beta-catenin. Cancer Res. 60:3338–3342.
2000.PubMed/NCBI
|
|
20.
|
Gressner AM, Weiskirchen R, Breitkopf K
and Dooley S: Roles of TGF-beta in hepatic fibrosis. Front Biosci.
7:d793–d807. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Sysa P, Potter JJ, Liu X and Mezey E:
Transforming growth factor-beta1 up-regulation of human alpha(1)(I)
collagen is mediated by Sp1 and Smad2 transacting factors. DNA Cell
Biol. 28:425–434. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Bataller R and Brenner DA: Hepatic
stellate cells as a target for the treatment of liver fibrosis.
Semin Liver Dis. 21:437–451. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Friedman SL: Molecular regulation of
hepatic fibrosis, an integrated cellular response to tissue injury.
J Biol Chem. 275:2247–2250. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Wu J and Zern MA: Hepatic stellate cells:
a target for the treatment of liver fibrosis. J Gastroenterol.
35:665–672. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Kwiecinski M, Noetel A, Elfimova N, et al:
Hepatocyte growth factor (HGF) inhibits collagen I and IV synthesis
in hepatic stellate cells by miRNA-29 induction. PLoS One.
6:e245682011. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Gressner AM and Weiskirchen R: Modern
pathogenetic concepts of liver fibrosis suggest stellate cells and
TGF-beta as major players and therapeutic targets. J Cell Mol Med.
10:76–99. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Geerts A, Schuppan D, Lazeroms S, De
Zanger R and Wisse E: Collagen type I and III occur together in
hybrid fibrils in the space of Disse of normal rat liver.
Hepatology. 12:233–241. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Milani S, Herbst H, Schuppan D, Surrenti
C, Riecken EO and Stein H: Cellular localization of type I III and
IV procollagen gene transcripts in normal and fibrotic human liver.
Am J Pathol. 137:59–70. 1990.PubMed/NCBI
|
|
29.
|
Sekiya Y, Ogawa T, Yoshizato K, Ikeda K
and Kawada N: Suppression of hepatic stellate cell activation by
microRNA-29b. Biochem Biophys Res Commun. 412:74–79. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Ogawa T, Iizuka M, Sekiya Y, Yoshizato K,
Ikeda K and Kawada N: Suppression of type I collagen production by
microRNA-29b in cultured human stellate cells. Biochem Biophys Res
Commun. 391:316–321. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Xu L, Hui AY, Albanis E, et al: Human
hepatic stellate cell lines, LX-1 and LX-2: new tools for analysis
of hepatic fibrosis. Gut. 54:142–151. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Kitada T, Seki S, Nakatani K, Kawada N,
Kuroki T and Monna T: Hepatic expression of c-Myb in chronic human
liver disease. Hepatology. 26:1506–1512. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Lee KS, Buck M, Houglum K and Chojkier M:
Activation of hepatic stellate cells by TGF alpha and collagen type
I is mediated by oxidative stress through c-myb expression. J Clin
Invest. 96:2461–2468. 1995. View Article : Google Scholar : PubMed/NCBI
|