Introduction
Since the first description of a ‘disappearing bone
disease’ (1) several rare
congenital osteolysis syndromes have been described with their
nosology being classified by Tyler and Rosenbaum (2) in 1976 and by the International
Classification of Constitutional Disorders of Bone (3). Autosomal dominant multicentric
carpotarsal osteolysis syndrome (MCTO; OMIM 166300) usually
presents in early childhood, often with a clinical appearance
mimicking juvenile idiopathic arthritis (JIA) (4). However, JIA can easily be
differentiated from MCTO by clinical, laboratory and radiological
findings (5). MCTO is
characterized by progressive and most often bilateral destruction
of the carpal and tarsal bones; however, osteolysis may also affect
other bones. Affected individuals may develop proteinuria and
progressive nephropathy leading to end-stage renal disease.
Craniofacial anomalies and mental impairment have also been
observed (5,6). Recently, Zankl et al
(7) reported an association of
MCTO with de novo missense mutations in the V-maf
musculoaponeurotic fibrosarcoma oncogene homolog B avian)
(MAFB) gene in 11 simplex cases. In two further multiplex
families, MAFB missense mutations segregated in an autosomal
dominant fashion. The protein encoded (MafB) by this gene is a
basic domain leucine zipper transcription factor with an
anti-osteoclastogenic function also involved in renal tubule
survival, tissue differentiation within the pancreas and
segmentation of the hindbrain (8–13).
The disturbance of these different functions may explain the
phenotypic features observed in association with MCTO.
In the present study, we identified a novel
p.Ser56Phe MAFB missense variant in a German individual with
MCTO. Surprisingly, an analysis of the family revealed presence of
this variant in three unaffected female family members
investigated.
Patients and methods
Patient description
The index case in this study is the second child of
unrelated parents. He has a three years older healthy sister. At
his first visit to the Genetics Department, University Hospital of
Bonn, Bonn, Germany, the index case was 17 years of age. His early
development was completely uneventful. By the age of four, he first
visited the Children’s Hospital for recurrent pain in his right
hand with swelling of his wrist, raising the suspicion of the onset
of juvenile rheumatoid arthritis. At the age of six he presented
with dysesthesia in his right leg with swelling of his ankle.
Subsequent diagnostic radiographic workup revealed several
carpotarsal osteolytic spots. Extensive diagnostic testing did not
reveal any immunological or infectious origin for his symptoms.
During the course of his extensive diagnostic workup to explain his
carpotarsal osteolysis, an electroencephalography revealed slow
waves on his right temporo-parieto-occipital recordings. Subsequent
brain magnetic resonance imaging revealed an Arnold-Chiari
malformation type I. During late puberty he developed androgenetic
alopecia and slight non-immunological bilateral exophthalmos. At 19
years of age, an X-ray revealed irregular and dysplastic, partially
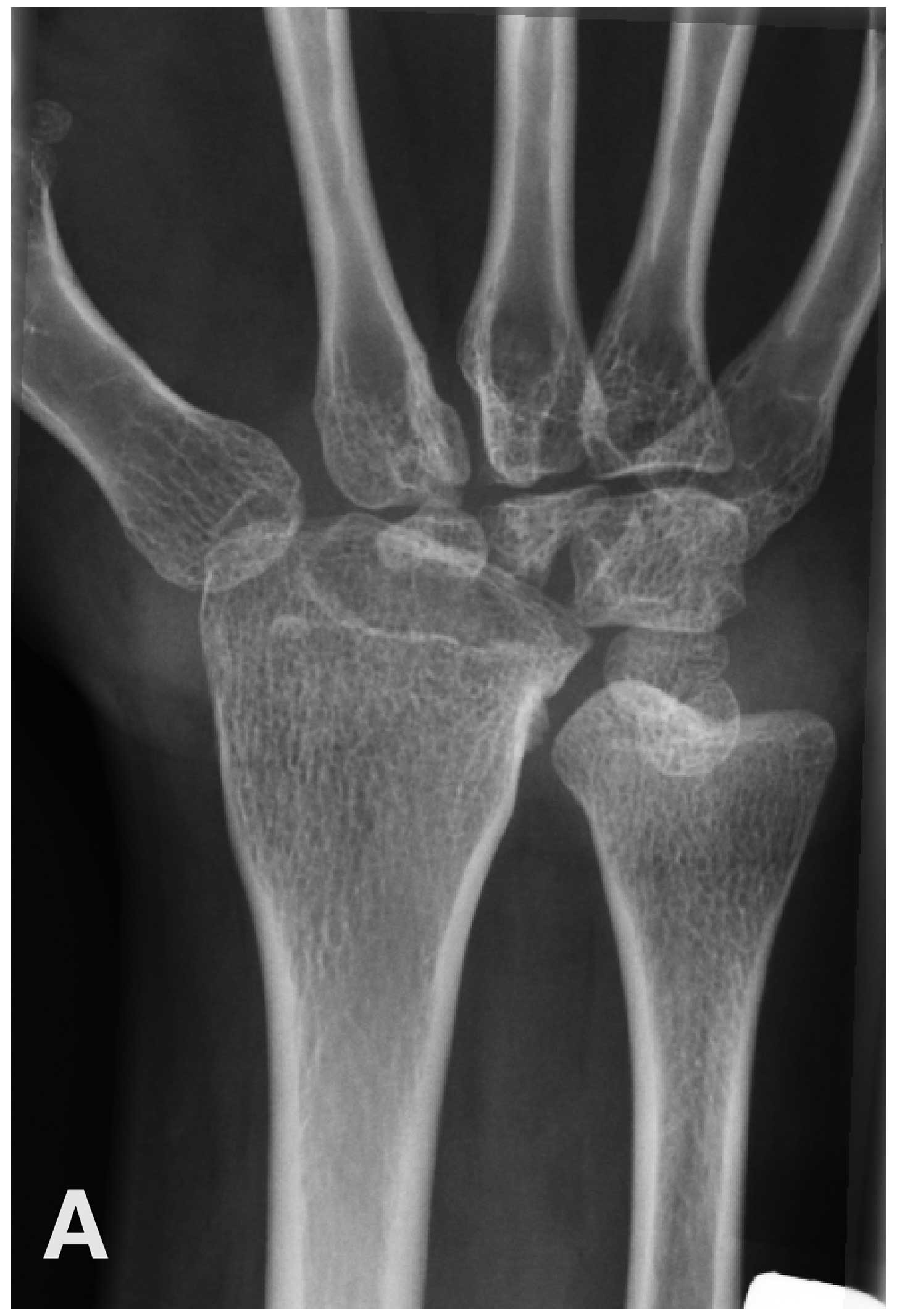
missing carpalia and dysplastic changes of the tarsalia (Fig. 1). Renal functional studies and
urine analysis showed normal renal function, without any sign of
proteinuria or nephropathy.
The mother did not report any history of swelling of
her wrists or ankles nor any radiographical signs of carpotarsal
osteolysis. In addition she did not present with any ulnar
deviation of her hands or wrist arthralgia. X-ray images, renal
functional analyses and urine analysis were normal. The sister and
maternal grandmother also showed no features characteristic of
MCTO.
Sample collection and DNA analysis
After informed consent was obtained, EDTA blood
samples were obtained from the family members. Genomic DNA was
isolated using the QIAamp DNA Blood kit and amplification of the
single MAFB exon was performed by polymerase chain reaction
(PCR) using HotStarTaq DNA polymerase (Qiagen, Hilden, Germany).
PCR primers (1F, 5′-GCTTGGCTCGGCGCGCTCC-3′; 1F2,
5′-CCGTGCGCGAGCTGAACCG-3′; 1R, 5′-GCGGCAGGGACAGGGTCCG-3′; 1R2,
5′-TGCGAGCCGATGAGCGCTTC-3′) also served as sequencing primers. For
mutational analysis, PCR-amplified DNA products were subjected to
direct automated sequencing using a 3130×l Genetic Analyzer
(Applied Biosystems, Inc., Foster City, CA, USA) and both strands
from the amplicons of the parents and patients were analyzed.
RNA analysis
RNA analyses were performed to investigate the
expression of both alleles in the patient and his mother. Blood
samples were collected in PAXgene tubes (PreAnalytiX,
Hombrechtikon, Switzerland) and RNA for reverse transcription
(RT)-PCR was prepared using the RNeasy Plus Micro kit (Qiagen) in
accordance with the manufacturer’s instructions. Allele-specific
amplification (ASA)-PCR and allele-specific quantitative PCR
(AS-qPCR) were carried out as previously described (14). ASA-PCR may lack reliability since
the mutated primer may still allow amplification of the normal
allele as shown by Hezard et al (15). These authors showed that allele
discrimination can be improved by incorporating an additional
mismatch in the antepenultimate position by substituting the
respective wild-type (WT) pyrimidine (purine) nucleotide by the
respective pyrimidine (purine) base. Therefore, to adjust for
similar annealing temperatures, ASA forward (F) primers and a
universal reverse (UR) and forward (UF) primer were designed as
follows (normal WT sequence, 5′-TGGAACTGAAAACCAGCGACCA-3′): WT-F,
5′-GAACTGAAAACCAGCGATCA-3′; mutant-G-F, 5′-GAACTGAAAACCAGC GATCG;
UR, 5′GGCACTAACCTCTCAATGCTG-3′; and UF,
5′-TGGAACTGAAAACCAGCGACC-3′. Reactions for AS-qPCR were performed
on an ABI PRISM 7900HT Fast Real-Time PCR System (Applied
Biosystems, Inc.) using SYBR-Green for detection. Each assay was
performed as previously described (14) and included DNA from four controls
(two male and two female samples) and the subjects’ DNA.
Results
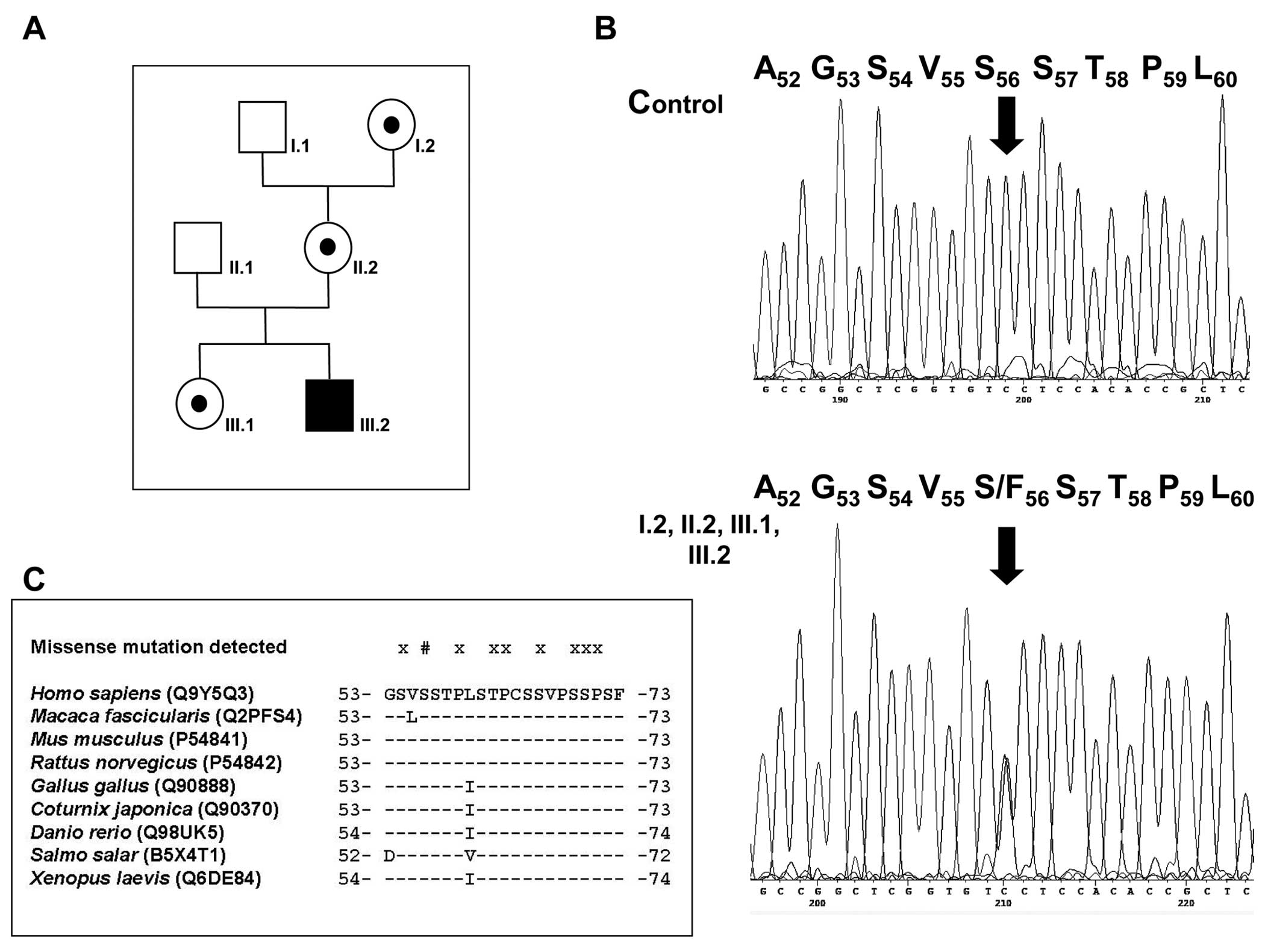
The screening of MAFB PCR products obtained
from the index case (Fig. 2A,
III.2) revealed a single heterozygous c.167C>T nucleotide
transition (Fig. 2B), predicting
a p.Ser56Phe substitution (GenBank accession no. NM_005461.3, with
the A of the start ATG as nucleotide 1). Gene analysis in the
unaffected mother of the patient (II.2) provided evidence that she
is a carrier of the mutant allele transmitted to her son. Moreover,
the healthy sister and the healthy maternal grandmother also showed
the heterozygous c.167C>T transition.
These findings led us to perform functional
analyses. To exclude the sole or predominant expression of the WT
allele in the mother, the RT-PCR products were first analyzed by
sequence analysis. Fluorescence intensity indicated the presence of
the WT and variant transcript in similar amounts in the samples of
the patient and his mother. To verify this finding we also
performed AS-qPCR.
Prior to qPCR it had to be validated that the ASA
primers only direct amplification on their complementary allele and
that the conditions chosen avoid any other unspecific
amplification. The ASA primers designed allowed the sole
amplification of the respective allele without any additional
unspecific priming, hence, qPCR was performed. This method involves
PCR amplification of the product of interest and, in a separate
reaction, amplification of an invariantly expressed internal
standard (housekeeping gene) to be used as a denominator to
normalize the amount of the target gene. Allele-specific qPCR also
showed similar amounts of both transcribed alleles in the patient
and his mother (data not shown).
Several clues point to a critical role for serine 56
located in the amino-terminal transcriptional activation domain of
MAFB: First, the c.167C>T transition has not been
deposited in the single nucleotide polymorphism (SNP, Build137)
database and in the Deep Catalog of Human Genetic Variation
(www.1000genomes.org), which argues against the
possibility that this substitution is just a rare benign variant.
In addition, the amino acid exchange substitutes a polar residue
for a non-polar one and pathogenicity prediction with several
algorithms (PolyPhen-2, SNPs&GO, PMut, SIFT) predicted this
variant to be disease-causing or at least possibly damaging.
Finally, Ser56 is absolutely conserved at its corresponding
position in species orthologues as part of the transactivation
motif at least as far down as Xenopus laevis (X.
laevis) (Fig. 2C).
Discussion
The patient presented in this study showed the
typical features of MCTO with wrist and ankle swelling, severe
osteolysis of almost all carpal and tarsal bones (Fig. 1) and bilateral exophthalmos. He
showed no proteinuria or nephropathy at the time of writing, but it
is as yet unclear, whether his renal function will remain normal.
However, complete absence of renal involvement has been frequently
observed (6) and was not reported
in 50% of the patients reported by Zankl et al (7). We further noted Arnold-Chiari
malformation type I and androgenetic alopecia but are not aware of
any other case showing the coincidence of MCTO with these
features.
The recent observations of Zankl et al
(7) suggested a heterozygous
de novo MAFB mutation in our isolated MCTO patient. As
expected, we observed a single heterozygous p.Ser56Phe amino acid
substitution that surprisingly was also present in three unaffected
females in this family. MAFB RNA analysis in the patient and
his mother revealed similar amounts of mutant and WT mRNAs
providing no evidence, that the phenotypic differences are a result
of a preferential transcription of the WT allele in the mother.
Nevertheless, several strands of evidence suggested
that the amino acid substitution observed represents a pathogenic
mutation. Aside from its absence in databases, comparative genomics
revealed apparent evolutionarily conservation of Ser56 at least as
far down as X. laevis (Fig.
2C). In addition, pathogenicity prediction revealed this
substitution to be disease-causing or at least possibly damaging.
Moreover, this novel variant resides in the transactivation domain
of MafB, affected by all the missense mutations (residues 54–71)
described to date (Fig. 2C)
(7).
Variable phenotypes and even intra-familial
variability was observed in the probands investigated by Zankl
et al (7). This suggests a
role for as yet unidentified modifier gene(s), epigenetic
mechanisms or environmental factors in disease penetrance which
could also explain the absence of the associated renal phenotype.
Gender differences in penetrance due to a protective modifier gene
may also be an explanation for the presence of three healthy
females in our family. Actually, incomplete penetrance occurring
only in female carriers has been described in familial hypokalemic
periodic paralysis due to mutations in the skeletal muscle calcium
channel α1 subunit gene (16).
Also, asymptomatic females carrying the same survival motor neuron
1 gene mutation as their affected relatives with recessive spinal
muscular atrophy have been observed (17). On the other hand, such a
protective effect in females should result in an unequal gender
ratio. However, a review of the data of all MCTO patients reported
to date showed no male predominance of the disease.
Similar to the data presented, several other bone
diseases exhibit variable expressivity and incomplete penetrance.
For example, heterozygous mutations in the sequestosome
(SQSTM)1 gene are now suggested not to cause Paget’s disease
of bone (OMIM 601530) but only predispose a carrier to the disease
(18). The fact that a single
mutant allele can be insufficient to cause the disorder has also
been observed in otosclerosis 1 (OMIM %166800) (19), characterized by alternating phases
of bone resorption and formation, as well as growth differentiation
factor 6-attributable oculo-skeletal phenotypes (20).
Finally, it would of interest to observe the effects
of the heterozygous p.Ser56Phe and other missense mutations in a
mouse model. Complete MafB deficiency has been shown to cause
defective respiratory rhythmogenesis and fatal central apnea at
birth (21). However, all mouse
models were generated by mutations not comparable with the missense
mutations observed in MCTO patients. In the first model,
MafB/Kreisler (kr), MAFB has been shown to be the
ortholog of the gene affected by the mouse Kreisler
mutation, that involves a submicroscopic inversion, leaving 30 kb
of 5′ sequence and the coding region intact (8,22).
Of note, in these homozygous mice, the expression of the
MafB/kr gene is still maintained outside of the hindbrain
indicating a selective, tissue-specific gene inactivation (22). Using N-ethyl-N-nitrosourea (ENU),
Cordes and Barsh (8) introduced
an A-to-G transition at nucleotide 743 resulting in a p.Asp248Ser
substitution. The mutation is located in the putative DNA-binding
domain and there seems to be a similar effect of the
krENU mutation which is suggested to retain
partial function in the absence of DNA binding, possibly through
interactions with other bZip transcription (co)factors (13). A previous study demonstrated that
krENU was able to form dimers with WT MafB and
transactivated a reporter gene (23). Finally, a targeting vector
(MafB/green fluorescent protein) was designed and RT-PCR
failed to detect transcripts in mutant renal cells and macrophages
(13). Again, all
MafB−/− mice died within 24 h after birth. A
phenotype strikingly similar to that of the Kreisler mutant,
was observed in these MafB−/− neonates but not in
their heterozygous siblings. Hence, it remains elusive, whether a
heterozygous missense mutation in the amino-terminal
transcriptional activation domain of mouse MafB mirrors the
phenotype observed in humans and whether incomplete penetrance can
also be observed in this model.
In conclusion, the novel MAFB variant
observed is expected to be responsible for the MCTO phenotype in
our patient. The effect of a modifier, incomplete penetrance,
epigenetic mechanisms or environmental factors may account for the
variable expressivity of the disease in the family presented in our
study. This not only increases the complexity of genetic counseling
but should also be considered in patient care, at least in renal
prognosis.
Acknowledgements
We thank the family for their kind
participation in this study. We thank Pia Uerdingen for her
excellent technical assistance. G.C.D. was supported by the
University of Bonn, BONFOR (grant no. O-149.0096).
References
|
1.
|
Jackson JBS: A boneless arm. Boston Med
Surg J. 18:368–369. 1838.
|
|
2.
|
Tyler T and Rosenbaum HD: Idiopathic
multicentric osteolysis. AJR Am J Roentgenol. 126:23–31. 1976.
View Article : Google Scholar
|
|
3.
|
Hall M: International nosology and
classification of constitutional disorders of bone (2001). Am J Med
Genet. 113:65–77. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Faber MR, Verlaak R, Fiselier TJ, Hamel
BC, Franssen MJ and Gerrits GP: Inherited multicentric osteolysis
with carpal-tarsal localisation mimicking juvenile idiopathic
arthritis. Eur J Pediatr. 163:612–618. 2004.PubMed/NCBI
|
|
5.
|
Pai GS and Macpherson RI: Idiopathic
multicentric osteolysis: report of two new cases and review of the
literature. Am J Med Genet. 29:929–936. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Carnevale A, Canún S, Mendoza L and del
Castillo V: Idiopathic multicentric osteolysis with facial
anomalies and nephropathy. Am J Med Genet. 26:877–886. 1987.
View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Zankl A, Duncan EL, Leo PJ, Clark GR,
Glazov EA, Addor MC, Herlin T, Kim CA, Leheup BP, McGill J, et al:
Multicentric carpotarsal osteolysis is caused by mutations
clustering in the amino-terminal transcriptional activation domain
of MAFB. Am J Hum Genet. 90:494–501. 2012. View Article : Google Scholar
|
|
8.
|
Cordes SP and Barsh GS: The mouse
segmentation gene kr encodes a novel basic domain-leucine zipper
transcription factor. Cell. 79:1025–1034. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Sadl V, Jin F, Yu J, Cui S, Holmyard D,
Quaggin S, Barsh G and Cordes S: The mouse Kreisler
(Krml1/MafB) segmentation gene is required for
differentiation of glomerular visceral epithelial cells. Dev Biol.
249:16–29. 2002.
|
|
10.
|
Nishimura W, Kondo T, Salameh T, El
Khattabi I, Dodge R, Bonner-Weir S and Sharma A: A switch from MafB
to MafA expression accompanies differentiation to pancreatic
beta-cells. Dev Biol. 293:526–539. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Artner I, Blanchi B, Raum JC, Guo M,
Kaneko T, Cordes S, Sieweke M and Stein R: MafB is required for
islet beta cell maturation. Proc Natl Acad Sci USA. 104:3853–3858.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Kim K, Kim JH, Lee J, Jin HM, Kook H, Kim
KK, Lee SY and Kim N: MafB negatively regulates RANKL-mediated
osteoclast differentiation. Blood. 109:3253–3259. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Moriguchi T, Hamada M, Morito N, Terunuma
T, Hasegawa K, Zhang C, Yokomizo T, Esaki R, Kuroda E, Yoh K, et
al: MafB is essential for renal development and F4/80 expression in
macrophages. Mol Cell Biol. 26:5715–5727. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Draaken M, Giesen CA, Kesselheim AL, Jabs
R, Aretz S, Kugaudo M, Chrzanowska KH, Krajewska-Walasek M and
Ludwig M: Maternal de novo triple mosaicism for two single
OCRL nucleotide substitutions (c.1736A>T, c1736A>G) in
a Lowe syndrome family. Hum Genet. 129:513–519. 2011.PubMed/NCBI
|
|
15.
|
Hezard N, Cornillet P, Droulle C, Gillot
L, Potron G and Nguyen P: Factor V Leiden: detection in whole blood
by ASA PCR using an additional mismatch in antepenultimate
position. Thromb Res. 88:59–66. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Kawamura S, Ikeda Y, Tomita K, Watanabe N
and Seki K: A family of hypokalemic periodic paralysis with CACNA1S
gene mutation showing incomplete penetrance in women. Intern Med.
43:218–222. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Oprea GE, Kröber S, McWhorter ML, Rossoll
W, Müller S, Krawczak M, Bassell GJ, Beattie CE and Wirth B:
Plastin 3 is a protective modifier of autosomal recessive spinal
muscular atrophy. Science. 320:524–527. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Leach RJ, Singer FR, Ench Y, Wisdom JH,
Pina DS and Johnson-Pais TL: Clinical and cellular phenotypes
associated with sequestosome 1 (SQSTM1) mutations. J
Bone Miner Res. 21(Suppl 2): P45–P50. 2006. View Article : Google Scholar
|
|
19.
|
Uppal S, Bajaj Y, Rustom I and Coatesworth
AP: Otosclerosis 1: the aetiopathogenesis of otosclerosis. Int J
Clin Pract. 63:1526–1530. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Asai-Coakwell M, French CR, Ye M, Garcha
K, Bigot K, Perera AG, Staehling-Hampton K, Mema SC, Chanda B,
Mushegian A, et al: Incomplete penetrance and phenotypic
variability characterize Gdf6-attributable oculo-skeletal
phenotypes. Hum Mol Genet. 18:1110–1121. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Blanchi B, Kelly LM, Viemari JC, Lafon I,
Burnet H, Bévengut M, Tillmanns S, Daniel L, Graf T, Hilaire G and
Sieweke MH: MafB deficiency causes defective respiratory
rhythmogenesis and fatal central apnea at birth. Nat Neurosci.
6:1091–1099. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Eichmann A, Grapin-Botton A, Kelly L, Graf
T, Le Douarin NM and Sieweke M: The expression pattern of the
mafB/kr gene in birds and mice reveals that the
kreisler phenotype does not represent a null mutant. Mech
Dev. 65:111–122. 1997.
|
|
23.
|
Sadl VS, Sing A, Mar L, Jin F and Cordes
SP: Analysis of hindbrain patterning defects caused by the
kreislerenu mutation reveals multiple roles of
Kreisler in hindbrain segmentation. Dev Dyn. 227:134–142.
2003. View Article : Google Scholar : PubMed/NCBI
|