Introduction
In a previous study, by combining differential
display, reverse transcription polymerase chain reaction (RT-PCR)
and DNA sequencing, we detected the overexpression of dihydrodiol
dehydrogenase (DDH) in primary non-small cell lung cancer (NSCLC)
specimens and lung cancer cell lines. We further found that DDH
overexpression correlated with a higher frequency of tumor
recurrence and distant metastasis (1). In retrospective studies, DDH
overexpression was shown to correlate with poor prognosis,
particularly in patients with late-stage disease (1,2).
By combining cDNA sequencing and two-dimensional gel
electrophoresis, we identified that the DDH in the cancer cells
belonged to aldo-keto reductase (AKR) family 1, member C1 (AKR1C1),
and to a lesser extent to AKR family 1, member C2 (AKR1C2) (for the
nomenclature of the respective enzymes, please refer to http://www.med.upenn.edu/akr/) (3). In vitro, the detection of DDH
overexpression in ethacrynic acid-induced drug-resistant colon
cancer cells and daunorubicin-resistant stomach cancer cells
suggested that the involvement of DDH in the resistance of cancer
cells to drugs may be a general phenomenon (4,5).
The fundamental nature of DDH to catabolize xenobiotic compounds
indicates that the enzyme may deactivate anticancer drugs with
similar polycyclic structures (1,6,7).
However, using cDNA microarray to investigate genes
which are overexpressed in cisplatin-resistant ovarian cancer
cells, we also identified DDH. Subsequent studies of ectopic DDH
expression confirmed our findings that DDH plays a role in the
resistance of cancer cells to cisplatin (8). The evidently different chemical
configurations and the evidently diverse resistance mechanisms
between daunorubicin and cisplatin, which are respectively
associated with cell membrane damage, inhibition of DNA
topoisomerase IIα activity and cross-linkage of double-stranded DNA
(9,10), nevertheless, raise the question as
to how the two distinct anticancer agents converge on DDH
expression. In particular in the latter case, in which cisplatin
introduces DNA cross-linkage, as well as DNA strand breaks,
indicates that the overexpression of DDH may also be involved in
resistance to radiation (11). A
previous study by Hung et al (12) demonstrated that cancer cells with
higher expression levels of DDH, were indeed more resistant to
irradiation. In fact, among the four subtypes of AKR1C, only DDH
(AKR1C1) has been frequently detected in cancer cells, including
bladder, esophageal, gastric, NSCLC, ovarian, prostate and uterine
cervical cancer cells, suggesting that DDH acts as an oncogene in
cancer progression (1,3,8,13–17).
Chen et al (18,19) found that DDH overexpression
suppressed the production of reactive oxygen species (ROS) and
increased cisplatin resistance in ovarian and lung cancer cell
lines. The silencing of DDH expression on the other hand, increased
ROS levels and cisplatin sensitivity, supporting the data from
other studies. Kruidering et al (20) demonstrated that cisplatin induced
ROS production in the mitochondria by inhibiting glutathione
reductase and activities of the respiratory chain. However, the
increase in ROS production alone did not sufficiently kill the
cancer cells, suggesting that other mechanisms, apart from the
decrease in intracellular ROS levels by DDH may play a role in the
inhibition of cell death. Nonetheless, no particular inhibitor
against DDH has been identified to date. In this study, we
therefore used DDH as a target enzyme in a live-cell enzyme-linked
immunosorbent assay (http://www.piercenet.com) to screen a panel of Chinese
medicinal herb extracts (CMHEs) in order to identify an inhibitor
of DDH expression. The function of the potentially effective
extracts, which were further fractionated by high-performance
liquid chromatography (HPLC), was determined by immunoblot analysis
and subsequent cell function analysis.
Materials and methods
Cell culture
Culture media and fetal calf serum (FCS) were from
Gibco Laboratories (Grand Island, NY, USA). All other materials
were of reagent grade and were obtained from Sigma (St. Louis, MO,
USA), and Merck (Darmstadt, Germany). The lung cancer cells, H125,
H226, H23, H838, H1437, H2009, H2087 and A549, the breast cancer
cells, ZR-75-1, BT-20, MCF-1, MCF-7 and T47D, as well as the
gastric cancer cells, AGS, KOTA-III, NUGC-1, NUGC-3 and SC-M1, were
purchased from the American Type Culture Collection (Manassas, VA,
USA), and were grown in monolayer in RPMI-1640 plus 10% FCS. All
cultures were incubated at 37°C and all media were supplemented
with 3 mM glutamine, penicillin (100 IU/ml) and streptomycin (100
μg/ml).
Live-cell enzyme-linked immunosorbent
assay (LCELISA) and colony forming assay for the determination of
drug and radiation sensitivity
In a 96-well plate, 2,000 lung cancer cells were
seeded into each well, and allowed to attach to the bottom of the
well for at least 18 h. Supernatants from 796 species of Chinese
herbs, which were prepared at 0.5 g/ml by collecting the
supernatant from 0.5% ethanol extracts of the herbal powder. The
extract was heated at 65°C for 30 min prior to collecting the
supernatant by filtering through a 0.45 nm aseptic disc. The
supernatant was respectively added to the wells at 1:200, 1:500 or
1:1250 dilutions, and incubated at 37°C for 72 h before fixing
cells with 4% paraformaldehyde in phosphate-buffered saline (PBS)
at room temperature for 15 min. The cells were perforated with 0.1%
Triton X-100 for 5 min prior to the addition of antibodies to DDH
to each well. The presence of DDH was detected by indirect
immunocytochemistry.
Drug and radiation sensitivity were measured by the
number of cells killed (21).
Cells were seeded at 100, 1,000 and 10,000 cells/6 cm plate 18 h
prior to the drug or radiation challenge. The cells were treated
with various doses of radiation or with various concentrations of
anticancer drugs, such as cisplatin, for 2 h before removing drugs.
The negative control groups included cells without radiation or
cells treated with the same dilution of DMSO that was used as the
solvent for the drug. The total number of survived cells was
determined seven to ten days following drug challenge by crystal
violet staining. The percentage survival of cells was quantified by
comparing with the control group.
Immunoblot analysis
The procedure for immunoblot analysis was carried
out as previously described (1,3).
Briefly, 5×106 cells were washed with PBS twice and
lysed in loading buffer [50 mM Tris (pH 6.8), 150 mM NaCl, 1 mM
disodium EDTA, 5% β-mercaptomethanol, 1 mM phenylmethylsulfonyl
fluoride, 10% glycerol, 1% SDS and 0.01% bromophenol blue].
Electrophoresis was carried out in a 10% polyacrylamide gel with
4.5% stacking gel. Following electrophoresis, the proteins were
transferred oonto a nitrocellulose membrane. The membrane was then
probed with specific antibodies. The signal was amplified by
biotin-labeled goat anti-mouse IgG and peroxidase-conjugated
streptavidin. Protein was visualized by exposing the membrane to an
X-Omat film (Eastman Kodak, Rochester, NY, USA) with enhanced
chemiluminescence reagent (Pierce, Rockford, IL, USA).
Electron microscopy
Electron microscopy was performed following a
previously published protocol (22,23). Briefly, the cells were fixed with
2.5% glutaraldehyde (EM grade; Sigma) in 100 mM phosphate buffer
(PB, pH 7.2) at 4°C for 18 h. The cells were rinsed with PB prior
to post-fixation with 1% osmium tetroxide. After removal of the
fixative with distilled water, the cells were suspended in 2%
molten agar (42°C), and the agar was then allowed to solidify. The
trimmed agar blocks were dehydrated in a serial dilution of ethanol
(absolute alcohol) for 15 min each, and then infiltrated with 100%
ethanol/LR white (1:1) mixture for 18 h. The blocks were changed to
pure LR white (Agar Scientific Ltd., Essex, UK) and infiltration
was continued at 4°C for 24 h, prior to transfer to a capsule
filled with LR white, and were then polymerized and solidified at
60°C for 48 h. The trimmed resin blocks were cut using an
ultramicrotome (Leica Ultracut R; Leica Microsystems GmbH, Vienna,
Austria), and the thin sections were transferred onto 200 mesh
copper grids. The specimens were stained with 2% uranyl acetate for
30 min, and 2.66% lead citrate (pH 12.0) for 10 min prior to
observation using an electron microscope (JEM-1400; Jeol USA, Inc.,
Peabody, MA, USA) at 100–120 kV.
Immunofluorescence staining and
immunofluorescence microscopy
For immunofluorescence microscopy, the cellular
uptake of MitoTracker® Green FM (Molecular Probes, Inc.,
Eugene, OR, USA) was used to label the mitochondria (22,24). The cells were then fixed with 4%
formaldehyde at room temperature for 15 min. After washing with
PBS, the cells were incubated with the primary antibodies to DDH as
previously described (1) or
ceramide (Enzo Life Sciences, Inc., Farmingdale, NY, USA) for 90
min and then washed with PBS. The secondary antibodies used were
rhodamine (TRITC)-conjugated rabbit anti-mouse IgG (Jackson
Laboratories, West Grove, PA, USA). The nuclei were stained with
4′,6-diamidino-2-phenylindole (DAPI). The slide was examined and
images were captured using an immunofluorescence microscope with an
UIS2 optical system (Olympus BX51; Olympus Corp., Tokyo, Japan).
The images were processed using Olympus DP2-BSW image capture
software (Olympus Corp.) and Adobe Photoshop 7.01 software (Adobe
Systems, Inc., San Jose, CA, USA).
The cells, which were transfected with
organelle-specific plasmids, human phosphatidylserine synthase 1
[FH-hPSS1, a marker enzyme for the endoplasmic reticulum (ER),
mitochondria-associated membrane (MAM) and microsomes] or
galactosyltransferase-conjugated green fluorescent protein (GT-GFP,
as a marker for the Golgi apparatus) for 24 h, were seeded onto
slides. After 48 h, the cells were fixed with 4% paraformaldehyde
for 15 min at room temperature and analyzed by fluorescence
microscopy. For the ceramide- and FH-hPSS1-expressing cells, the
cells were permeabilized with 0.1% Triton X-100 for 15 min at room
temperature, and subsequently stained with mouse anti-HA tag (Santa
Cruz Biotechnology, Inc., Santa Cruz, CA, USA), and
fluorescein-conjugated anti-mouse IgG antibodies (Invitrogen,
Carlsbad, CA, USA).
Statistical analysis
The two-way ANOVA test was performed using GraphPad
Prism 5 statistical software (GraphPad Software. Inc., San Diego,
CA, USA). A p-value <0.05 was considered to indicate a
statistically significant difference.
Results
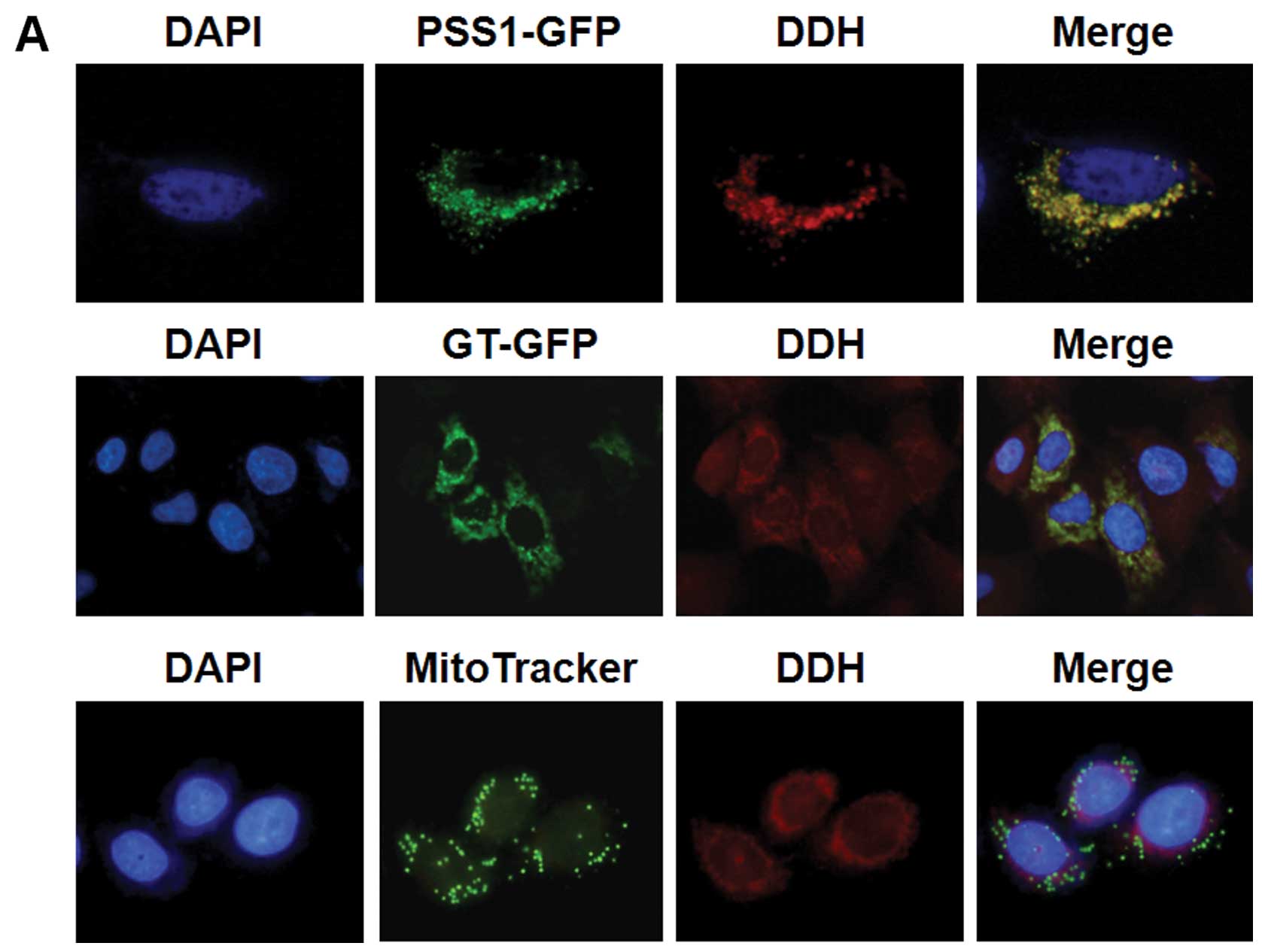
Selection of cancer cell lines with high
levels of DDH expression
Among the eight lung cancer cell lines screened by
immunoblot analysis, DDH was highly expressed in the H838, H1437
and A549 cells, and weakly expressed in the H226 and H2087 cells.
DDH was not detected in the H23, H125 and H2009 cells (Fig. 1A). Among the five breast cancer
cell lines, DDH was detected in the BT-20, MCF-1 and MCF-7 cells
(Fig. 1B), and among the five
gastric cancer cell lines, DDH was detected in the KATO-III, NUGC-1
and SC-M1 cells (Fig. 1C). We
therefore selected to use the H838, BT-20 and KATO-III cells in the
screening of the 796 CMHEs, which may contain vital ingredients
that could suppress DDH protein expression, but not immediately
kill the cells.
 | Figure 1Selection of cancer cell lines with a
high expression of dihydrodiol dehydrogenase (DDH). (A) Among the
eight lung cancer cell lines examined by immunoblot analysis, DDH
was highly expressed in the H838, H1437 and A549 cells, and weak
expressed in the H226 and H2087 cells. DDH was not detected in the
H23, H125 and H2009 cells. (B) Among the five breast cancer cell
lines, DDH was detected in the BT-20, MCF-1 and MCF-7 cells. (C)
Among the five gastric cancer cell lines, DDH was detected in the
KATO-III, NUGC-1 and SC-M1 cells. The H838, BT-20 and KATO-III
cells were thus selected for use in screening Chinese medicinal
herb extracts (CMHEs), which may contain ingredients that inhibit
DDH protein expression, but not immediately kill the cells. Our aim
was to find CMHE ingredients, which can inhibit DDH activity, and
those that are specific for cancer progression. The addition of
these ingredients could then increase cytosensitivity to anticancer
drugs and radiation. |
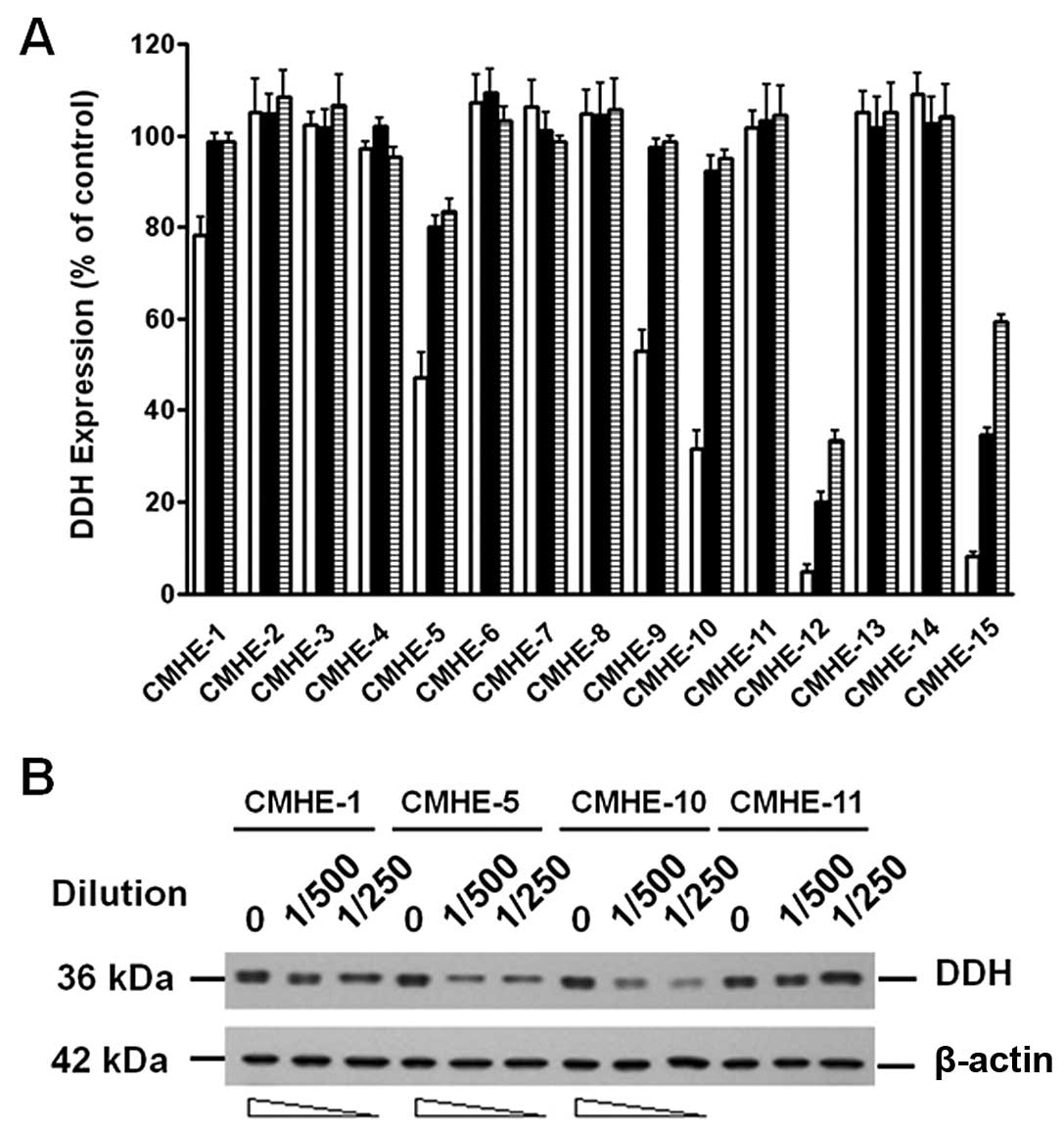
Effects of CMHE on the suppression of DDH
protein expression as determined by LCELISA and immunoblot
analysis
Using LCELISA to screen the 796 CMHEs, we identified
49 extracts that suppressed DDH protein expression (Fig. 2A). The results were confirmed by
immunoblot analysis (Fig. 2B).
Although some CMHEs suppressed DDH protein expression and induced
cell death, most of the CMHEs induced DDH expression and enhanced
cell growth.
Isolation and characterization of pure
compounds in CMHEs which suppress DDH expression
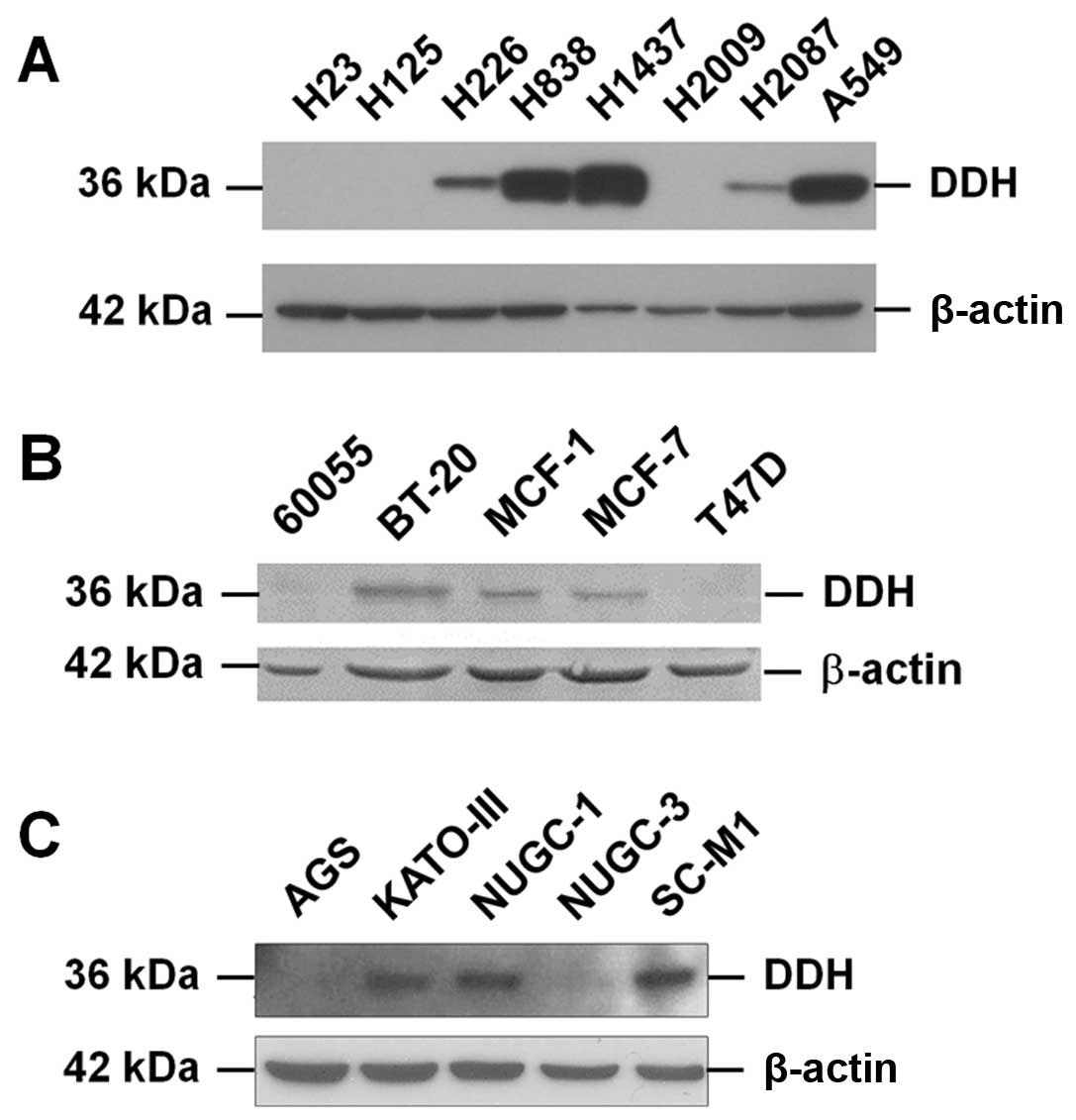
As shown in Fig.
2A, as CMHE-12 [from Nothapodytes foetida (Wight)
Sleumer] and CMHE-15 (from Koelreuteria henryi Dummer) had a
greater suppressive effect on DDH expression, we thus fractionated
the pure compounds using HPLC from these two extracts and
characterized the anti-DDH activities. The major compound isolated
from CMHE-12, which markedly inhibited DDH expression, turned out
to be camptothecin (25), which
induced the cleavage of poly(ADP-ribose) polymerase (PARP)
(Fig. 3A) and apoptosis. From
CMHE-15, among the final six fractions, four compounds were
identified by nuclear magnetic resonance (NMR) (Fig. 3B); however, only two compounds
contained effective ingredients (Fig.
3C). Similar to CMHE-12, compound G
(apigenin-4′-O-β-glucopyranoside) induced PARP cleavage and
apoptosis, but did not markedly inhibit DDH expression, nor
synergistically enhance the cytotoxicity of cisplatin, adriamycin,
vincristine, etoposide or radiation (data not shown). Compound B,
which intermediately suppressed DDH protein expression, however,
did not extensively induce PARP cleavage, but synergistically
enhanced the cytotoxic effects of the anticancer drugs and
irradiation. Using NMR, compound B was characterized as
kaempferol-3-O-glucoside (Fig.
3C), also known as astragalin, an abundant ingredient in the
Chinese medicinal herb, Astragalus membranaceus (Huang-qi)
(26). Although by conformation,
compound I (austrobailignan-1) resembled epipodophyllotoxins, it
did not induce PARP cleavage.
 | Figure 3Isolation and characterization of
pure compounds in the Chinese medicinal herb extract (CMHE) which
markedly suppressed dihydrodiol dehydrogenase (DDH) protein
expression. As CMHE-12 and -15 had the strongest inhibitory effect
on DDH protein expression, the active ingredients in these two
plant extracts were fractionated using high-performance liquid
chromatography (HPLC), and characterized by LCELISA as well as
immunoblot analysis. (A) From CMHE-12, the major ingredient
isolated was identified to be camptothecin, which not only reduced
DDH protein expression, but also induced the cleavage of
poly(ADP-ribose) polymerase (PARP), a marker of apoptosis. (B)
Among the six final fractions of CMHE-15, five contained
ingredients which effectively inhibited DDH expression. (C) The
identity of four compounds was determined by nuclear magnetic
resonance. Compound C (kaempferol-3-O-a-arabinoside) and compound I
(austrobailignan) did not effectively inhibit DDH or induce
apoptosis. Although compound G (apigenin-4′-O-b-D-glucopyranoside)
markedly induced the cleavage of PARP, it did not effectively
inhibit DDH expression nor did it synergistically enhance the
effects of the anticancer drugs or radiation (data not shown). As
compound B (kaempferol-3-O-glucoside, astragalin), intermediately
suppressed DDH protein expression, but did not extensively induce
PARP cleavage, its cytological effects were further examined. |
Cytological effects of astragalin
Astragalin, which intermediately inhibited DDH
protein expression, did not induce the cleavage of PARP (Fig. 4A), but reduced the levels of
ATPase family AAA domain containing 3A (ATAD3A) and dynamin-related
protein-1 (DRP-1), and induced the conversion of LC-3-I to LC3-II
(Fig. 4B). Although astragalin
increased the levels of pro-caspase-3, pro-caspase-8 and Bax
(Fig. 4C), no evident activation
of pro-caspase-3 or pro-caspase-8 cleavage was observed. On the
other hand, astragalin reduced the mitochondrial membrane potential
(MMP) (Fig. 4D), suggesting that
astragalin induces cell death through mechanisms other than the
apoptotic pathway. In fact, the addition of astragalin increased
the intracellular numbers of autophagic vacuoles (Fig. 4E), indicating that astragalin
indeed induced autophagy (Fig.
4F). In this way, the addition of astragalin enhanced the
cytosensitivity of the cancer cells to the anticancer drugs and
radiation (Fig. 4G).
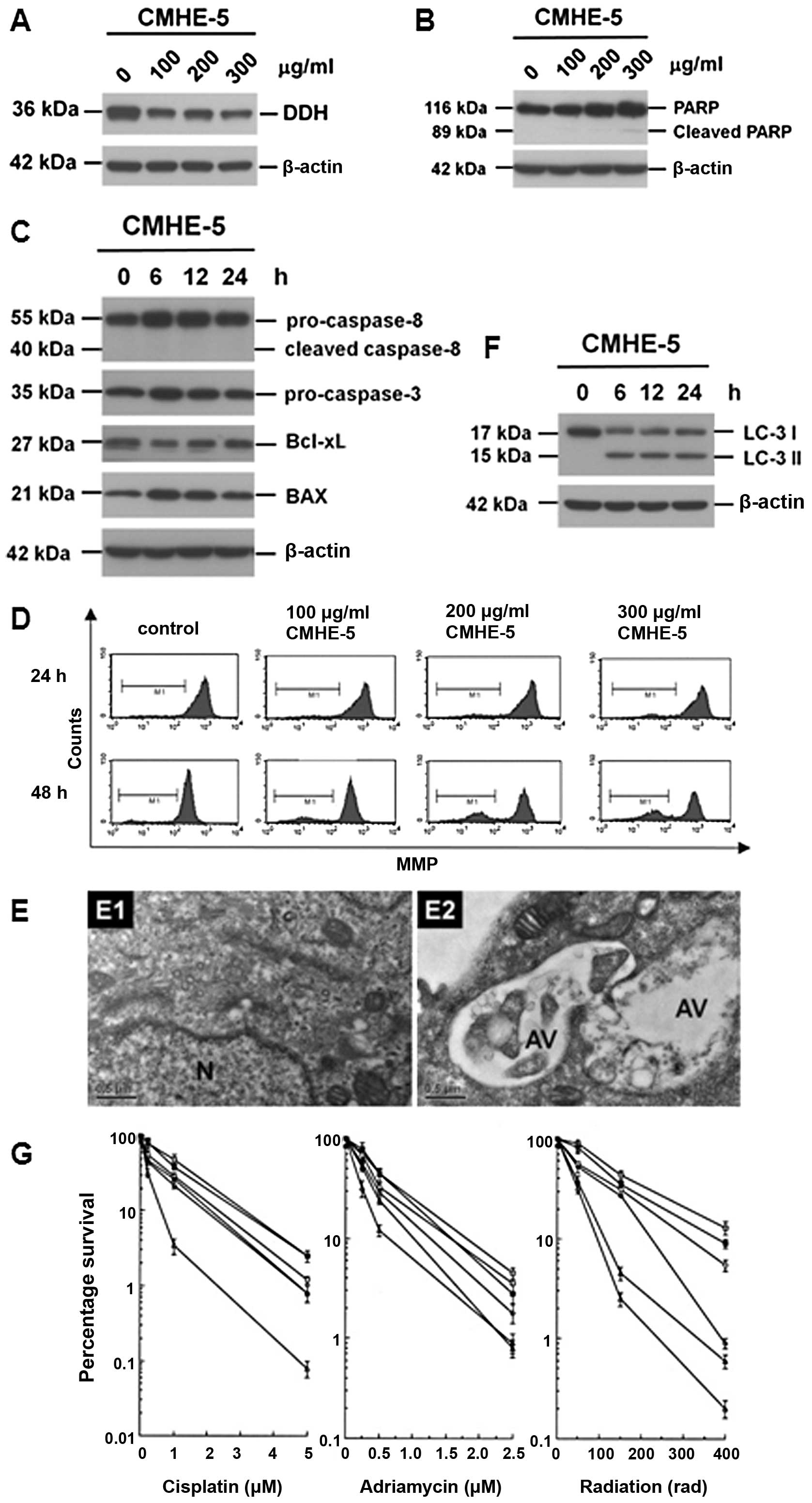 | Figure 4Cytological changes induced by
astragalin. (A) Astragalin inhibited dihydrodiol dehydrogenase
(DDH) expression in a time-dependent manner. However, it did not
induce the extensive cleavage of poly(ADP-ribose) polymerase
(PARP). (B) Astragalin also suppressed the expressions of ATPase
family AAA domain containing 3A (ATAD3A) and dynamin-related
protein–1 (DRP-1), and increased the conversion of LC-3-I to
LC3-II, suggesting that astragalin may be able to induce autophagy.
(C) Astragalin markedly increased the protein levels of
pro-caspase-3, pro-caspase-8 and Bax, but it did not induce the
cleavage of pro-caspase-3 or -8. (D) Treatment with astragalin
reduced mitochondrial membrane potential (MMP), supporting the
previous observations that astragalin may be able to induce cell
death, other than apoptosis. (E) Astragalin increased intracellular
numbers of autophagic vacuoles as determined by electron
microscopy. (F) Astragalin increased the conversion of LC3-I to
LC-3II, an authentic autophagic marker as determined by immunoblot
analysis. (G) Astragalin increased the cytosensitivity of cancer
cells to the anticancer drugs, such as cisplatin and adriamycin, as
well as radiation. |
Astragalin reduces ceramide distribution
to the plasma membrane, but increases intracellular autophagic
vesicles
As shown in Fig.
5A, using immunofluorescence microscopy, DDH was localized to
the ER, MAM, microsomes (Fig. 5A,
top panel) and Golgi apparatus (Fig.
5A, middle panel), but not on the mitochondria (Fig. 5A, lower panel). The addition of
astragalin, however, did not affect DDH localization to the
organelles (data not shown).
On the other hand, astragalin altered the
intracellular distribution of ceramide. Ceramide (27), which is normally localized on the
intracellular vesicular structures and plasma membrane (Fig. 5B, left panel), was accumulated on
MAM- or Golgi-like structures (Fig.
5B, center panel) 6 h following treatment with astragalin. At
18 h post-astragalin treatment, ceramide was amassed on the
autophagic vesicles (Fig. 5B,
right panel), suggesting that the inhibition of DDH expression
disrupted the intracellular transportation/distribution of
ceramide.
Discussion
Our data demonstrate that extracts from
Taiwan-endemic roadside trees, Koelreuteria henryi Dummer,
contained at least two ingredients, which respectively induced
apoptosis and autophagy, thus enhancing the cytotoxic effects of
the anticancer drugs and irradiation. Using HPLC and NMR, one of
the active compounds isolated from the plant extracts was
kaempferol-3-O-glucoside (or astragalin), a major ingredient in the
traditional Chinese medicinal herb, Astragalus membranaceus
(Huang-qi). Unlike herbal Astragalus membranaceus though,
Koelreuteria henryi Dummer is deciduous and the contents of
astragalin and apigenin-4′-O-β-glucopyranoside fluctuate as the
seasons change. Apigenin-4′-O-β-glucopyranoside extensively induced
caspase-dependent apoptosis. However, astragalin only induced
autophagy possibly by interrupting the protein and lipid flow among
the organelles, and this concept was clearly supported by the
immunofluorescence microscopic observations that astragalin
markedly altered the intracellular distribution of ceramide, which
was accumulated on the MAM- and Golgi-like finite
configurations.
The identification of DDH on the microsomes, Golgi
apparatus, ER and MAM, but not on the mitochondria consolidated our
theory, which supports not only previous studies, in which DDH has
been shown to deactivate doxorubicin, an anticancer drug with a
polycyclic hydrophobic side-chain (4,5),
but also evidence that DDH catabolizes hydrophobic xenobiotic
compounds, in particular polycyclic aromatic hydrocarbons (PAHs),
which are more likely to be associated with lipid membranes
(4–7). Furthermore, the results corresponded
well with the observations that DDH had the worst effect on tumor
progression when the enzyme was concurrently overexpressed with
microsomal epoxide hydrolase (28). As reported previously, Chen et
al (18,19) found that higher DDH levels
correlated with the reduction of mitochondrial ROS concentrations.
The silencing of DDH, on the other hand, increased ROS levels.
Moreover, Nie et al (29)
showed that elevated intracellular ROS levels increased the
oxidation of cys62 in Bax, which then induced the
membrane perforation of the mitochondria and triggered apoptosis
(30–32). However, by demonstrating that DDH
was not located on the mitochondria and that changes in MMP did not
occur after 24 h of astragalin treatment, our data suggest that the
effects of DDH on ROS production may be indirect and that the cell
death mediated by astragalin may be autophagic.
Recently, we found a novel import passage of the
mitochondria, which originates from the ER and via transport
vesicles (22,24). This pathway requires DRP-1
(33), ATAD3A (24) and mitofusin-2 (Mfn2) (22,24). The knockdown of the ATAD3A gene
markedly reduced the protein levels of DDH and global DNA repair,
but increased autophagy (22,24). The silencing of DRP-1 expression
induced a comparable autophagic response (33). Of note, apart from DDH, astragalin
inhibited the expression of DRP-1 and ATAD3A. A poor supply of
cytoplasmic DRP-1, on the other hand, increases mitochondrial
fusion and the bulging of MAM, a specific region of the ER, part of
which tethers the ER and mitochondria together by Mfn2 on both
organelles to coordinate Ca2+ flow (34,35), and from which the newly
synthesized proteins, phospholipids and sphingolipids are
respectively via transport vesicles imported to the mitochondria
(22,24) or exported to the Golgi and plasma
membrane (3).
In fact, the cellular location of ceramide, which is
synthesized specifically on the inner leaflet of the ER and is
finally located on the outer leaflet of the plasma membrane
following export through the Golgi apparatus and transport vesicles
(27), has casted an intriguing
question as to its biological function. In particular, the
exogenously added ceramide induced autophagy (36). It is worth noting that ceramide is
a hydrophobic compound, and is associated with membrane structure
or carrier proteins. Under normal conditions, it is rapidly
converted to sphingosine by cerases or sphingomyelin by
sphingomyelin synthase, preventing the accidental induction of
differentiation or cell death (27,36).
Previous studies have demonstrated that the
silencing of DDH expression reduces the levels of vacuolar protein
sorting (Vps)4 (37), an
essential ATPase for the movement of multivesicular body (MVB) in
the endosomal sorting complex required for transport (ESCRT), and
increases those of autophagy-related gene (Atg) 16L2 (38). Of note, ATAD3A shares some
homology with Vps4 in the ATPase domain and with Atg16L2 in
coiled-coil domains. The silencing of Vps4 induces autophagy as
well. Although these results are yet to be verified, these data,
together with evidence of DDH collaborating with epoxide hydrolase
to fully detoxify hydrophobic PAHs into catechol (4–7,39–41), suggest that these enzymes are
located in the near vicinity of membranous structures to guard the
proper intracellular trafficking of proteins and lipids. Since the
type of epoxide hydrolase, which has been commonly detected in
cancer cells, is microsomal epoxide hydrolase (28), it is possible that DDH is also
located in the immediate vicinity of these subcellular structures,
which are originally from the ER and MAM. Our results demonstrated
that DDH was not only located on the ER and MAM, but also on the
Golgi apparatus, indicating that it may have a biological role
apart from PAH detoxification. The interruption of these processes,
i.e., by RNAi or small molecules derived from CMHEs, would
certainly affect the enzyme activity involved in lipid flow and
thus lead to the accumulation of toxic lipids in the organelles and
interorganelle transport vesicles, inducing aberrant autophagy and
inhibiting tumor progression.
In conclusion, our results revealed that DDH can
indeed be used as a target enzyme for screening CMHEs, which
contained potential ingredients able to inhibit DDH expression. The
inhibition of DDH function reduced the expression of anti-apoptotic
proteins, such as Bcl-xL, but increased the expression of
pro-apoptotic proteins, such as Bax, and caspase precursors,
including pro-caspase-3 and -8. However, the suppression of DDH
alone did not induce apoptosis, but induced autophagy, thus
enhancing the sensitivity to anticancer drugs and radiation. This
weakening activity was specific for cancer cells, which not only
highly expressed DDH, but also DRP-1, ATAD3A and eukaryotic
elongation factor 2 (eEF2) (24,33,42,43), a condition that was not detected
in non-transformed cells. Moreover, some of the purified
prospective ingredients concurrently inhibited both DDH and ATAD3A,
and the latter is an ATPase which is closely associated with the
intracellular transport of proteins, phospholipids and
sphingolipids. The silencing of ATAD3A induced massive autophagy
and enhanced cytosensitivity to anticancer drugs and radiation,
suggesting that these prospective ingredients may not only
selectively target DDH, but also a spectrum of cancer
progression-related genes, which are involved in the interorganelle
transportation of essential proteins and lipids.
Acknowledgements
The present study was supported by grants from the
Department of Health, Executive Yuan, Taipei, Taiwan to the China
Medical University Hospital, the Cancer Research of Excellence
program (DOH102-TD-C-111-005), Taichung, Taiwan, and the National
Science Council (NSC, 101-2320-B-005-002), Taipei, Taiwan.
References
|
1
|
Hsu NY, Ho HC, Chow KC, et al:
Overexpression of dihydrodiol dehydrogenase as a prognostic marker
of non-small cell lung cancer. Cancer Res. 61:2727–2731.
2001.PubMed/NCBI
|
|
2
|
Chen CY, Hsu CP, Hsu NY, Shih CS, Lin TY
and Chow KC: Expression of dihydrodiol dehydrogenase in the
resected stage I non-small cell lung cancer. Oncol Rep. 9:515–519.
2002.PubMed/NCBI
|
|
3
|
Huang KH, Chiou SH, Chow KC, et al:
Overexpression of aldo-keto reductase 1C2 is associated with
disease progression in patients with prostatic cancer.
Histopathology. 57:384–394. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Shen H, Kauvar L and Tew KD: Importance of
glutathione and associated enzymes in drug response. Oncol Res.
9:295–302. 1997.PubMed/NCBI
|
|
5
|
Ax W, Soldan M, Koch L and Maser E:
Development of daunorubicin resistance in tumour cells by induction
of carbonyl reduction. Biochem Pharmacol. 59:293–300. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Shou M, Harvey RG and Penning TM:
Contribution of dihydrodiol dehydrogenase to the metabolism of
(+/−)-trans-7,8-dihydroxy-7,8-dihydrobenzo[a]pyrene in fortified
rat liver subcellular fractions. Carcinogenesis. 13:1575–1582.
1992.PubMed/NCBI
|
|
7
|
Flowers-Geary L, Harvey RG and Penning TM:
Cytotoxicity of polycyclic aromatic hydrocarbon o-quinones in rat
and human hepatoma cells. Chem Res Toxicol. 6:252–260. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Deng HB, Parekh HK, Chow KC and Simpkins
H: Increased expression of dihydrodiol dehydrogenase induces
resistance to cisplatin in human ovarian carcinoma cells. J Biol
Chem. 277:15035–15043. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chow KC, MacDonald TL and Ross WE: DNA
binding by epipodophyllotoxins and N-acyl anthracyclines:
implications for mechanism of topoisomerase II inhibition. Mol
Pharmacol. 34:467–473. 1988.PubMed/NCBI
|
|
10
|
Hill BT, Shellard SA, Hosking LK,
Fichtinger-Schepman AM and Bedford P: Enhanced DNA repair and
tolerance of DNA damage associated with resistance to
cis-diammine-dichloroplatinum (II) after in vitro exposure of a
human teratoma cell line to fractionated X-irradiation. Int J
Radiat Oncol Biol Phys. 19:75–83. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sorenson CM and Eastman A: Mechanism of
cis-diamminedichloroplatinum(II)-induced cytotoxicity: role of G2
arrest and DNA double-strand breaks. Cancer Res. 48:4484–4488.
1988.PubMed/NCBI
|
|
12
|
Hung JJ, Chow KC, Wang HW and Wang LS:
Expression of dihydrodiol dehydrogenase and resistance to
chemotherapy and radiotherapy in adenocarcinoma cells of lung.
Anticancer Res. 26:2949–2955. 2006.PubMed/NCBI
|
|
13
|
Wang LS, Chow KC, Wu YC, Lin TY and Li WY:
Inverse expression of dihydrodiol dehydrogenase and
glutathione-S-transferase in patients with esophageal squamous cell
carcinoma. Int J Cancer. 111:246–251. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chow KC, Lu MP and Wu MT: Expression of
dihydrodiol dehydrogenase plays important roles in apoptosis- and
drug-resistance of A431 squamous cell carcinoma. J Dermatol Sci.
41:205–212. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ueda M, Hung YC, Chen JT, et al: Infection
of human papillomavirus and overexpression of dihydrodiol
dehydrogenase in uterine cervical cancer. Gynecol Oncol.
102:173–181. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tai HL, Lin TS, Huang HH, et al:
Overexpression of aldo-keto reductase 1C2 as a high-risk factor in
bladder cancer. Oncol Rep. 17:305–311. 2007.PubMed/NCBI
|
|
17
|
Chang HC, Chen YL, Chan CP, et al:
Overexpression of dihydrodiol dehydrogenase as a prognostic marker
in resected gastric cancer patients. Dig Dis Sci. 54:342–347. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chen J, Adikari M, Pallai R, Parekh HK and
Simpkins H: Dihydrodiol dehydrogenases regulate the generation of
reactive oxygen species and the development of cisplatin resistance
in human ovarian carcinoma cells. Cancer Chemother Pharmacol.
61:979–987. 2008. View Article : Google Scholar
|
|
19
|
Chen J, Emara N, Solomides C, Parekh H and
Simpkins H: Resistance to platinum-based chemotherapy in lung
cancer cell lines. Cancer Chemother Pharmacol. 66:1103–1111. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kruidering M, Van de Water B, de Heer E,
Mulder GJ and Nagelkerke JF: Cisplatin-induced nephrotoxicity in
porcine proximal tubular cells: mitochondrial dysfunction by
inhibition of complexes I to IV of the respiratory chain. J
Pharmacol Exp Ther. 280:638–649. 1997.
|
|
21
|
Chow KC, King CK and Ross WE: Abrogation
of etoposide-mediated cytotoxicity by cycloheximide. Biochem
Pharmacol. 37:1117–1122. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chiang SF, Huang CY, Lin TY, Chiou SH and
Chow KC: An alternative import pathway of AIF to the mitochondria.
Int J Mol Med. 29:365–372. 2012.PubMed/NCBI
|
|
23
|
Yoon Y, Pitts KR, Dahan S and McNiven MA:
A novel dynamin-like protein associates with cytoplasmic vesicles
and tubules of the endoplasmic reticulum in mammalian cells. J Cell
Biol. 140:779–793. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Fang HY, Chang CL, Hsu SH, et al: The
ATPase family, AAA domain containing 3A is a novel anti-apoptotic
factor in lung adenocarcinoma cells. J Cell Sci. 123:1171–1180.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Aiyama R, Nagai H, Nokata K, Shinohara C
and Sawada S: A camptothecin derivative from Nothapodytes
foetida. Phytochemistry. 27:3663–3664. 1988. View Article : Google Scholar
|
|
26
|
Kwon HJ and Park YD: Determination of
astragalin and astragaloside content in Radix Astragali using
high-performance liquid chromatography coupled with pulsed
amperometric detection. J Chromatogr A. 1232:212–217. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zheng W, Kollmeyer J, Symolon H, et al:
Ceramides and other bioactive sphingolipid backbones in health and
disease: lipidomic analysis, metabolism and roles in membrane
structure, dynamics, signaling and autophagy. Biochim Biophys Acta.
1758:1864–1884. 2006. View Article : Google Scholar
|
|
28
|
Lin TS, Huang HH, Fan YH, Chiou SH and
Chow KC: Genetic polymorphism and gene expression of microsomal
epoxide hydrolase in non-small cell lung cancer. Oncol Rep.
17:565–572. 2007.PubMed/NCBI
|
|
29
|
Nie C, Tian C, Zhao L, Petit PX, Mehrpour
M and Chen Q: Cysteine 62 of Bax is critical for its conformational
activation and its proapoptotic activity in response to
H2O2-induced apoptosis. J Biol Chem.
283:15359–15369. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Szabò I, Soddemann M, Leanza L, Zoratti M
and Gulbins E: Single-point mutations of a lysine residue change
function of Bax and Bcl-xL expressed in Bax- and Bak-less mouse
embryonic fibroblasts: novel insights into the molecular mechanisms
of Bax-induced apoptosis. Cell Death Differ. 18:427–438. 2011.
|
|
31
|
Griparic L, van der Wel NN, Orozco IJ,
Peters PJ and van der Bliek AM: Loss of the intermembrane space
protein Mgm1/OPA1 induces swelling and localized constrictions
along the lengths of mitochondria. J Biol Chem. 279:18792–18798.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Fang HY, Chen CY, Chiou SH, et al:
Overexpression of OPA1 protein increases cisplatin resistance via
inactivation of caspase-dependent apoptosis in lung adenocarcinoma
cells. Hum Pathol. 43:105–114. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chiang YY, Chen SL, Hsiao YT, et al:
Nuclear expression of dynamin-related protein 1 in lung
adenocarcinomas. Mod Pathol. 22:1139–1150. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
de Brito OM and Scorrano L: Mitofusin 2
tethers endoplasmic reticulum to mitochondria. Nature. 456:605–610.
2008.PubMed/NCBI
|
|
35
|
Merkwirth C and Langer T: Mitofusin 2
builds a bridge between ER and mitochondria. Cell. 135:1165–1167.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Scarlatti F, Bauvy C, Ventruti A, et al:
Ceramide-mediated macroautophagy involves inhibition of protein
kinase B and up-regulation of beclin 1. J Biol Chem.
279:18384–18391. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hill CP and Babst M: Structure and
function of the membrane deformation AAA ATPase Vps4. Biochim
Biophys Acta. 1823:172–181. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ishibashi K, Fujita N, Kanno E, et al:
Atg16L2, a novel isoform of mammalian Atg16L that is not essential
for canonical autophagy despite forming an Atg12-5-16L2 complex.
Autophagy. 7:1500–1513. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Vogel K, Bentley P, Platt KL and Oesch F:
Rat liver cytoplasmic dihydrodiol dehydrogenase. Purification to
apparent homogeneity and properties. J Biol Chem. 255:9621–9625.
1980.PubMed/NCBI
|
|
40
|
Glatt HR, Vogel K, Bentley P and Oesch F:
Reduction of benzo(a)-pyrene mutagenicity by dihydrodiol
dehydrogenase. Nature. 277:319–320. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Penning TM: Dihydrodiol dehydrogenase and
its role in polycyclic aromatic hydrocarbon metabolism. Chem Biol
Interact. 89:1–34. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wang HW, Lin CP, Chiu JH, et al: Reversal
of inflammation-associated dihydrodiol dehydrogenases (AKR1C1 and
AKR1C2) overexpression and drug resistance in nonsmall cell lung
cancer cells by wogonin and chrysin. Int J Cancer. 120:2019–2027.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Chen CY, Fang HY, Chiou SH, et al:
Sumoylation of eukaryotic elongation factor 2 is vital for protein
stability and anti-apoptotic activity in lung adenocarcinoma cells.
Cancer Sci. 102:1582–1589. 2011. View Article : Google Scholar : PubMed/NCBI
|