Introduction
Hypoxic pulmonary hypertension (HPH) is an important
pathophysiological process and plays a key role in the development
of a variety of pulmonary diseases, including chronic obstructive
pulmonary disease (COPD), pulmonary fibrosis and pulmonary heart
disease. Multiple pathogenic pathways have been implicated in the
development of HPH, including those at the molecular and genetic
levels of smooth muscle and endothelial cells and adventitia. The
HPH ‘phenotype’ is characterized by a decreased ratio of
apoptosis/proliferation in pulmonary artery smooth muscle cells
(PASMCs) and a thickened, disordered adventitia (1). The increased proliferation and
reduced apoptosis of PASMCs is a component of pulmonary
hypertension, increased pulmonary vascular resistance and pulmonary
arterial pressure, which is associated with COPD and right
ventricular failure (2). Since
the proliferation of SMCs is an essential feature of vascular
proliferative disorders, considerable efforts have been made to
develop a therapeutic strategy that effectively suppresses SMC
proliferation.
Peroxisome proliferator-activated receptor γ (PPARγ)
agonists act as negative regulators of SMC growth (3). They are potent inhibitors of
vascular SMC proliferation in vitro and in vivo
(3). PPARγ agonists can stimulate
the production and secretion of apolipoprotein E (ApoE), which has
anti-proliferative effects on human PASMCs (4). Nisbet et al (5) found that rosiglitazone (a PPARγ
agonist) attenuated hypoxia-induced pulmonary vascular remodelling
and hypertension by suppressing oxidative and proliferative
signals. These studies provide the mechanisms underlying the
therapeutic effects of PPARγ activation in pulmonary
hypertension.
Studies have also suggested that PPARγ agonists
upregulate phosphatase and tensin homologue deleted on chromosome
10 (PTEN) expression in allergen-induced asthmatic lungs (6). Zhang et al (7) demonstrated that rosiglitazone
inhibits the migration of human hepatocellular BEL-7404 cells by
upregulating PTEN. The tumor suppressor gene, PTEN, encodes a
dual-specificity phosphatase that recognizes protein and
phosphatidylinositol substrates and modulates cellular functions,
such as migration and proliferation. However, the role of PPARγ in
regulating PTEN expression in PASMCs under hypoxic conditions has
not yet been elucidated. Therefore, the present study aimed to
examine the molecular mechanisms through which PPARγ activation
modulates PTEN expression in PASMCs under hypoxic conditions, since
hypoxia is a proliferative stimulator of PASMCs (5).
In the present study, we investigated the effects of
troglitazone (a PPARγ agonist) on PTEN gene expression and the
apoptosis of PASMCs under hypoxic conditions. We found that
troglitazone increased PTEN expression in PASMCs in a
concentration-dependent manner and increased the apoptosis of
PASMCs under hypoxic conditions.
Materials and methods
Cell culture of PASMCs
Human PASMCs were purchased from ScienCell Research
Laboratories (Carlsbad, CA, USA). The PASMCs were cultured and
expanded in SMC growth medium (GM): smooth muscle cell basal medium
(BM; ScienCell Research Laboratories) supplemented with SMC growth
supplement (ScienCell Research Laboratories), 10% fetal bovine
serum (FBS) and 1% penicillin/streptomycin (P/S). The cells were
cultured at 37ºC in a 5% CO2 incubator, and the cell
culture medium was changed every 2 days. Cells were regularly
passaged every 4–5 days.
Hypoxia treatment
The PASMCs were cultured under hypoxic conditions in
order to determine the anti-proliferative effects of troglitazone
on PASMCs. A hypoxic environment for PASMC culture was created in
an incubator: 5% CO2+94% N2+1% O2.
The cells were collected for gene and protein expression
experiments after 72 h. Generally, the PASMCs were seeded in plates
in BM overnight before being used in the experiments.
PASMC culture with PTEN inhibitor or
PPARγ antagonist
The PASMCs were seeded at 1×104
cells/well in a 12-well plate and cultured in GM supplemented with
2.5 μM dipotassium
bisperoxo(5-hydroxypyridine-2-carboxyl)oxovanadate (V)
[bpV(HOpic)], a PTEN inhibitor, or 1 μM
2-chloro-5-nitro-N-phenylbenzamide (GW9662), a PPARγ antagonist
(both were purchased from EMD4Biosciences, Darmstadt, Germany).
bpV(HOpic) and GW9662 were added to the cell culture medium 1 h
prior to the addition of troglitazone. The PASMCs were cultured in
an incubator under hypoxic conditions for 72 h. Cell proliferation
was determined using the CyQUANT® Cell Proliferation
assay kit (Invitrogen, Carlsbad, CA, USA). Proteins were extracted
from the treated cells using PhosphoSafe™ Extraction Reagent (EMD
Millipore, Billerica, MA, USA). Following quantification, the
proteins were used for western blot analysis.
Quantitative reverse transcription PCR
(qRT-PCR) analysis
PASMCs were analyzed by qRT-PCR to determine gene
expression at 0.5, 1, 2, 4, 8 and 24 h after treatment with
troglitazone. Total RNA was isolated as previously described
(8). DNase I was used to remove
DNA contamination. cDNA was synthesized using the
Maxima® First Strand cDNA Synthesis kit (Fermentas,
Hanover, MD, USA).
To quantify gene expression levels,
Maxima® SYBR-Green qPCR Master Mix (2X) (Fermentas) was
used. The qPCR thermal cycling protocol was programmed for 40
cycles: 1 cycle of initial denaturation for 10 min, then
denaturation at 95ºC for 15 sec, annealing for 30 sec and extension
at 72ºC for 30 sec. The primers used are listed in Table I.
 | Table IPrimers used for RT-PCR. |
Table I
Primers used for RT-PCR.
| Gene | Primer sequence | Annealing temp.
(ºC) | Size (bp) |
|---|
| GAPDH | Forward
5′-AGCCACATCGCTCAGACAC-3′
Reverse 5′-TAAAAGCAGCCCTGGTGAC-3′ | 60 | 90 |
| PTEN | Forward
5′-TCACCAACTGAAGTGGCTAAAGA-3′
Reverse 5′-CTCCATTCCCCTAACCCGA-3′ | 60 | 155 |
| AKT-1 | Forward
5′-TAACCTTTCCGCTGTCGC-3′
Reverse 5′-ATGTTGTAAAAAAACGCCG-3′ | 58 | 125 |
| AKT-2 | Forward
5′-GGTCGCCAACAGCCTCAA-3′
Reverse 5′-CACTTTAGCCCGTGCCTTG-3′ | 58 | 127 |
Cell proliferation
PASMC proliferation was determined using the
CyQUANT® Cell Proliferation assay kit (Invitrogen).
Briefly, 1×104 PASMCs/well were seeded into 24-well
plates and cultured with BM for 24 h. The cell culture medium was
changed to GM supplemented with various concentrations of
troglitazone (stock solution 20 mM in 100% DMSO) for 72 h in an
incubator under hypoxic conditions at 37ºC. Subsequently, the cell
culture supernatant was removed. The cells were washed with PBS and
frozen at −80ºC for 1 h. Each well was then incubated with 200 μl
CyQUANT® cell-lysis buffer containing DNase-free RNase
(1.35 U/ml) for 1 h at room temperature to eliminate the RNA
component of the fluorescence signal. Subsequently, 200 μl
cell-lysis buffer containing 2X solution of CyQUANT® GR
dye were added to each well for 10 min. The fluorescence intensity
was measured using a Tecan fluorescence microplate reader (Tecan
Infinite M200 microplate reader; LabX, Canada) with 480 nm
excitation and 520 nm emission.
Lactate dehydrogenase (LDH) release for
cytotoxic determination
The cytotoxicity of troglitazone towards PASMCs was
determined using CytoTox-ONE™ Homogeneous Membrane Integrity assay
(Promega, Madison, WI, USA). Briefly, 5×104 PASMCs/well
were cultured in a 12-well plate with GM supplemented with
troglitazone for 72 h under normoxic conditions at 37ºC.
Subsequently, cell culture supernatant was collected and mixed with
CytoTox-ONE™ Reagent for 20 min. After the addition of stop
solution, the fluorescence signal was measured at an excitation
wavelength of 560 nm and an emission wavelength of 590 nm.
Western blot analysis of PASMCs
Protein expression in the treated and non-treated
PASMCs was determined by western blot analysis as previously
described (9). Cells were lysed
with PhosphoSafe™ Extraction Reagent (EMD Millipore) and the
protein concentration was determined using Bradford reagent
(Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Proteins were separated and electrophoretically
blotted onto a nitrocellulose membrane. After washing with 10 mM
Tris/HCl wash buffer (pH 7.6) containing 0.05% Tween-20, the
membrane was incubated in blocking buffer (5% non-fat dry milk, 10
mM Tris pH 7.5, 100 mM NaCl, 0.1% Tween-20) for 3 h at room
temperature. Subsequently, the blots were incubated with diluted
primary antibodies: AKT (1:1,000), phosphorylated (Ser 473) AKT
(p-AKT) (1:500), glyceraldehyde-3-phosphate dehydrogenase (GAPDH)
(1:1,000) and PTEN (1:200) (all purchased from Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA) overnight at 4ºC.
Subsequently, anti-rabbit IgG conjugated with HRP (dilution,
1:3,000–1:8,000) was used to detect the binding of antibodies. The
binding of the specific antibody was visualized using the
SuperSignal Chemiluminescent Substrate kit and exposed to X-ray
film (both from Pierce Biotechnology, Inc., Rockford, IL, USA).
Apoptosis assay
Proteins from the treated and non-treated PASMCs
were used to determine the apoptosis of PASMCs by determining the
activities of caspase-3, -8 and -9.
Caspase-3 and -8 activities were determined using
caspase-3 and -8 assay kits, Fluorimetric (Sigma-Aldrich, Sigma,
St. Louis, MO, USA). The fluorescence intensity of caspase-3 was
recorded at a wavelength of 360 nm for excitation and a wavelength
of 460 nm for emission, and that of caspase-8 was recorded at an
excitation of 360 nm and an emission of 440 nm. The activity of
caspase was calculated as fluorescence intensity (FI)/min/ml =
ΔFlt/(t × v), where ΔFlt is the difference in fluorescence
intensity between time zero and time t minutes; t is the reaction
time in min; and v is the volume of the sample in ml.
Similarly, caspase-9 activity was determined using
the caspase-9 assay kit, Fluorimetric (QIA72; EMD4Biosciences). The
fluorescence intensity was recorded at a wavelength of 400 nm for
excitation and a wavelength of 505 nm for emission. The same
formula as the one used for the calculation of caspase-3 activity
was used to calculate caspase-9 activity.
TUNEL assay
To detect the apoptosis of PASMCs following
treatment with troglitazone, the PASMCs were cultured in BM and
incubated for 72 h under hypoxic conditions. TUNEL assay was
performed using the In situ Cell Death Detection kit (Roche)
as per the manufacturer's instructions. In brief, the cells were
fixed with 4% paraformaldehyde for 20 min at 25ºC. After washing
with PBS for 30 min, the cells were incubated in permeabilization
solution (0.1% Triton X-100, 0.1% sodium citrate) for 10 min at
4ºC. After washing, the cells were incubated with rabbit
anti-calponin antibody overnight at 4ºC. On the second day,
reaction mixture from the kit supplemented with goat anti-rabbit
IgG conjugated with FITC was applied to the cells for 1 h at 37ºC
in the dark. The samples were counterstained with DAPI to visualize
the nuclei after TUNEL staining. The apoptotic index was calculated
as TUNEL+ cells/total cells/low magnification field.
Statistical analysis
All statistical analyses were performed using SPSS
software (version 10.0). All experiments were performed at least 3
times. The data are presented as the means ± standard error means
(SEM) and analyzed with the method of analysis of variance (ANOVA)
using the Bonferroni test. A Student's t-test was performed to
determine statistical difference. All tests were performed with a
significance level of 5%.
Results
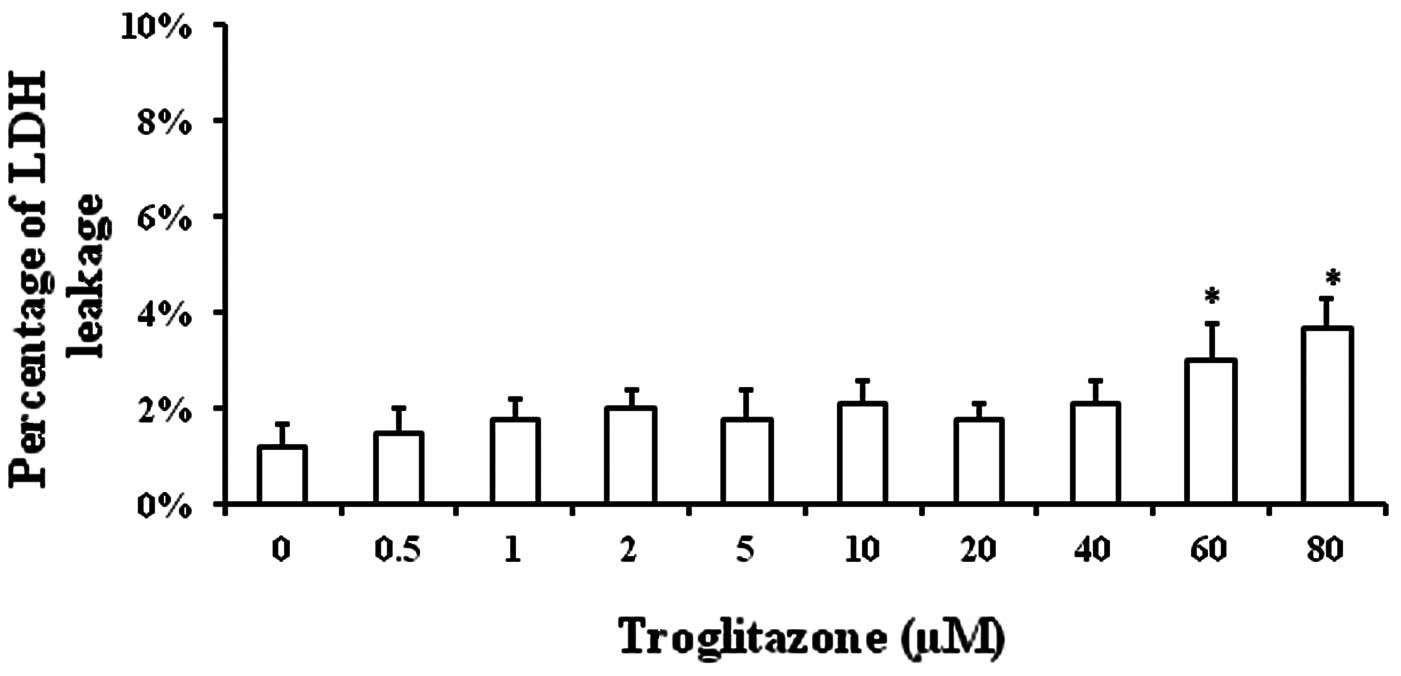
Toxicity of troglitazone towards
PASMCs
The toxicity of troglitazone towards the cells was
determined in order to exclude the possibility that the decrease in
PASMC proliferation was due to the toxicity of troglitazone towards
PASMCs. PASMCs were cultured in GM supplemented with troglitazone
for 48 h in an incubator under normal conditions. The supernatant
was collected to measure LDH concentration (an index of cell
injury). Significant cell injury was only observed when the
troglitazone concentration was increased up to 60 and 80 μM
(Fig. 1).
The percentage of LDH in the supernatant was
1.2±0.5% when the cells were cultured in GM without troglitazone,
while it was 1.5±0.5% with 0.5 μM troglitazone, 1.8±0.4% with 1 μM,
2±0.4% with 2 μM, 1.8±0.6% with 5 μM, 2.1±0.5% with 10 μM, 1.8±0.3%
with 20 μM, 2.1±0.5% with 40 μM, 3±0.8% with 60 μM and 3.7±0.6%
with 80 μM troglitazone (Fig. 1).
These results demonstrated that troglitazone was well tolerated by
PASMCs.
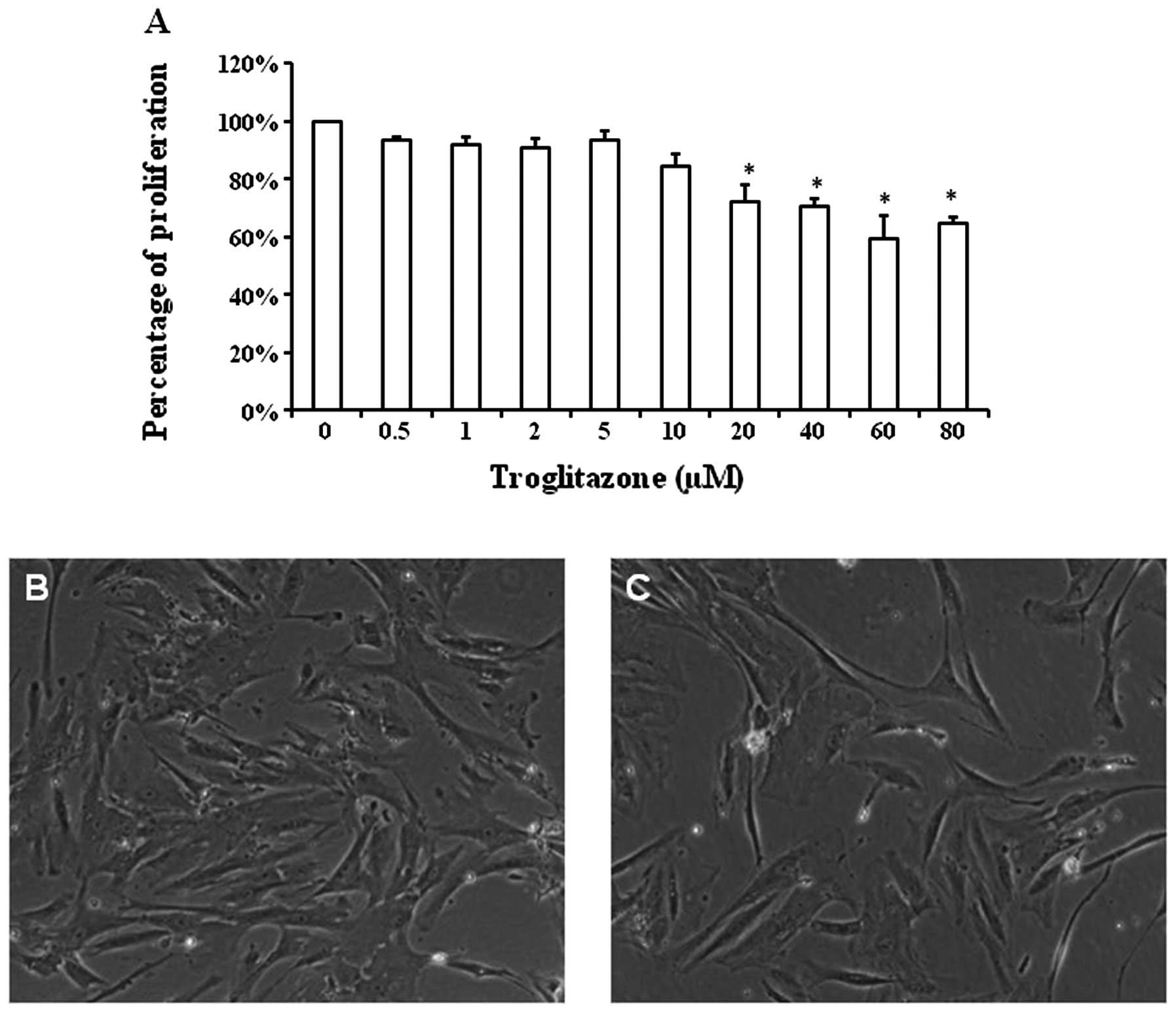
Concentration-dependent effects of
troglitazone on PASMC proliferation
The proliferation rate of the PASMCs was altered in
a concentration-dependent manner by troglitazone in the cells
cultured in GM under hypoxic conditions (Fig. 2A). The cell number of PASMCs
treated with 0.5 μM troglitazone was reduced to 93.3±1.2% compared
with that of the PASMCs not treated with troglitazone (considered
as 100%). The percentage cell number of PASMCs cultured with
troglitazone was 91±2.7% with 1 μM of troglitazone, 91.1±3.5% with
2 μM, 93.5±2.8% with 5 μM, 84.3±4.2 with 10 μM, 72±5.8% with 20 μM,
70.4±3% with 40 μM, 59.3±8.3% with 60 μM and 64.6±2.1% with 80 μM
troglitazone. Starting at the concentratikno of 20 μM, troglitazone
significantly reduced the proliferation of PASMCs under hypoxic
conditions (P<0.05).
Typical images of PASMC density following culture in
GM with or without troglitazone are shown in Fig. 2B and C. Starting at the
concentration of 20 μM, troglitazone significantly reduced the
PASMC proliferation rate. Thus, we selected the concentration of 20
μM troglitazone for the remaining experiments.
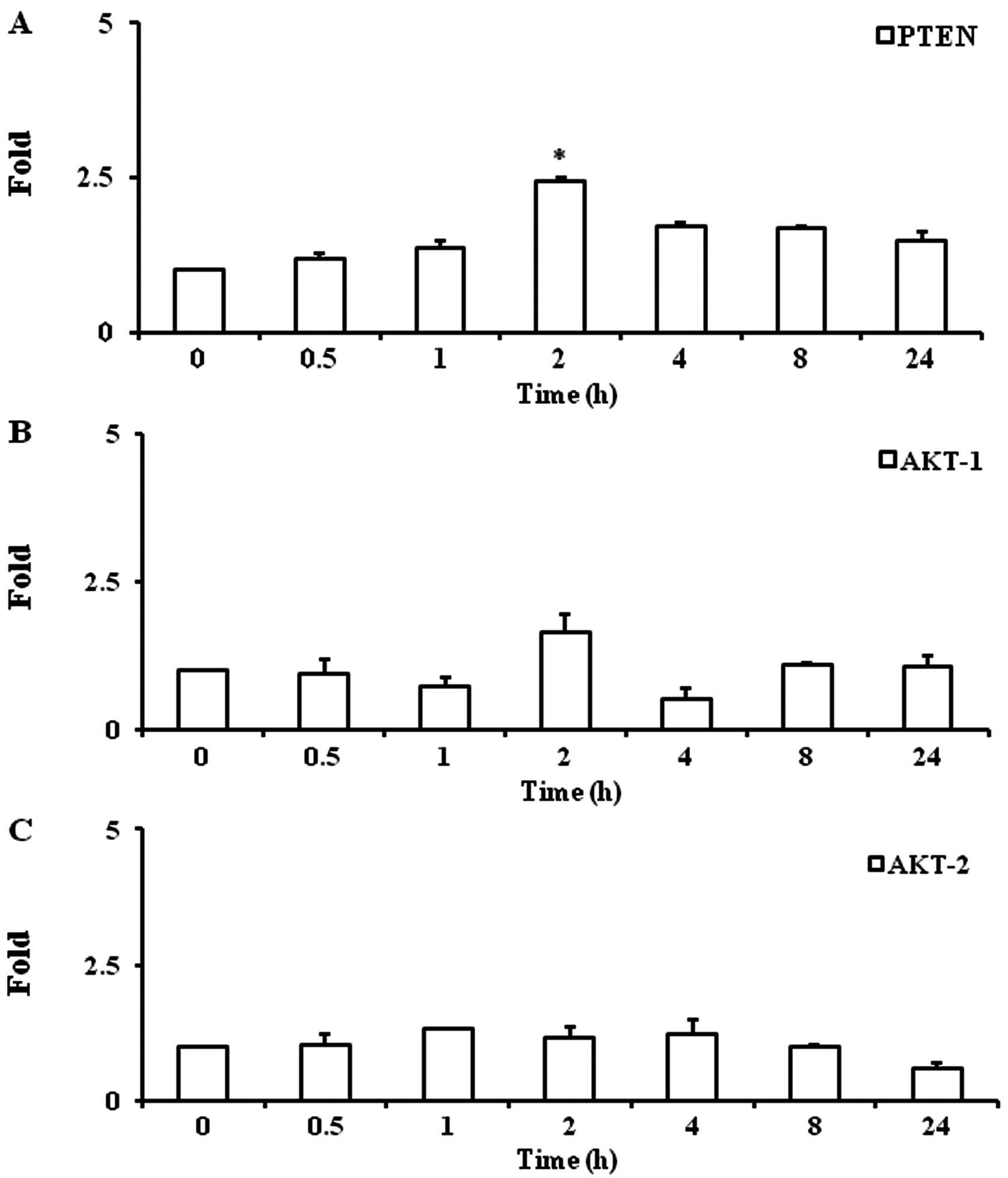
Troglitazone increases PTEN gene
expression
Troglitazone significantly increased PTEN gene
expression by 2.4±0.1-fold at 2 h as compared with the baseline
levels (P<0.05) under hypoxic conditions. Upregulated PTEN gene
expression was maintained up to 8 h (1.7±0.02-fold, P<0.05)
(Fig. 3A).
Troglitazone does not alter gene
expression of AKT-1 and AKT-2
Troglitazone did not significantly alter the gene
expression of AKT-1 and AKT-2 in the PASMCs between any 2 time
points (Fig. 3B and C).
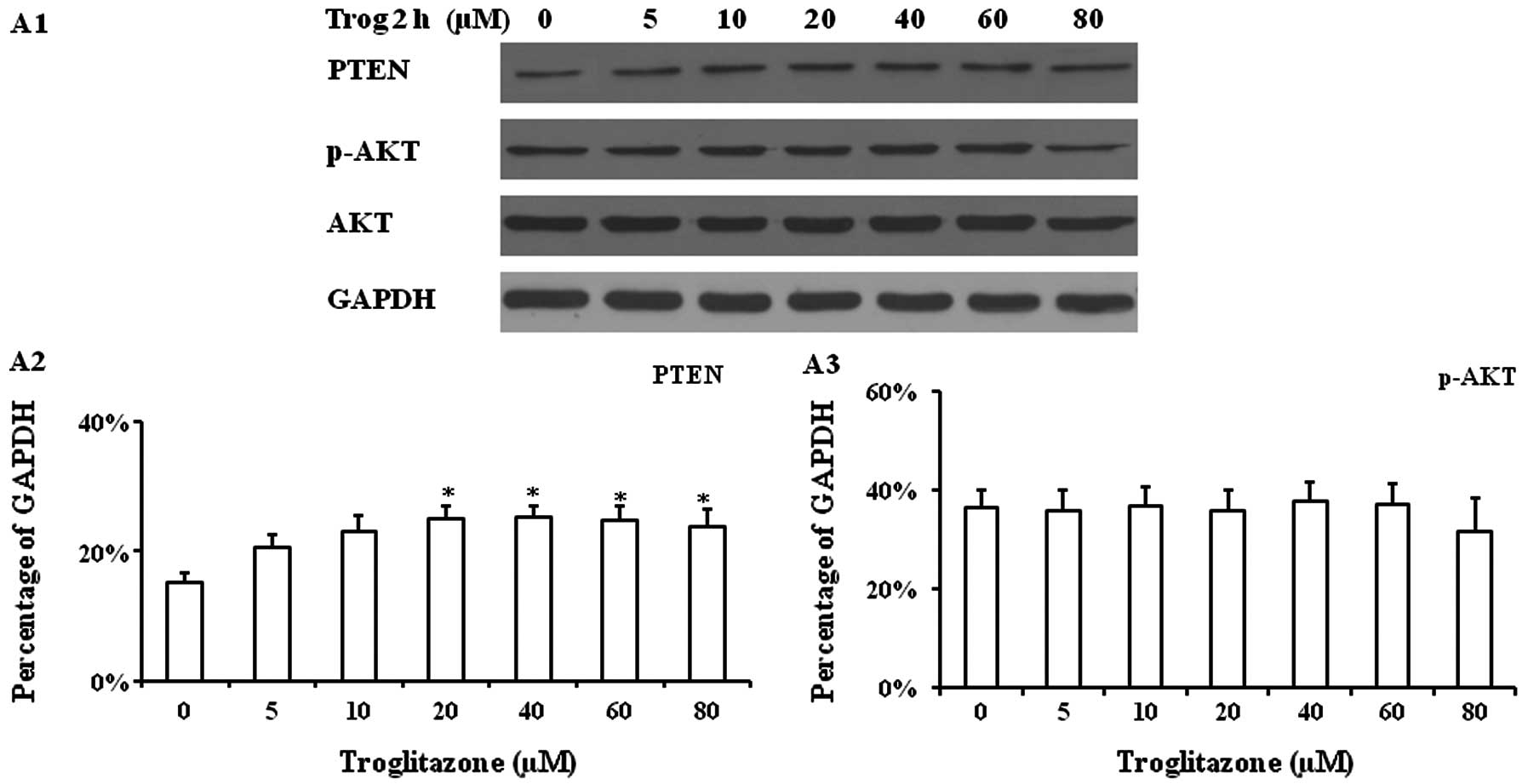
Western blot analysis of cultured PASMCs
supplemented with troglitazone
Troglitazone increased PTEN protein expression in
PASMCs cultured under hypoxic conditions for 2 h. A significantly
increased PTEN protein expression level was observed when the cells
were treated with troglitazone at concentrations between 20–80 μM,
while no significant changes in p-AKT and AKT expression were
observed (Fig. 4A). The PASMCs
were then cultured with 20 μM troglitazone for 24 h and
DMSO-treated cells (without troglitazone) were used as controls,
since troglitazone was diluted in DMSO. In the cells treated with
troglitazone, a significantly increased PTEN protein expression was
observed within 2 h compared with the cells treated with DMSO
alone. The highest PTEN protein expression level was observed
within 4 h after the addition of troglitazone, which was
significantly higher than that at 0.5 h (Fig. 4B). Troglitazone also progressively
reduced p-AKT expression within 2 h; these levels were
significantly lower than those from the cells treated with DMSO
alone.
To determine whether the upregulated PTEN protein
expression induced by troglitazone was mediated by the PPARγ
signaling pathway and inhibited by the PTEN inhibitor, PASMCs were
pre-treated with 2.5 μM bpV(HOpic) (a PTEN inhibitor) or 1 μM
GW9662 (PPARγ antagonist) for 1 h prior to the addition of
troglitazone. It was found that the upregulated protein expression
of PTEN was reduced by pre-treatment with bpV(HOpic) and GW9662 for
up to 24 h (Fig. 4C and D).
Effects of bpV(HOpic) and GW9662 on
proliferation of PASMCs treated with troglitazone
Typical images of the PASMC growth profile are shown
in Fig. 5A–G. Troglitazone
significantly reduced the proliferation rate of the PASMCs under
hypoxic conditions. bpV(HOpic) was used to determine whether the
PTEN inhibitor reversed the inhibitory effects of troglitazone on
PASMC proliferation. It was found that troglitazone did not reduce
the proliferation rate of the PASMCs that were pre-treated with
bpV(HOpic). These results suggest that troglitazone inhibits PASMC
proliferation by upregulating PTEN expression, which may be
reversed by the PTEN inhibitor (Fig.
5H).
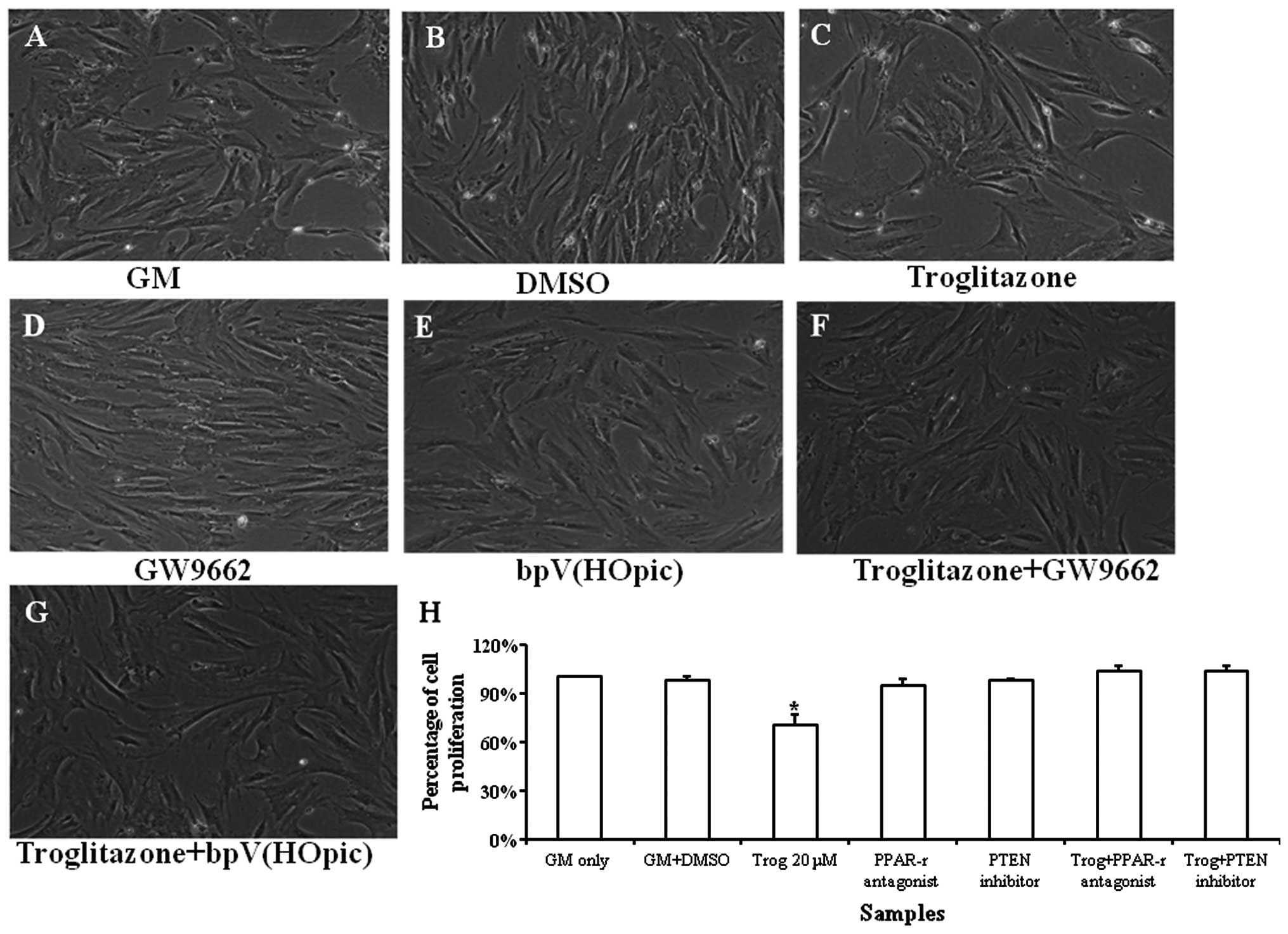 | Figure 5Growth profile of pulmonary artery
smooth muscle cells (PASMCs) cultured in growth medium (GM)
supplemented with troglitazone, GW9662 and bpV(HOpic) for 72 h
under hypoxic conditions. Phase contrast images of (A) PASMCs
cultured in GM only, (B) GM supplemented with DMSO, (C) GM
supplemented with troglitazone, (D) GW9662, (E) bpV(HOpic), (F)
troglitazone + GW9662, (G) troglitazone + bpV(HOpic). (H)
Quantification of PASMCs treated with troglitazone, GW9662 and
bpV(HOpic). The cell number of PASMCs cultured in GM was regarded
as 100%. *P<0.05 vs. any other samples.
Magnification, ×100; Trog, troglitazone. The experiment was
performed in triplicate. |
GW9662 was also used to determine whether the
upregulation in PTEN expression stimulated by troglitazone was
mediated by the PPARγ signaling pathway. It was found that GW9662
reversed the inhibitory effects of troglitazone on PASMC
proliferation. These results suggest that troglitazone upregulates
PTEN expression through the PPARγ signaling pathway (Fig. 5).
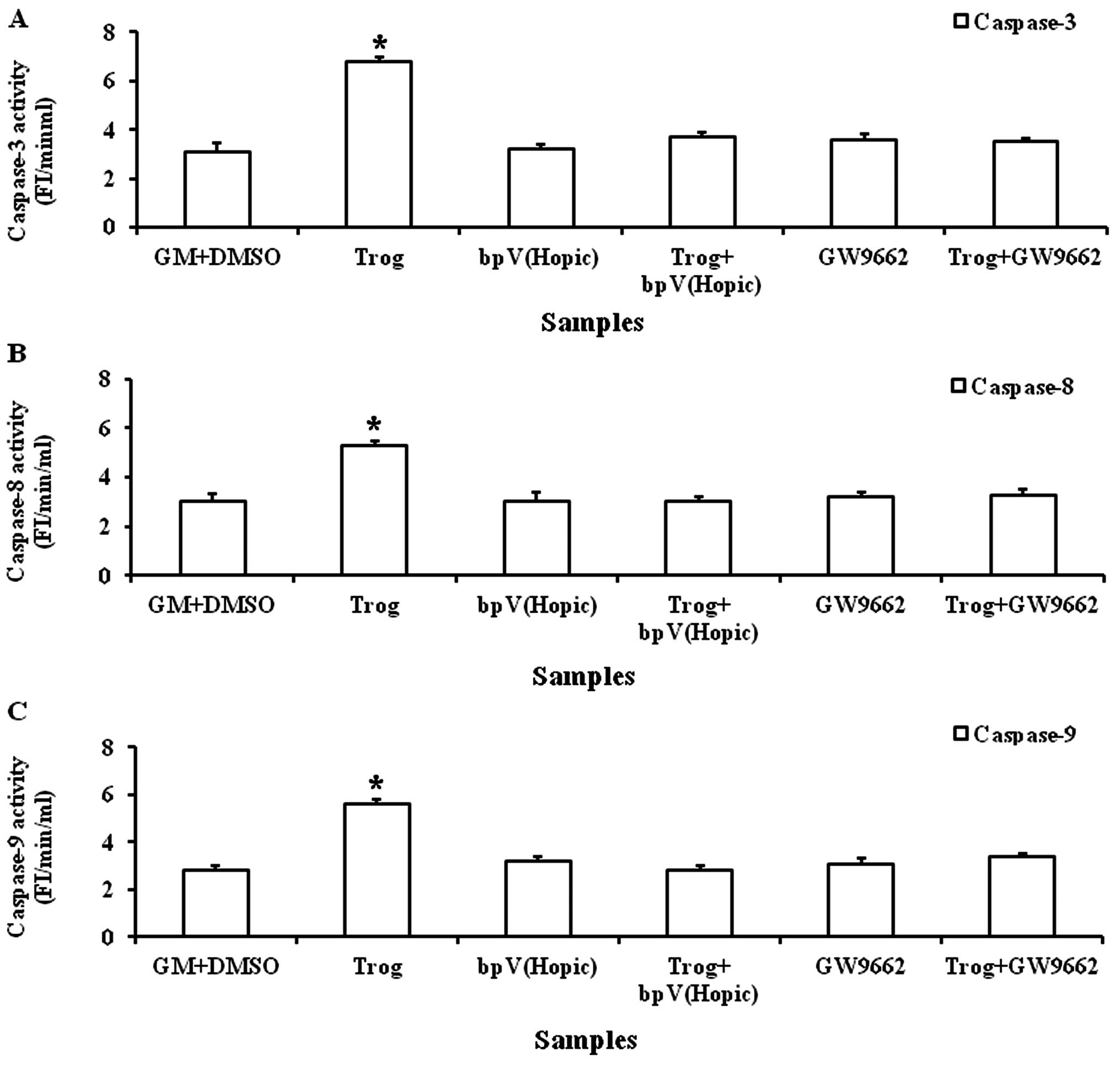
Troglitazone increases caspase-3, -8 and
-9 activities
A significantly increased caspase-3 activity was
observed when the PASMCs were cultured in GM (6.5±0.15 FI/min/ml)
supplemented with troglitazone for 8 h (Fig. 6A). Similarly, a significant
increase in caspase-8 and -9 activities was observed when the
PASMCs were cultured in GM (5.3±0.15 and 6.1±0.27 FI/min/ml)
supplemented with troglitazone for 8 h (Fig. 6B and C).
Troglitazone increases the apoptosis of
PASMCs under hypoxic conditions
TUNEL staining revealed that the apoptosis of PASMCs
cultured in BM increased after 72 h under hypoxic conditions
(Fig. 6D and E). Quantification
of TUNEL+ cells suggested that troglitazone
significantly increased the apoptotic PASMC number compared with
the cells cultured in BM alone (P<0.05).
Discussion
The present study demonstrates that troglitazone, a
high-affinity synthetic ligand of PPARγ upregulates PTEN gene and
protein expression levels in PASMCs under hypoxic conditions in a
concentration-dependent manner. In addition, we demonstrate that
troglitazone increases the apoptosis of PASMCs under hypoxic
conditions.
Pulmonary vascular remodelling in pulmonary arterial
hypertension is characterized by changes in pulmonary vascular
structure (1,2). This is partially caused by an
imbalance in PASMC proliferation and apoptosis (10). The increased proliferation and
decreased apoptosis of PASMCs result in the thickening of the
pulmonary vasculature, which subsequently increases pulmonary
vascular resistance and pulmonary arterial pressure (11).
Our results demonstrated that troglitazone at the
concentration of 20–80 μM effectively inhibited PASMC proliferation
under hypoxic conditions. The anti-proliferative effects of
troglitazone may be related to its role in the upregulation of PTEN
and the induction of the apoptosis of PASMCs under hypoxic
conditions. PPARγ expression has been found in pulmonary vascular
endothelial cells and SMCs (12,13). However its expression is reduced
in lung vascular cells of rats with pulmonary hypertension and in
patients with advanced primary or secondary pulmonary hypertension
(12). The targeted depletion of
PPARγ from SMCs has been shown to result in spontaneous pulmonary
hypertension in mice (4).
However, the activation of PPARγ with thiazolidinediones attenuates
vascular remodeling and pulmonary hypertension caused by
monocrotaline (14) or hypobaric
hypoxia (15) in rats.
Thiazolidinediones also reduce pulmonary hypertension in
ApoE-deficient mice fed high-fat diets (16) and attenuate hypoxia-induced
pulmonary hypertension and Nox4 expression and activity in mice
(5).
However, the role of PPARγ in the regulation of PTEN
expression of PASMCs under hypoxic conditions has not been examined
yet. Our results demonstrate that PPARγ upregulates PTEN gene and
protein expression levels. Studies have shown that PPARγ agonists
upregulate the transcription of PTEN in cancer cells. PPARγ
agonists have been used as tumor suppressors in breast cancer by
upregulating PTEN gene expression (17), while cells lacking PTEN or PPARγ
are unable to induce PTEN-mediated cellular events in the presence
of lovastatin or rosiglitazone. Zhang et al (7) showed that PTEN is required for the
rosiglitazone-induced inhibition of BEL-7404 cells. Rosiglitazone
upregulated PTEN expression in a concentration- and time-dependent
manner, which was mediated by PPARγ. Furthermore, PTEN
overexpression resulted in the inhibition of BEL-7404 cell
migration, and PTEN knockdown blocked the effects of rosiglitazone
on cell migration.
It is known that PPARγ agonists can stimulate the
production and secretion of ApoE, which has anti-proliferative
effects on human PASMCs (4).
Nisbet et al (5) found
that rosiglitazone attenuated hypoxia-induced pulmonary vascular
remodelling and hypertension by suppressing oxidative and
proliferative signals. They found that rosiglitazone attenuated the
reduction in PTEN expression. However, the present study
demonstrates that troglitazone upregulates PTEN gene and protein
expression levels in human PASMCs under hypoxic conditions. This
may provide new insight into the mechanisms behind the
anti-proliferative effects of PPARγ on PASMCs under hypoxic
conditions.
The data from the present study also suggest that
troglitazone increases the apoptosis of PASMCs under hypoxic
conditions. It is known that chronic hypoxia can prolong the growth
of human vascular SMCs by inducing telomerase activity and telomere
stabilization (18). The
upregulation of PTEN negatively regulates the AKT/PKB signaling
pathway and leads to a decrease in p-AKT levels and, consequently,
to a decrease in AKT-mediated proliferation (19). Thus, a reduced p-AKT expression in
PASMCs was found after the addition of troglitazone in this study.
By reducing the cellular levels of p-AKT, PTEN downregulates cell
division and increases apoptosis. This may result in increased
activities of caspase-3, -8 and -9, thus inducing the apoptosis of
PASMCs under hypoxic conditions.
In conclusion, the present study highlights that
troglitazone increases PTEN gene expression at the transcriptional
level under hypoxic conditions in a concentration-dependent manner.
Troglitazone increases the apoptosis of PASMCs under hypoxic
conditions. The increased PTEN expression and apoptosis of PASMCs
provide new insight into the mechanisms behind the
anti-proliferative effects of troglitazone on PASMCs.
References
|
1
|
McLaughlin VV, Archer SL, Badesch DB, et
al: ACCF/AHA 2009 expert consensus document on pulmonary
hypertension: a report of the American College of Cardiology
Foundation Task Force on Expert Consensus Documents and the
American Heart Association: developed in collaboration with the
American College of Chest Physicians, American Thoracic Society,
Inc., and the Pulmonary Hypertension Association. Circulation.
119:2250–2294. 2009.
|
|
2
|
Pietra GG, Edwards WD, Kay JM, et al:
Histopathology of primary pulmonary hypertension. A qualitative and
quantitative study of pulmonary blood vessels from 58 patients in
the National Heart, Lung, and Blood Institute, Primary Pulmonary
Hypertension Registry. Circulation. 80:1198–1206. 1989. View Article : Google Scholar
|
|
3
|
Nakaoka T, Gonda K, Ogita T, et al:
Inhibition of rat vascular smooth muscle proliferation in vitro and
in vivo by bone morphogenetic protein-2. J Clin Invest.
100:2824–2832. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hansmann G, de Jesus Perez VA, Alastalo
TP, et al: An antiproliferative BMP-2/PPARgamma/apoE axis in human
and murine SMCs and its role in pulmonary hypertension. J Clin
Invest. 118:1846–1857. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Nisbet RE, Bland JM, Kleinhenz DJ, et al:
Rosiglitazone attenuates chronic hypoxia-induced pulmonary
hypertension in a mouse model. Am J Respir Cell Mol Biol.
42:482–490. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lee KS, Park SJ, Hwang PH, Yi HK, Song CH,
Chai OH, Kim JS, Lee MK and Lee YC: PPAR-gamma modulates allergic
inflammation through up-regulation of PTEN. FASEB J. 19:1033–1035.
2005.PubMed/NCBI
|
|
7
|
Zhang W, Wu N, Li ZX, Wang LY, Jin JW and
Zha XL: PPARgamma activator rosiglitazone inhibits cell migration
via upregulation of PTEN in human hepatocarcinoma cell line
BEL-7404. Cancer Biol Ther. 5:1008–1014. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ye L, Haider KhH, Esa WB, et al:
Liposome-based vascular endothelial growth factor-165 transfection
with skeletal myoblast for treatment of ischaemic limb disease. J
Cell Mol Med. 14:323–336. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ye L, Lee KO, Su LP, et al: Skeletal
myoblast transplantation for attenuation of hyperglycaemia,
hyperinsulinaemia and glucose intolerance in a mouse model of type
2 diabetes mellitus. Diabetologia. 52:1925–1934. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang S, Fantozzi I, Tigno DD, et al: Bone
morphogenetic proteins induce apoptosis in human pulmonary vascular
smooth muscle cells. Am J Physiol Lung Cell Mol Physiol.
285:L740–L754. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Barst RJ, McGoon M, Torbicki A, Sitbon O,
Krowka MJ, Olschewski H and Gaine S: Diagnosis and differential
assessment of pulmonary arterial hypertension. J Am Coll Cardiol.
43:40S–47S. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ameshima S, Golpon H, Cool CD, et al:
Peroxisome proliferator-activated receptor gamma (PPARgamma)
expression is decreased in pulmonary hypertension and affects
endothelial cell growth. Circ Res. 92:1162–1169. 2003. View Article : Google Scholar
|
|
13
|
Michalik L, Auwerx J, Berger JP, et al:
International Union of Pharmacology. LXI Peroxisome
proliferator-activated receptors. Pharmacol Rev. 58:726–741. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Matsuda Y, Hoshikawa Y, Ameshima S, et al:
Effects of peroxisome proliferator-activated receptor gamma ligands
on monocrotaline-induced pulmonary hypertension in rats. Nihon
Kokyuki Gakkai Zasshi. 43:283–288. 2005.(In Japanese).
|
|
15
|
Crossno JT Jr, Garat CV, Reusch JE, et al:
Rosiglitazone attenuates hypoxia-induced pulmonary arterial
remodeling. Am J Physiol Lung Cell Mol Physiol. 292:L885–L897.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hansmann G, Wagner RA, Schellong S, et al:
Pulmonary arterial hypertension is linked to insulin resistance and
reversed by peroxisome proliferator-activated receptor-gamma
activation. Circulation. 115:1275–1284. 2007.
|
|
17
|
Teresi RE, Shaiu C, Chen C, Chatterjee K,
Waite KA and Eng C: Increased PTEN expression due to
transcriptional activation of PPARgamma by Lovastatin and
Rosiglitazone. Int J Cancer. 118:2390–2398. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Minamino T, Mitsialis SA and Kourembanas
S: Hypoxia extends the life span of vascular smooth muscle cells
through telomerase activation. Mol Cell Biol. 21:3336–3342. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Maehama T and Dixon JE: The tumor
suppressor, PTEN/MMAC1, dephosphorylates the lipid second
messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem.
273:13375–13378. 1998. View Article : Google Scholar : PubMed/NCBI
|