Introduction
Cardiovascular diseases such as myocardial
infarction and stroke, which cause serious damage to human health,
are mainly caused by the destabilization and rupture of
atherosclerotic plaques (1,2).
However, there is currently a lack of efficient therapeutic methods
for the treatment of atherosclerotic plaque stability. The
hallmarks of rupture-prone plaques are a large lipid core, a thin
fibrous cap and loss of collagen caused by leukocyte infiltration
and a decrease of smooth muscle cells (SMCs) (3,4).
The stability of the thickness and structure of the fibrous cap are
crucial for plaque stability (5).
Therefore, reduction of acute coronary events through stabilization
of atherosclerotic plaques has become a new treatment target.
Macrophages are largely accumulated in
atherosclerotic plaques, which is crucial in the occurrence of
atherosclerosis as well as in the process of atherosclerotic plaque
rupture (6). Macrophages express
inflammatory factors including matrix metalloproteinases (MMPs)
(7), monocyte chemoattractant
protein 1 (MCP-1) (8) and tissue
factor (TF) (9), which promote
the destabilization and rupture of atherosclerotic plaques.
Therefore, macrophage removal has been considered to be beneficial
for plaque stability (10).
Previous studies reported that everolimus, which is
a rapamycin derivative and an inhibitor of mammalian target of
rapamycin (mTOR), selectively eliminated macrophages in
atherosclerotic plaques, suggesting that mTOR is important in the
destabilization and rupture of atherosclerotic plaques (11). mTOR is a member of the
phosphoinositol kinase-related kinase (PIKK) family which mediates
gene translation in the presence of nutrients including insulin,
mitogens and growth factors involved in cell growth,
differentiation, migration and survival (12,13). Inhibition of mTOR has been shown
to have anti-cell proliferative effects in vitro and in
vivo (14), leading to cell
apoptosis (15,16) and autophagic cell death (17,18). Thus, mTOR has become a new target
for the treatment of diseases such as cancer (19). mTOR also affects inflammatory cell
activity and cytokine release (20). Inhibition of mTOR by sirolimus or
everolimus suppresses atherosclerosis and shows a decrease in
macrophages via the induction of autophagy (11,21). Autophagy is considered to be a
conserved pathway for the destruction of damaged or unwanted
intracellular material, including proteins and entire organelles
contributing to cell homeostasis, and is closely associated with
various diseases (22,23). Given this, targeting mTOR for
macrophage autophagy is a promising strategy for the treatment of
atherosclerotic plaque stability.
Molecular-targeted therapy is also considered to
have potential applications in disease treatment. RNA interference
(RNAi) is a simple and effective method for blocking gene
expression and has great advantages over traditional methods for
cardiovascular disease therapy. In the present study, we used small
hairpin RNA (shRNA) lentivirus (LV)-mediated-targeted mTOR in a
mouse model that specifically inhibited mTOR expression in
vivo. Results showed that the transfection of LV-mediated mTOR
shRNA effectively inhibited mTOR mRNA and protein expression in the
carotid artery of the mouse model. Silencing of mTOR ameliorated
the dysregulated blood lipid metabolism caused by a high-fat diet
(HFD) including the downregulation of total cholesterol,
triglycerides and LDL cholesterol (LDL-C) and the upregulation of
HDL cholesterol (HDL-C) in the atherosclerotic mouse model.
Silencing of mTOR increased plaque stability by decreasing the
plaque area and increasing the fibrous cap. In addition, silencing
of mTOR expression led to a decrease of macrophages throughout the
atherosclerotic plaque. Furthermore, inhibition of mTOR induced
macrophage autophagy by promoting autophagy-related protein 13
(Atg13) dephosphorylation and upregulating light chain 3-I/light
chain 3-II (LC3-I/LC3-II). MMP-2, MCP-1 and TF, all of which were
proposed to promote plaque destabilization, were also decreased by
the silencing of mTOR. These results demonstrated that the
suppression of mTOR in an atherosclerotic mouse model by
LV-mediated RNAi inhibited atherosclerosis and stabilized plaques
via a decrease of macrophages by autophagy. The profound effects
and underlying mechanism should be further investigated. However,
to the best of our knowledge, the present study identified a new
method for the stabilization of atherosclerotic plaques.
Materials and methods
Animals
Male apolipoprotein E-deficient mice (3 weeks old,
weighing 25–30 g) were obtained from Changzhou Cavens Laboratory
Animal Co., Ltd. (Changzhou, China). Mice were raised under
standard conditions of room temperature, dark-light cycles and
humidity and fed on HFD (20% fat, 20% sugar and 1.25% cholesterol)
with free access to water. Experiments were conducted under a
protocol approved by the Institutional Animal Care and Use
Committee of the Xi’an Jiaotong University. All efforts were made
to minimize suffering.
Construction and production of LV shRNA
vectors
LV shRNA vectors were constructed as described
previously (24). Briefly, DNA
fragments containing GACAGCACA as the loop for shRNA and shRNA
sequences against mTOR were synthesized and cloned into human U6
promoter-containing pBluescript SK(+) plasmid (pU6). shRNA
fragments (shmTOR) were then subcloned into the plasmid pRRL (Irvin
S.Y. Chen, UCLA) with a human PSMA promoter, with restriction sites
for BamHI and HindIII (Takara, Tokyo, Japan).
Non-specific shRNA sequences (shCON) cloned into the vectors were
obtained as the control (25). A
total of 20 μg of lentiviral vector carrying shRNA, 15 μg of
packaging vectors pCMVR (Beijing Zhongyuan Ltd., Beijing, China), 2
μg of pCMV-VSVG (Sidansai Stem Cell Technology Co., Ltd., Shanghai,
China) and 100 μl Lipofectamine® 2000 (Invitrogen,
Carlsbad, CA, USA) were mixed and incubated with 293T cells at
37°C, 5% CO2 for 48 h. The cell supernatants were
collected and concentrated using a 0.45 μm filter (Amicon Ultra-15
100K; Millipore, Billerica, MA, USA). The viral titer was
determined by p24 ELISA kit (Cell Biolabs, Inc., San Diego, CA,
USA) and recombinant virus was stored at −80°C until use.
Carotid collar placement and LV
transfection
Atherosclerotic lesions were induced according to a
previously reported study (26).
In brief, a restraint perivascular silica collar (0.3 mm in
internal diameter and 3 mm in length) was placed around the left
common carotid artery to induce the formation of atherosclerotic
plaque. Sodium pentobarbital (40 mg/kg; no. 1; Biochem & Pharm,
Shanghai, China) was used to anesthetize mice by subcutaneous
injection. The common carotid arteries were dissected without
causing damage to the carotid bodies and the vagus nerves. Collars
were placed around the right common carotid arteries and their
axial edges were approximated by placement of 3 circumferential
silk ties. Then, the entry wound was closed and the mice were
recovered under normal conditions for subsequent experiments. Eight
weeks after surgery, the collars were removed and mice were
transfected with LV through the right common carotid arteries. Mice
were randomly divided into the control group, where mice received
200 μl phosphate-buffered saline (PBS) injection; the LV-shCON
group, where mice received 200 μl recombinant LV (5×1010
plaque-forming units of virus) expressing no-specific shRNA; and
the LV-shmTOR group, where mice received 200 μl (5×1010
pfu) recombinant LV expressing mTOR shRNA. Another injection was
performed after 2 weeks and mice were euthanized for analysis 2
weeks after the second injection.
Quantitative real-time PCR
Two weeks after the second injection, mice were
euthanized and total RNA was extracted from the right common
carotid arteries using TRIzol reagent (Invitrogen) following the
manufacturer’s protocol. Up to 5 μg of the total RNA was
reverse-transcribed into cDNA using M-MLV reverse transcriptase
(Clontech, Palo Alto, CA, USA). The cDNAs were used as templates
for quantitative real-time PCR. The PCR primers for mTOR were
5′-ctgggactcaaatgtg tgcagttc-3′ (forward) and
5′-gaacaatagggtgaatgatccggg-3′ (reverse); while those for β-actin
were 5′-caacttgatgtatgtat gaaggctttggt-3′ (forward) and
5′-acttttattggtctcaagtcagtgtacag-3′ (reverse). The qRT-PCR system
contained 5 μl SsoFast™ EvaGreen® Supermix (Bio-Rad), 1
μl of cDNA (diluted in 1:50) and 2 μl of each of the forward and
reverse primers (1 μM) to a final volume of 10 μl. The PCR
procedure was as follows: 94°C for 4 min; 94°C for 20 sec, 55°C for
30 sec and 72°C for 20 sec; 2 sec for plate reading for 35 cycles;
and melt curve from 65 to 95°C. β-actin was used as the control for
normalizing gene expression. Three independent experiments were
performed. All the values obtained were normalized to mouse
β-actin. The data obtained were calculated by 2−ΔΔCt and
treated for statistical analysis as described previously (27), followed by an unpaired sample
t-test.
Western blotting
Two weeks after the second injection, mice were
euthanized and tissues from the right common carotid arteries were
frozen in liquid nitrogen and treated with ice-cold lysis buffer,
prior to being homogenized and centrifuged. The supernatant was
collected and protein concentration was determined by the Bradford
method. A total of 30 μg of protein was fractionated by 12%
SDS-PAGE electrophoresis and transferred to a nitrocellulose
membrane (Amersham, Little Chalfont, UK). The membrane was treated
using the following procedure: shaking and blocking at room
temperature with 2% non-fat dry milk in PBS for 1 h, followed by
incubation in primary antibody blocking solution at 4°C overnight.
Subsequently, the membrane was incubated in HRP-labeled secondary
antibodies diluted at 1:3,000 in the blocking buffer for 4 h.
4-Chloro-1-naphthol (4-CN) was used as an HRP substrate for
visualizing the target protein.
Detection of blood lipid
Mice were sacrificed by pentobarbital overdose (200
mg/kg) and blood was collected from mouse orbital veins. Blood
samples were centrifuged and serum was collected. Levels of total
cholesterol, triglyceride, LDL-C and HDL-C in serum samples were
measured according to kit standard procedures (Shanghai Hushang
Biotechnology Co. Ltd., Shanghai, China) by an Automatic
Biochemistry Analyzer (Hitachi, Tokyo, Japan).
Hematoxylin and eosin (H&E)
staining
Mouse hearts were perfused with PBS and then 4%
paraformaldehyde for 30 min under physiological pressure. The
carotid artery was isolated and fixed with 4% paraformaldehyde for
12 h, embedded in paraffin and cut into 5-μm serial sections.
Corresponding sections were stained with hematoxylin (Sigma, St.
Louis, MO, USA) for 10 min at room temperature. The sections were
then washed with running water. Subsequently, the sections were
washed with Scott promote blue liquid for 1 min, 1% hydrochloric
acid alcohol differentiation liquid for 20 sec and Scott promote
blue liquid for 1 min. Sections were then stained with eosin (0.5%;
Merck, Whitehouse Station, NJ, USA) for 30 sec. The sections were
washed with running water and sealed for observation. For the
lesion area analysis and calculation (plaque area, fibrous cap,
cap-to-core ratio, and intima-media thickness), the sections were
observed by Image-Pro Plus 5.0 software (Media Cybernetics, Inc.,
Bethesda, MD, USA).
Statistical analysis
Assays were performed in triplicate and data were
presented as mean ± SEM. Differences between groups were analyzed
by the Student’s t-test. P<0.05 was considered statistically
significant. Statistical analyses were performed using SPSS v11.5
(SPSS Inc., Chicago, IL, USA).
Results
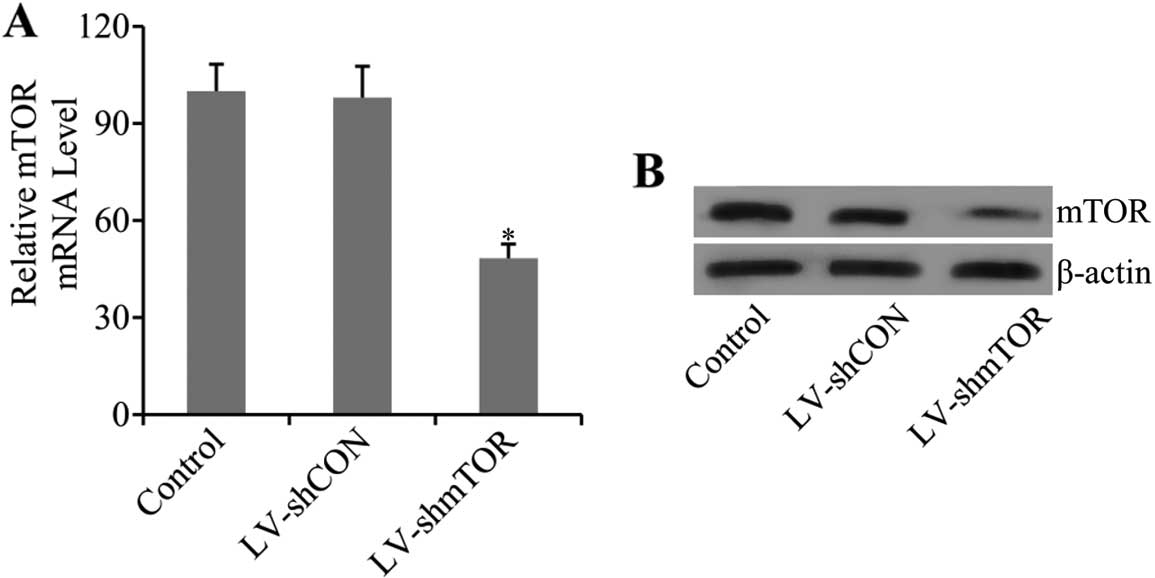
Efficient knockdown of mTOR by
LV-mediated shRNA
To examine whether transfection of LV-mediated shRNA
of mTOR inhibited the expression of mTOR, real-time PCR (qRT-PCR)
and western blot analysis were performed. Results showed that the
mRNA level of mTOR was markedly decreased (48.36±4.32) in
LV-shmTOR-transfected mice (P<0.05), while control LV-shCON
transfection had no effect on mTOR mRNA expression (Fig. 1A). Furthermore, the results were
confirmed by western blot analysis. As shown in the results, the
protein expression level of mTOR was also significantly decreased
in carotid artery tissues of LV-shmTOR-infected mice compared with
the mice infected with LV-shCON or uninfected control mice
(Fig. 1B).
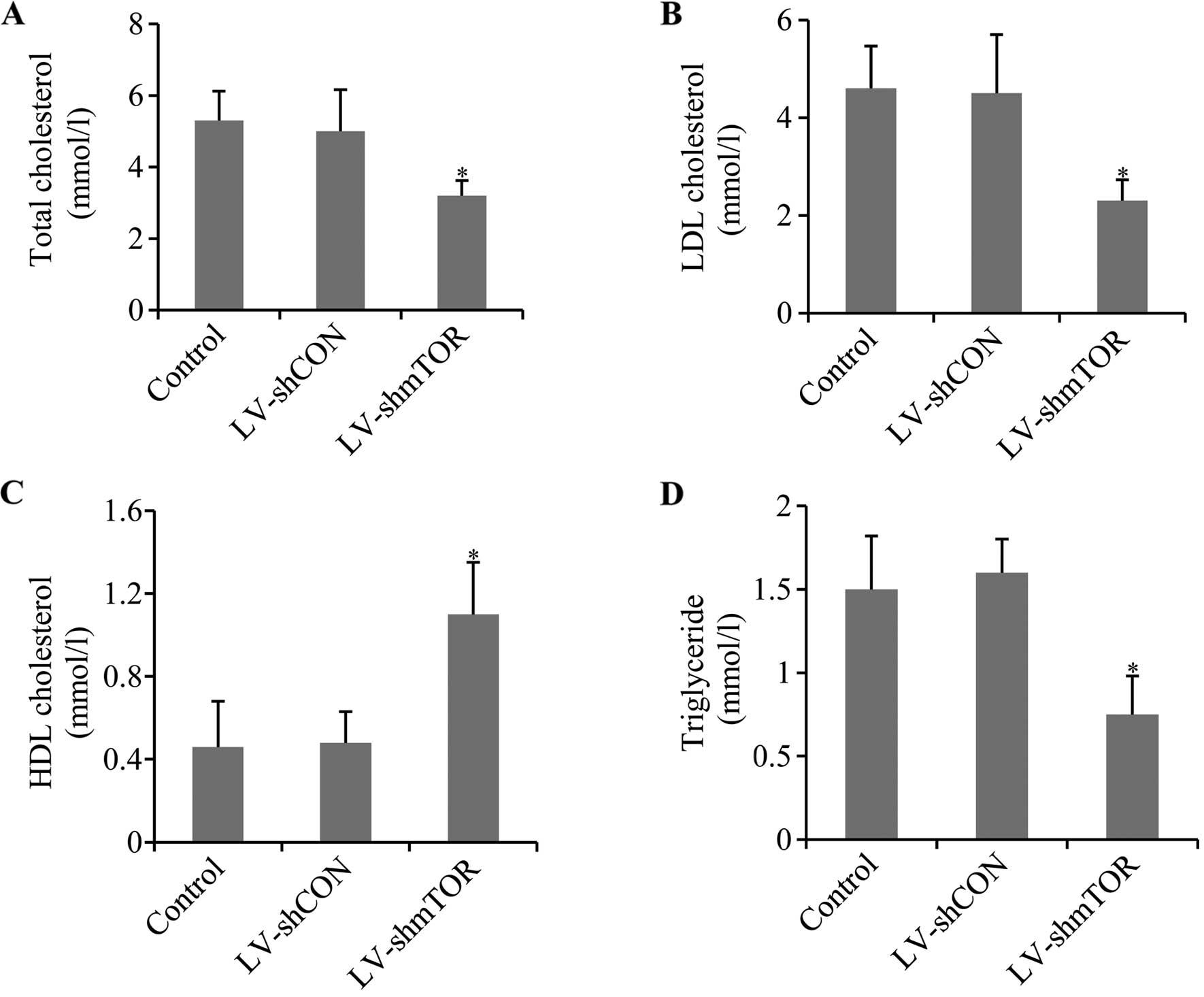
Knockdown of mTOR ameliorated
dysregulated blood lipid metabolism
To evaluate the effect of mTOR knockdown on the mice
of atherosclerosis, serum levels of total cholesterol,
triglyceride, LDL-C and HDL-C was determined. Knocking down of mTOR
led to a significant decrease in the total cholesterol, LDL-C and
triglycerides in LV-shmTOR-infected mice compared to the control,
whereas LV-shCON had no effect. Otherwise, knockdown of mTOR
increased HDL-C level in LV-shmTOR-infected mice, compared with
mice infected with LV-shCON or uninfected control mice (P<0.05)
(Fig. 2).
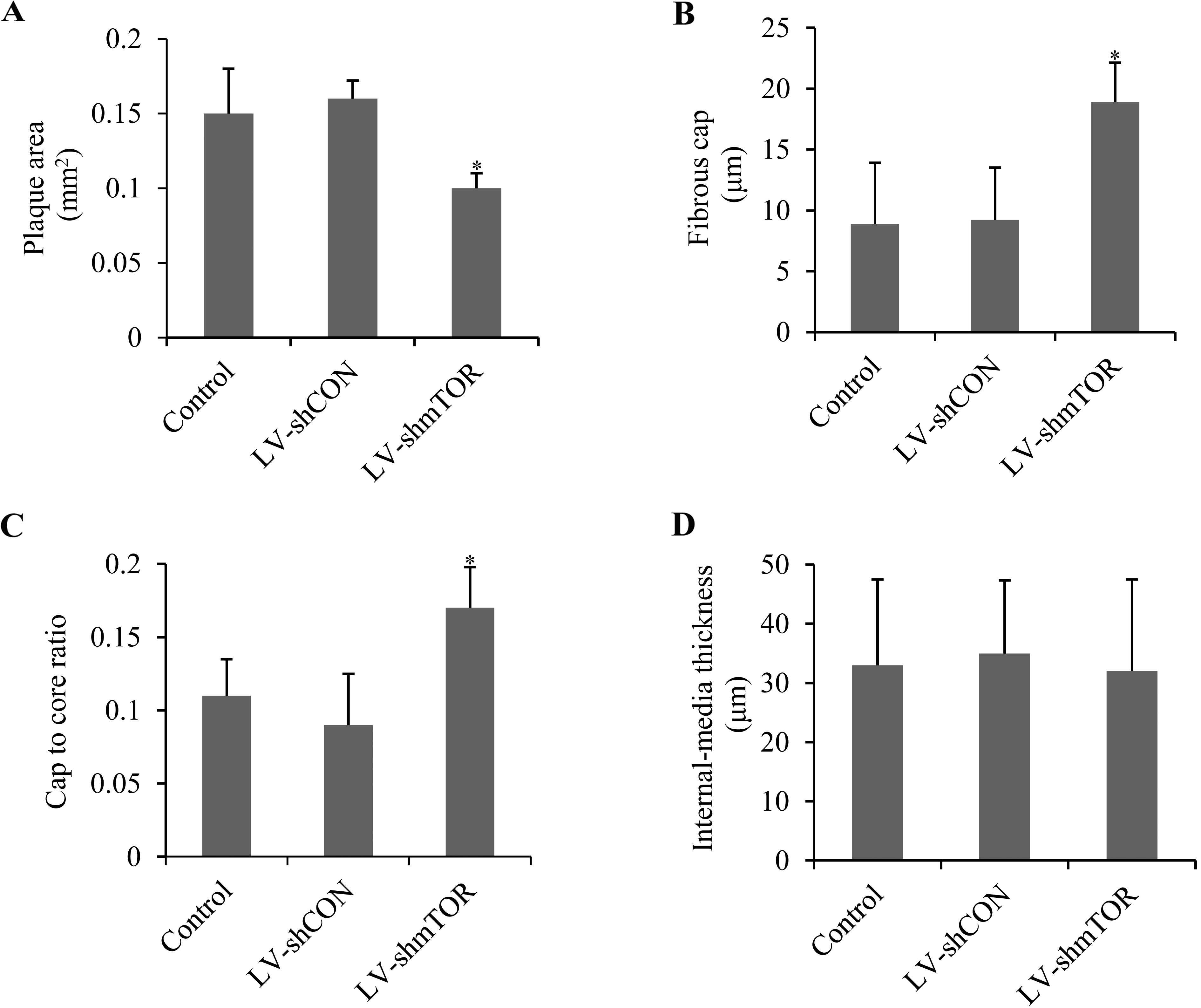
Knockdown of mTOR stabilized aortic
atherosclerotic plaque
To evaluate the role of mTOR on plaque stability,
plaque area, fibrous cap, cap-to-core ratio and intima-media
thickness were analyzed by H&E staining. LV-shmTOR-infected
mice demonstrated a significant decrease in the plaque area
compared with control or LV-shCON-infected mice (0.11±0.08 vs.
0.15±0.12 or 0.16±0.10 mm2; P<0.05) (Fig. 3A). Fibrous cap in
LV-shmTOR-infected mice (18.93±3.02 μm) showed a marked increase
(P<0.05), compared with that in control mice (8.9±5.75 μm) or
LV-shCON infected mice (9.2±5.45 μm) (Fig. 3B). The cap-to-core ratio was also
increased in LV-shmTOR-infected mice (0.17±0.028, P<0.05),
compared to the control mice (0.11±0.025) or LV-shCON-infected mice
(0.09±0.032) (Fig. 3C). However,
the intima-media thickness showed no significant alterations in the
three groups (Fig. 3D).
Knockdown of mTOR decreased macrophages
within atherosclerotic plaques
To evaluate the effect of LV-mediated RNAi on
macrophages within atherosclerotic plaques, western blotting was
performed using macrophage marker CD14 antibody. Results showed
that CD14 was decreased by the knockdown of mTOR in
LV-shmTOR-infected mice, compared to the control mice and
LV-shCON-infected mice (Fig. 4),
suggesting that macrophages were decreased in mTOR
siRNA-transfected mice. The results suggested that macrophages
could be decreased by silencing mTOR throughout the atherosclerotic
plaque.
Knockdown of mTOR promoted macrophage
autophagy
To explore the underlying mechanism of mTOR in
macrophage decrease, the protein expression level of Atg13, which
is involved in macrophage autophagy, was analyzed (28). Dephosphorylated Atg13 was reported
to induce autophagy. The results showed that silencing mTOR
promoted Atg13 dephosphorylation, suggesting that macrophage
autophagy activity was increased. LC3 protein is the marker of
autophagosome formation and consists of the isoforms LC3-II and
LC3-I. An increased LC3-I/LC3-II ratio indicates the accumulation
of autophagosomes (29). In mTOR
siRNA-transfected mice, LC3-II was upregulated while LC3-I was
downregulated, which led to an increase in the LC3-I/LC3-II ratio
(Fig. 5), further confirming that
autophagy was induced.
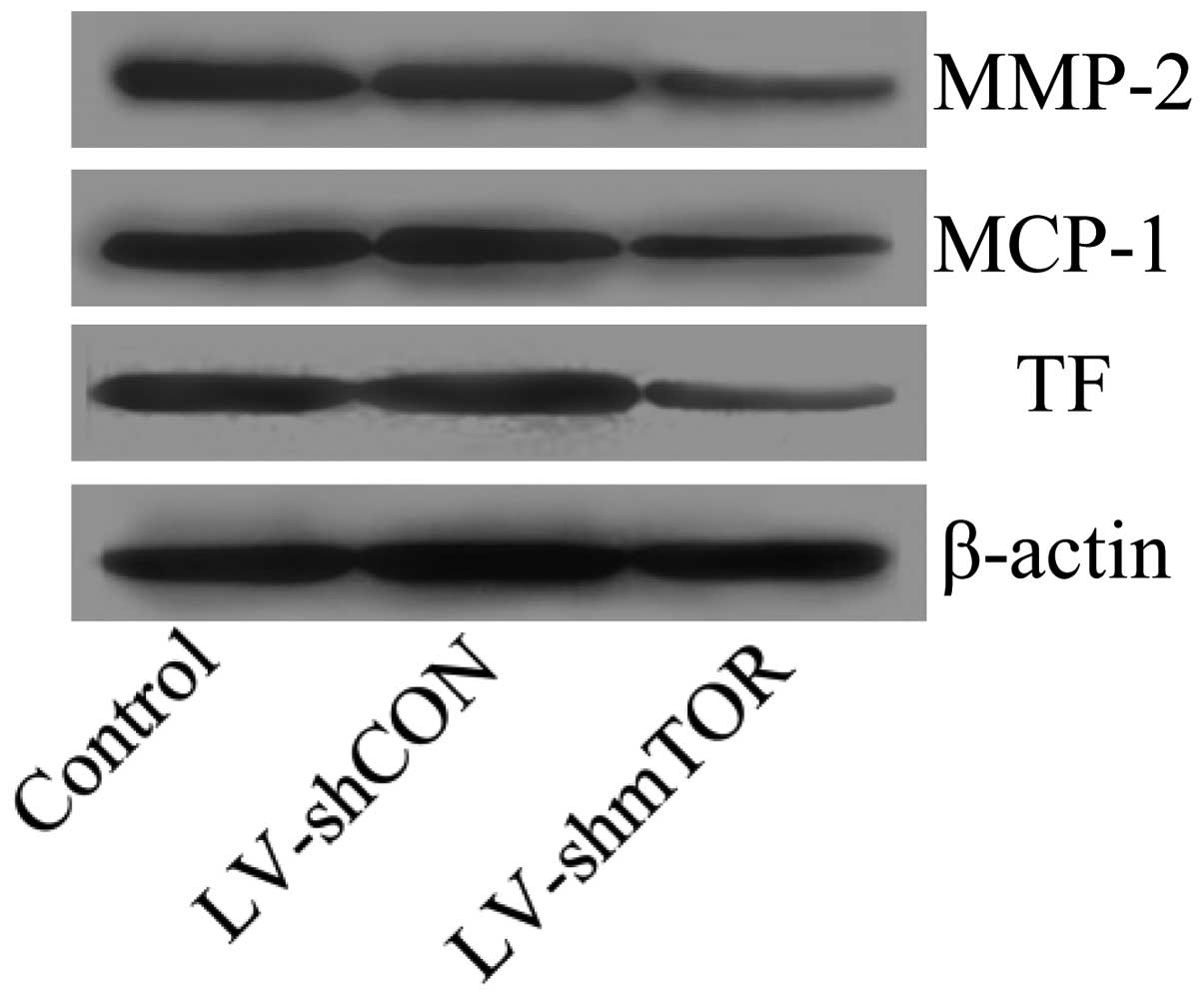
Knockdown of mTOR suppressed the
expression of genes promoting plaque destabilization
Inflammatory factors including MCP-1, MMPs and TF
have been reported to promote plaque rupture. To investigate the
effect of inhibition of mTOR on the expression of MCP-1, MMPs and
TF, western blot analysis was performed. Results showed that the
inhibition of mTOR significantly suppressed the protein expression
of MCP-1, MMP-2 and TF (Fig.
6).
Discussion
In the present study, we demonstrated that knockdown
of mTOR by LV-mediated RNAi in atherosclerotic plaques showed
beneficial effects for maintaining atherosclerotic plaque
stability. Thus, targeting mTOR by RNAi for atherosclerosis therapy
is a promising method that should be further investigated.
The dysregulation of lipid metabolism has been
proposed to be one of the major causes contributing to the
processes underlying the formation and development of
atherosclerosis. High levels of cholesterol, triglycerides and
LDL-C aggravate atherosclerotic progression, while high levels of
HDL-C are able to reduce the risk of atherosclerotic progression
(30). It has also been reported
that inhibition of mTOR restricts lipid accumulation in macrophages
and facilitates macrophage cholesterol efflux, while promoting
reverse cholesterol transport (31–33). Results of the present study
demonstrate that silencing of mTOR in vivo by RNAi
alleviated the dysregulated lipid metabolism by decreasing the
level of cholesterol, triglyceride and LDL-C, and increasing the
level of HDL-C in the atherosclerotic mouse model.
Accordingly, suppression of mTOR alleviated lipid
metabolism and prevented macrophage accumulation (31–33). It has been demonstrated that
administration of the mTOR inhibitor everolimus leads to inhibition
of protein synthesis and results in the selective elimination of
macrophages in atherosclerotic plaques by autophagy-mediated cell
death (11,34,35). Furthermore, in vitro
studies demonstrated that mTOR gene silencing by mTOR-specific
siRNA induced macrophage cell death. Consistent with previous
studies (11), we found that
knockdown of mTOR in an atherosclerotic mouse model by RNAi
resulted in a decrease of macrophages. As macrophages have been
suggested to contribute to plaque destabilization (10), these results suggest that
targeting mTOR for inhibition of macrophages is a promising for the
treatment of atherosclerotic plaque stability.
However, the mechanism of mTOR-mediated macrophage
death remains elusive. As mTOR regulates gene translation,
inhibition of mTOR by everolimus showed a strong inhibition on
protein synthesis by dephosphorylation of p70S6 kinase and 4E-BP1,
as well as hyperphosphorylation of eIF2α and eEF2, which were the
protein effectors (34). Thus,
suppression of the protein synthesis by mTOR inhibition may be
responsible for the induction of macrophage death. Findings of
studies have suggested that mTOR regulates proteins involved in
autophagy including Atg13 (28).
Accordingly, the inhibition of mTOR rapidly dephosphorylated Atg13,
which then formed a complex with Atg1 and led to the induction of
autophagy. The LC3 protein is the marker of autophagosome formation
and an increase of the LC3-I/LC-II ratio indicated the accumulation
of autophagosome (29). In the
present study, we found that inhibition of mTOR in vivo by
RNAi led to dephosphorylation of Atg13, suggesting that autophagy
activity was increased. In addition, the LC3-I protein level was
decreased whereas the LC3-II protein level was increased by
silencing mTOR, which confirmed the accumulation of autophagosomes.
Thus, in accordance with previous studies (28), our findings suggest that the
inhibition of mTOR led to autophagy by dephosphorylation of Atg13.
However, in addition to Atg13, mTOR regulates other
autophagy-specific genes such as Atg8 and Atg14 (36,37). Therefore, the precise mechanism of
mTOR inhibition-mediated autophagy needs further clarification.
Enhanced inflammation activity was also a hallmark
of plaque rupture. As macrophages were decreased subsequent to mTOR
inhibition, we detected the effects on macrophage-secreted
inflammatory factors, including MMP-2, MCP-1 and TF, which have
been suggested to be responsible for atherosclerotic plaque
destabilization and rupture and are considered a clinical detection
marker (8,38). One of the causes of plaque rupture
was the fibrous cap becoming thin (39). The fibrous cap is mainly composed
of extracellular matrix and the decrease of extracellular matrix
was suggested to be associated with fibrous cap thinning (40). Increasing evidences have reported
that MMPs are hyper-expressed by various cells including
macrophages upon various cytokines affection in atherosclerosis,
which largely degrades the extracellular matrix (41). Studies have demonstrated that
MMP-2 was significantly upregulated in unstable plaques (7), suggesting that MMP-2-mediated
degradation of the extracellular matrix was an important risk
factor for unstable plaques. MCP-1 is a type of chemokine that
mainly recruits monocytes (42)
and is highly expressed in macrophage-enriched plaques, suggesting
critical roles in the processes of atherosclerosis and plaque
instability (43). More recently,
it has been demonstrated that silencing of MCP-1 prevents
vulnerable plaque disruption (44). TF was the regulator of coagulation
and hemostasis, and is mainly secreted by macrophages, which
activate the coagulation system when the fibrous cap is broken,
resulting in thrombosis (9,45).
Thus, stability of the fibrous cap is critical for plaque
stability. The fibrous cap is capable of preventing blood entering
the arterial lumen from tissues under the fibrous cap (46). Results of the present study show
that the knockdown of mTOR promotes plaque stability by decreasing
the plaque area and increasing the fibrous cap. Knockdown of mTOR
also suppressed MMP-2, MCP-1 and TF protein expression levels,
which was beneficial for plaque stability.
The method of RNAi which efficiently and selectively
silences target gene expression has been applied in experimental
studies for the treatment of various diseases (47,48). In the present study, we
successfully inhibited mTOR expression in vivo in an
atherosclerotic mouse model by RNAi mediated by LV. Inhibition of
mTOR expression showed protective effects on the stability of
atherosclerotic plaques via the removal of macrophages induced by
autophagy. In addition, the genes that promoted plaque instability,
including MMP-2, MCP-1 and TF, were downregulated. These results
indicate that mTOR is a promising target for the treatment of
atherosclerotic plaque destabilization and rupture. However, the
underlying mechanism of mTOR in regulating plaque stability remains
to be clarified. The RNAi-mediated silencing of gene expression
with specificity for target genes has advantages over traditional
drug inhibitors. Thus, the investigation of mTOR in vitro
and in vivo models with regard to RNAi should be
conducted.
Acknowledgements
The present study was supported by grants from the
National Natural Science Foundation of China (no., 81070045).
Abbreviations:
|
mTOR
|
mammalian target of rapamycin
|
|
shRNA
|
small hairpin RNA
|
|
RNAi
|
RNA interference
|
|
LV
|
lentivirus
|
|
MMPs
|
matrix metalloproteinases
|
|
MCP-1
|
monocyte chemoattractant protein 1
|
|
TF
|
tissue factor
|
References
|
1
|
Davies MJ and Thomas AC: Plaque fissuring
- the cause of acute myocardial infarction, sudden ischaemic death,
and crescendo angina. Br Heart J. 53:363–373. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Falk E, Shah PK and Fuster V: Coronary
plaque disruption. Circulation. 92:657–671. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Davies MJ, Richardson PD, Woolf N, Katz DR
and Mann J: Risk of thrombosis in human atherosclerotic plaques:
role of extracellular lipid, macrophage, and smooth muscle cell
content. Br Heart J. 69:377–381. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Virmani R, Burke AP, Farb A and Kolodgie
FD: Pathology of the vulnerable plaque. J Am Coll Cardiol.
47:C13–C18. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mizuno Y, Jacob RF and Mason R:
Inflammation and the development of atherosclerosis. J Atheroscler
Thromb. 18:351–358. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Libby P, Ridker PM and Maseri A:
Inflammation and atherosclerosis. Circulation. 105:1135–1143. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Shah PK, Falk E, Badimon JJ, et al: Human
monocyte-derived macrophages induce collagen breakdown in fibrous
caps of atherosclerotic plaques. Potential role of matrix-degrading
metalloproteinases and implications for plaque rupture.
Circulation. 92:1565–1569. 1995.
|
|
8
|
Niu J and Kolattukudy PE: Role of MCP-1 in
cardiovascular disease: molecular mechanisms and clinical
implications. Clin Sci (Lond). 117:95–109. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lwaleed BA, Cooper AJ, Voegeli D and
Getliffe K: Tissue factor: a critical role in inflammation and
cancer. Biol Res Nurs. 9:97–107. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Boyle JJ: Macrophage activation in
atherosclerosis: pathogenesis and pharmacology of plaque rupture.
Curr Vasc Pharmacol. 3:63–68. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Verheye S, Martinet W, Kockx MM, et al:
Selective clearance of macrophages in atherosclerotic plaques by
autophagy. J Am Coll Cardiol. 49:706–715. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Thomas G and Hall MN: TOR signalling and
control of cell growth. Curr Opin Cell Biol. 9:782–787. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Dennis PB, Fumagalli S and Thomas G:
Target of rapamycin (TOR): balancing the opposing forces of protein
synthesis and degradation. Curr Opin Genet Dev. 9:49–54. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Easton JB and Houghton PJ: Therapeutic
potential of target of rapamycin inhibitors. Expert Opin Ther
Targets. 8:551–564. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Woltman AM, van der Kooij SW, Coffer PJ,
et al: Rapamycin specifically interferes with GM-CSF signaling in
human dendritic cells, leading to apoptosis via increased p27KIP1
expression. Blood. 101:1439–1445. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Huang S, Shu L, Dilling MB, et al:
Sustained activation of the JNK cascade and rapamycin-induced
apoptosis are suppressed by p53/p21(Cip1). Mol Cell. 11:1491–1501.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ravikumar B, Vacher C, Berger Z, et al:
Inhibition of mTOR induces autophagy and reduces toxicity of
polyglutamine expansions in fly and mouse models of Huntington
disease. Nat Genet. 36:585–595. 2004. View
Article : Google Scholar
|
|
18
|
Noda T and Ohsumi Y: Tor, a
phosphatidylinositol kinase homologue, controls autophagy in yeast.
J Biol Chem. 273:3963–3966. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Abraham RT and Gibbons JJ: The mammalian
target of rapamycin signaling pathway: twists and turns in the road
to cancer therapy. Clin Cancer Res. 13:3109–3114. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Suzuki T, Kopia G, Hayashi S, et al:
Stent-based delivery of sirolimus reduces neointimal formation in a
porcine coronary model. Circulation. 104:1188–1193. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Elloso MM, Azrolan N, Sehgal SN, et al:
Protective effect of the immunosuppressant sirolimus against aortic
atherosclerosis in apo E-deficient mice. Am J Transplant.
3:562–569. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mizushima N and Komatsu M: Autophagy:
renovation of cells and tissues. Cell. 147:728–741. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yang Z and Klionsky DJ: Eaten alive: a
history of macroautophagy. Nat Cell Biol. 12:814–822. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chen Y, Lin MC, Yao H, et al:
Lentivirus-mediated RNA interference targeting enhancer of zeste
homolog 2 inhibits hepatocellular carcinoma growth through
down-regulation of stathmin. Hepatology. 46:200–208. 2007.
View Article : Google Scholar
|
|
25
|
Jiang L, Lai YK, Zhang J, et al: Targeting
S100P inhibits colon cancer growth and metastasis by
lentivirus-mediated RNA interference and proteomic analysis. Mol
Med. 17:709–716. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
von der Thusen JH, van Berkel TJ and
Biessen EA: Induction of rapid atherogenesis by perivascular
carotid collar placement in apolipoprotein E-deficient and
low-density lipoprotein receptor-deficient mice. Circulation.
103:1164–1170. 2001.PubMed/NCBI
|
|
27
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2−(Delta Delta C(T)) method. Methods. 25:402–408. 2001.
|
|
28
|
Levine B and Klionsky DJ: Development by
self-digestion: molecular mechanisms and biological functions of
autophagy. Dev Cell. 6:463–477. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Mizushima N: Methods for monitoring
autophagy. Int J Biochem Cell Biol. 36:2491–2502. 2004. View Article : Google Scholar
|
|
30
|
Toth PP: High-density lipoprotein as a
therapeutic target: clinical evidence and treatment strategies. Am
J Cardiol. 96:50K–58K. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ma KL, Ruan XZ, Powis SH, Moorhead JF and
Varghese Z: Anti-atherosclerotic effects of sirolimus on human
vascular smooth muscle cells. Am J Physiol Heart Circ Physiol.
292:H2721–H2728. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ma KL, Varghese Z, Ku Y, et al: Sirolimus
inhibits endogenous cholesterol synthesis induced by inflammatory
stress in human vascular smooth muscle cells. Am J Physiol Heart
Circ Physiol. 298:H1646–H1651. 2010. View Article : Google Scholar
|
|
33
|
Mathis AS, Jin S, Friedman GS, et al: The
pharmacodynamic effects of sirolimus and sirolimus-calcineurin
inhibitor combinations on macrophage scavenger and nuclear hormone
receptors. J Pharm Sci. 96:209–222. 2007. View Article : Google Scholar
|
|
34
|
Croons V, Martinet W, Herman AG,
Timmermans JP and De Meyer GR: Selective clearance of macrophages
in atherosclerotic plaques by the protein synthesis inhibitor
cycloheximide. J Pharmacol Exp Ther. 320:986–993. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Martinet W, Verheye S, De Meyer I, et al:
Everolimus triggers cytokine release by macrophages: rationale for
stents eluting everolimus and a glucocorticoid. Arterioscler Thromb
Vasc Biol. 32:1228–1235. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Huang WP, Scott SV, Kim J and Klionsky DJ:
The itinerary of a vesicle component, Aut7p/Cvt5p, terminates in
the yeast vacuole via the autophagy/Cvt pathways. J Biol Chem.
275:5845–5851. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chan TF, Bertram PG, Ai W and Zheng XF:
Regulation of APG14 expression by the GATA-type transcription
factor Gln3p. J Biol Chem. 276:6463–6467. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Crouzet J, Faucher JF, Toubin M, Hoen B
and Estavoyer JM: Serum C-reactive protein (CRP) and procalcitonin
(PCT) levels and kinetics in patients with leptospirosis. Eur J
Clin Microbiol Infect Dis. 30:299–302. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Bea F, Blessing E, Bennett B, et al:
Simvastatin promotes atherosclerotic plaque stability in
apoE-deficient mice independently of lipid lowering. Arterioscler
Thromb Vasc Biol. 22:1832–1837. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zaman AG, Helft G, Worthley SG and Badimon
JJ: The role of plaque rupture and thrombosis in coronary artery
disease. Atherosclerosis. 149:251–266. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Morancho A, Rosell A, García-Bonilla L and
Montaner J: Metalloproteinase and stroke infarct size: role for
anti-inflammatory treatment? Ann NY Acad Sci. 1207:123–133. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Deshmane SL, Kremlev S, Amini S and Sawaya
BE: Monocyte chemoattractant protein-1 (MCP-1): an overview. J
Interferon Cytokine Res. 29:313–326. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Rull A, Beltrán-Debón R, Aragonès G, et
al: Expression of cytokine genes in the aorta is altered by the
deficiency in MCP-1: effect of a high-fat, high-cholesterol diet.
Cytokine. 50:121–128. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Liu XL, Zhang PF, Ding SF, et al: Local
gene silencing of monocyte chemoattractant protein-1 prevents
vulnerable plaque disruption in apolipoprotein E-knockout mice.
PLoS One. 7:e334972012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Jude B, Zawadzki C, Susen S and Corseaux
D: Relevance of tissue factor in cardiovascular disease. Arch Mal
Coeur Vaiss. 98:667–671. 2005.PubMed/NCBI
|
|
46
|
Kume N: Molecular mechanisms of coronary
atherosclerotic plaque formation and rupture. Nihon Rinsho.
68:637–641. 2010.(In Japanese).
|
|
47
|
Song E, Lee SK, Wang J, et al: RNA
interference targeting Fas protects mice from fulminant hepatitis.
Nat Med. 9:347–351. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
48
|
Paroo Z and Corey DR: Challenges for RNAi
in vivo. Trends Biotechnol. 22:390–394. 2004. View Article : Google Scholar : PubMed/NCBI
|