Introduction
Stroke is regarded as the second most common cause
of mortality worldwide (1).
Tissue plasminogen activator for removal of the thrombus, which
must be administered during the early stages, within 4–5 h of
stroke onset, is the currently approved pharmacotherapy
administered after ischemic stroke (1–3).
Reperfusion following cerebral ischemia plays an important role in
the recovery of blood flow in order to reduce neuronal damage;
however, cerebral ischemia and reperfusion trigger multiple cell
signaling pathways, leading to cell survival or cell damage
(4).
Accumulating evidence suggests that, apart from
necrosis, apoptosis is an additional cause of hypoxic-ischemic
neuronal loss in the nervous system. The ischemic core undergoes
necrotic injury within minutes after ischemic stroke; however, the
ischemic penumbra, the border of the necrotic core, undergoes
apoptosis within several hours or days with the activation of
multiple death pathways (5,6).
Neuronal cells in the distal penumbra area tolerate a longer
duration of ischemia, compared with those of the core, and cerebral
blood flow is less severely affected (7). Damage to the penumbra caused by
ischemic stroke may be reversible, comprising an energy-dependent
apoptosis when blood flow is restored (5,8).
Due to the contribution of neuronal cell loss,
apoptosis is a fundamental target for neuroprotective strategies in
the management of cerebral ischemia (9,10).
The overload of intracellular calcium by excessive overstimulation
of ionotropic glutamate receptors following cerebral ischemia can
cause irreversible neuronal cell death through intracellular
apoptotic signaling cascades (11–13). Calpain, caspases, phospholipase A2
and endonucleases, calcium-dependent effective proteins, lead to
cell death (14,15).
The major apoptotic signaling pathways may be
classified as the extrinsic death receptor (DR) and intrinsic
mitochondrial apoptotic pathways (4). DR apoptotic pathways are regulated
by the activation of DRs with their ligands on the cell membrane,
finally resulting in the activation of caspases (16). The mitochondrial apoptotic pathway
is mediated by the activation of pro-apoptotic proteins, such as
Bid and Bax, and Bcl-2 family proteins (17). Both the extrinsic and intrinsic
apoptotic pathways play a vital role in the execution of cell
death, recruiting downstream apoptotic molecules (4).
Acupuncture, a traditional therapeutic treatment in
Oriental medicine, has long been used in clinical practice for the
prevention and treatment of stroke. In particular,
electroacupuncture (EA), which involves the application of
electrical stimulation with acupuncture needles, has been widely
used both in clinical practice and in studies using animal stroke
models in Korea. Accumulating evidence has indicated that treatment
with EA can effectively attenuate cerebral infarction and exerts
anti-apoptotic and/or neuroprotective effects after
hypoxic-ischemic insults (3,18–20).
The regulation of apoptotic signaling pathways
following focal or global ischemia may result in the attenuation of
neuronal cell death and may lead to the development of novel
therapeutic strategies for the treatment of cerebral ischemia.
Treatment with EA may reverse the damage caused to the ischemic
penumbra by inhibiting the activation of apoptotic pathways;
however, the underlying mechanisms remain unclear. According to our
results, treatment with EA at the acupoints corresponding to Baihui
and Qihai resulted in a significantly reduced infarct volume and
improvement of neurological outcome after stroke, consistent with
previous studies (21,22). Therefore, we hypothesized that
treatment with EA can alleviate cell death resulting from ischemic
injury through the main routes of the apoptotic pathway,
particularly in the cortex of the brain, which tolerates a longer
duration of ischemia, which may be reversible by treatment with EA.
We investigated the apoptotic signaling pathways involved in the
beneficial effects of EA in a rat model of middle cerebral artery
occlusion (MCAO).
Materials and methods
Antibodies
DR4, DR5, tumor necrosis factor-related
apoptosis-inducing ligand (TRAIL), Fas, Fas ligand (FasL), Bcl-2,
Bcl-xL, Bax, Bad, Bid, X-linked inhibitor of apoptosis protein
(XIAP), caspase-3, caspase-8, caspase-9, poly[ADP-ribose]
polymerase (PARP)-1, β-catenin and phospholipase C γ1 (PLCγ1) and
secondary antibodies were supplied by Santa Cruz Biotechnology
(Santa Cruz, CA, USA). cIAP-1 and cIAP-2 were supplied by
Calbiochem (San Diego, CA, USA).
Animals
Male Sprague-Dawley rats averaging 150–180 g in
weight were obtained from DooYeol Biotech (Seoul, Korea). The rats
were housed at 22°C under alternating 12 h cycles of dark and
light, and were fed a commercial diet and allowed access to tap
water ad libitum, commencing one week before the study began
and continuing throughout the study. All experiments were approved
by the Pusan National University Animal Care and Use Committee in
accordance with the National Institutes of Health Guidelines. Each
group included six rats and all treatments were administered under
isoflurane (Choongwae, Seoul, Korea) anesthesia, using a VIP 3000
calibrated vaporizer (Midmark, Orchard Park, OH, USA). The rats
were randomly assigned to the sham-operated, MCAO and MCAO + EA
groups. The rats in the MCAO group were subjected to MCAO only and
the rats in the MCAO + EA group received EA stimulation immediately
after reperfusion. The sham-operated rats underwent the same
procedure without MCAO.
Focal cerebral ischemia
Focal cerebral ischemia was induced by occluding the
middle cerebral artery using the intraluminal filament technique.
Rats used in the experiment were anesthetized using 2.0% isoflurane
(Choongwae) and cerebral blood flow was monitored using a PeriFlux
System 5000 laser-Doppler flowmeter (Perimed, Stockholm, Sweden).
Under a SXZ16 operating microscope (Olympus, Tokyo, Japan), the
right external carotid artery and the internal carotid artery were
bound with 4-0 silk sutures (Mersilk; Ethicon Inc., Somerville, NJ,
USA). The middle cerebral artery was occluded by a 2.0–3.0 cm
length of 4-0 nylon suture with a silicon-coated tip. After 90 min
of MCAO, the filament was withdrawn and reperfusion was confirmed
using laser Doppler examination.
EA stimulation
Under light isoflurane anesthesia, two bilateral
stainless-steel 0.2 mm-diameter needles were inserted to a depth of
approximately 3 mm at the acupoints corresponding to Baihui (GV20,
the midpoint of the line connecting the apexes of both ears on the
parietal bone) and Qihai (CV6, the lower abdomen and on the
anterior midline) in men, and were connected to a Pulsemaster
Multi-channel Stimulator SYS-A300 electrical stimulator (World
Precision Instruments, Berlin, Germany). EA treatment was performed
with 2 Hz stimulation for 30 min and intensity was set at 1 mA. EA
was administered twice per day at 12-h intervals immediately after
occlusion. Animals that did not receive EA were subjected to the
same anesthesia as those that received EA.
Determination of infarct area
For quantification of ischemic damage, the rats were
anesthetized by an intraperitoneal injection of 8% chloral hydrate
solution (300 mg/kg) and the areas of cerebral infarction were
evaluated by 2,3,5-triphenyltetrazolium chloride (TTC;
Sigma-Aldrich, St. Louis, MO, USA) staining three days after
occlusion. The brains were cut into eight 2-mm-thick sections and
stained with 2% TTC in phosphate-buffered saline (PBS) for 30 min
at room temperature. The sections were then stored in formaldehyde
solution (Duksan Pure Chemicals, Ansan, Korea). Photographs of each
brain section were acquired using a digital camera by arranging the
sections in turn and the infarct area was measured using an
I-Solution™ Image Analyzer [Image & Microscope Technology
(IMT), Vancouver, BC, Canada]. The infarct area was calculated by
the direct measurement of the areas of each section.
Measurement of apoptotic neuronal
death
The rats received intracardial perfusion with 200 ml
of PBS followed by 100 ml of fixative containing 4%
paraformaldehyde in PBS under chloral hydrate anesthesia and the
cerebral cortex of the ipsilateral ischemic area was removed. The
removed tissue was kept in the same fixative for 24 h, followed by
immersion in 30% sucrose for 48 h at 4°C for cryoprotection. Frozen
sections (14-μm-thick) were then prepared. Apoptotic neuronal death
was characterized by staining with Hoechst 33342 (Invitrogen,
Carlsbad, CA, USA) and a terminal deoxynucleotidyl
transferase-mediated dUTP nick end-labeling (TUNEL) assay. Tissues
were stained with Hoechst 33342 at room temperature for 30 min and
fragmented or condensed DNA was counted. TUNEL assays were
performed using a Fluorometric TUNEL System (Promega, Madison, WI,
USA) and TUNEL-positive cells were counted. Quantitative analysis
was performed in a blinded manner by counting the number of
apoptotic cells in five random regions using a fluorescence
microscope (Carl Zeiss, Göttingen, Germany). Data are presented as
the total number of apoptotic cells.
Western blot analysis
Cortical tissues were washed in cold HEPES buffer
and homogenized in lysis buffer [200 mM Tris (pH 8.0), 150 mM NaCl,
2 mM EDTA, 1 mM NaF, 1% NP40, 1 mM phenylmethanesulfonylfluoride
(PMSF), 1 mM Na3Vo4 and protease inhibitor
cocktail]. Equal amounts of protein were then separated by 10%
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
(SDS-PAGE); the resolved proteins were then transferred onto
nitrocellulose membranes (Whatman, Dassel, Germany). The membranes
were incubated with antibodies overnight at 4°C. The membranes were
subsequently incubated with horseradish peroxidase-conjugated
secondary antibody. β-actin was used as a loading control for all
experiments. Densitometric analysis was performed using an
ImageQuant LAS-4000 imaging system (Fujifilm, Tokyo, Japan) for the
quantification of immunoreactivity corresponding to the bands.
Caspase assay
Colorimetric assay kits (R&D Systems,
Minneapolis, MN, USA), which utilize synthetic tetrapeptides
[Asp-Glu-Val-Asp (DEAD) for caspase-3; Ile-Glu-Thr-Asp (IETD) for
caspase-8; and Leu-Glu-His-Asp (LEHD) for caspase-9] labeled with
p-nitroaniline (pNA), were used for the determination of caspase
activity. Briefly, 150 μg protein in the cerebral cortex were mixed
with extraction buffer [40 mM HEPES (pH 7.4), 20% glycerol (v/v), 1
mM EDTA, 0.2% NP-40 and 10 mM DL-DTT] containing 100 μM fluorogenic
peptide substrate, diluted with extraction buffer setting a volume
of 100 μl per sample in a microtiter plate, followed by incubation
at 37°C for 2 h in the dark. For the determination of caspase
activity, changes in absorbance were measured at a wavelength of
405 nm using an ELISA reader.
Data analyses
All data are expressed as the means ± SEM and the
SigmaStat statistical program Version 11.2 (Systat Software, San
Jose, CA, USA) was used for data analysis. For statistical analysis
of the data, the Student’s t-test was used when comparing two
groups, and one-way ANOVA via Tukey’s post hoc comparison was used
when comparing more than two groups. A p<0.05 was considered to
indicate a statistically significant difference.
Results
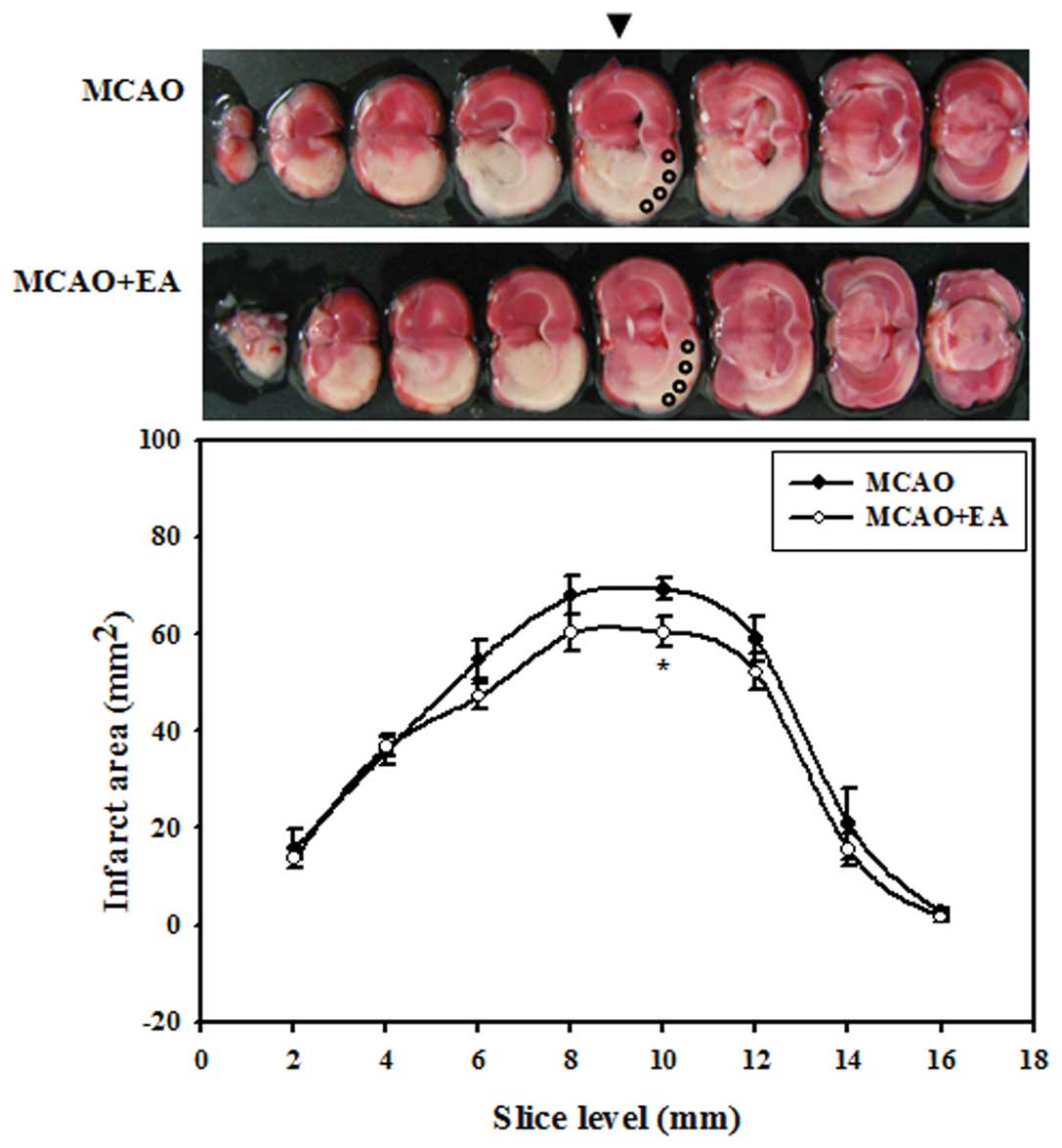
Effects of EA on the infarct area
We performed EA on the rats with MCAO at vessel
reperfusion and 12 h after occlusion twice the first day and twice
per day at 12-h intervals thereafter. To determine which region of
the brain shows beneficial effects by EA, we analyzed the infarct
area of each coronal slice between the rats in the MCAO and MCAO +
EA group three days after occlusion. Treatment with EA resulted in
the reduction of the infarct area from the anterior region to the
posterior region of the brain. In addition, a significant decrease
in the infarct area was observed at the middle region of the brain,
mainly in the fifth slice level at 10 mm in distance from the
frontal pole. These results suggested that EA stimulation exerted a
significant neuroprotective effect against focal cerebral ischemia,
particularly in the middle region of the brain. We used cortical
tissue obtained from the fifth slice level within circles measuring
2 mm in diameter for the analysis of apoptosis (Fig. 1).
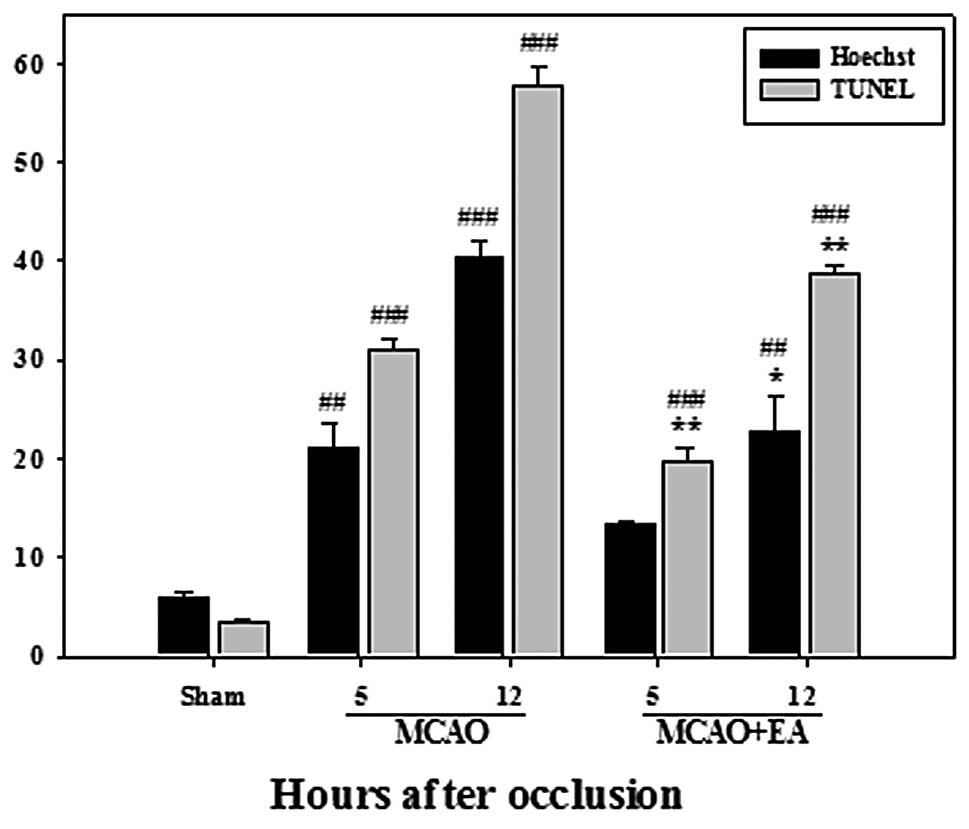
Effects of EA on apoptotic cell
death
We performed Hoechst 33342 and TUNEL staining as a
marker of apoptotic cells. The cells with fragmented or condensed
DNA and TUNEL-positive cells were counted; the data are presented
as apoptotic neurons as a percentage of total neurons. The number
of apoptotic cells significantly increased at 5 and 12 h after
occlusion in the rats with MCAO, compared with the sham-operated
rats. However, the number of apoptotic cells markedly decreased
following EA stimulation (Fig.
2). These changes indicate that progressive cell loss in the
cerebral cortex of an ischemic cerebrum may occur due to apoptosis,
not necrosis, and may be alleviated by EA stimulation.
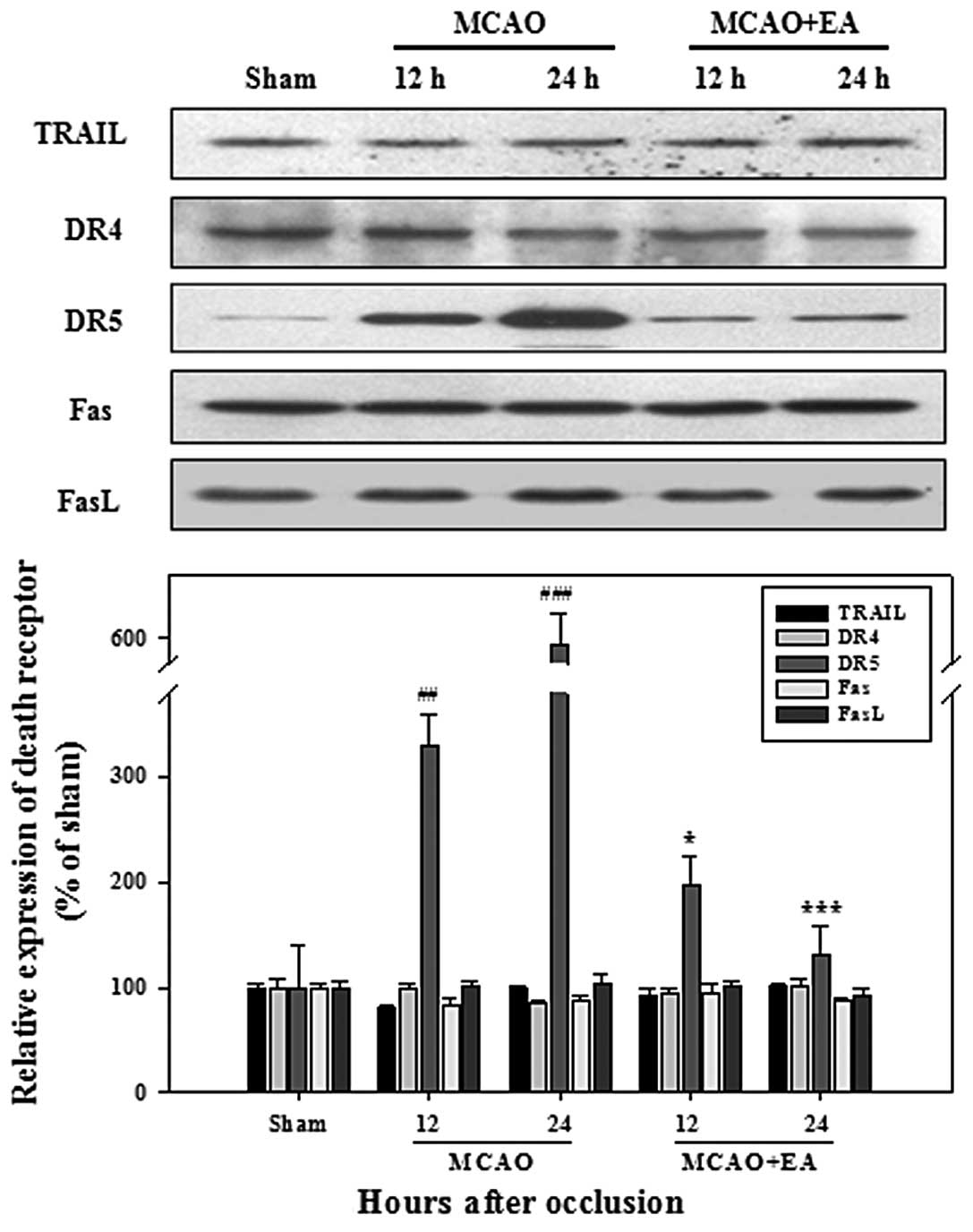
Effects of EA on apoptotic signaling
pathways
We performed western blot analysis to determine
whether neuronal apoptosis occurs through the death receptor and
mitochondrial pathways. As regards the expression of death ligands
and receptors, no significant changes were observed between the
rats in the MCAO and MCAO + EA group, apart from DR5. The
expression of DR5 was significantly higher in the rats with MCAO,
compared with the sham-operated rats at 12 and 24 h after
occlusion; however, this increase was reversed following treatment
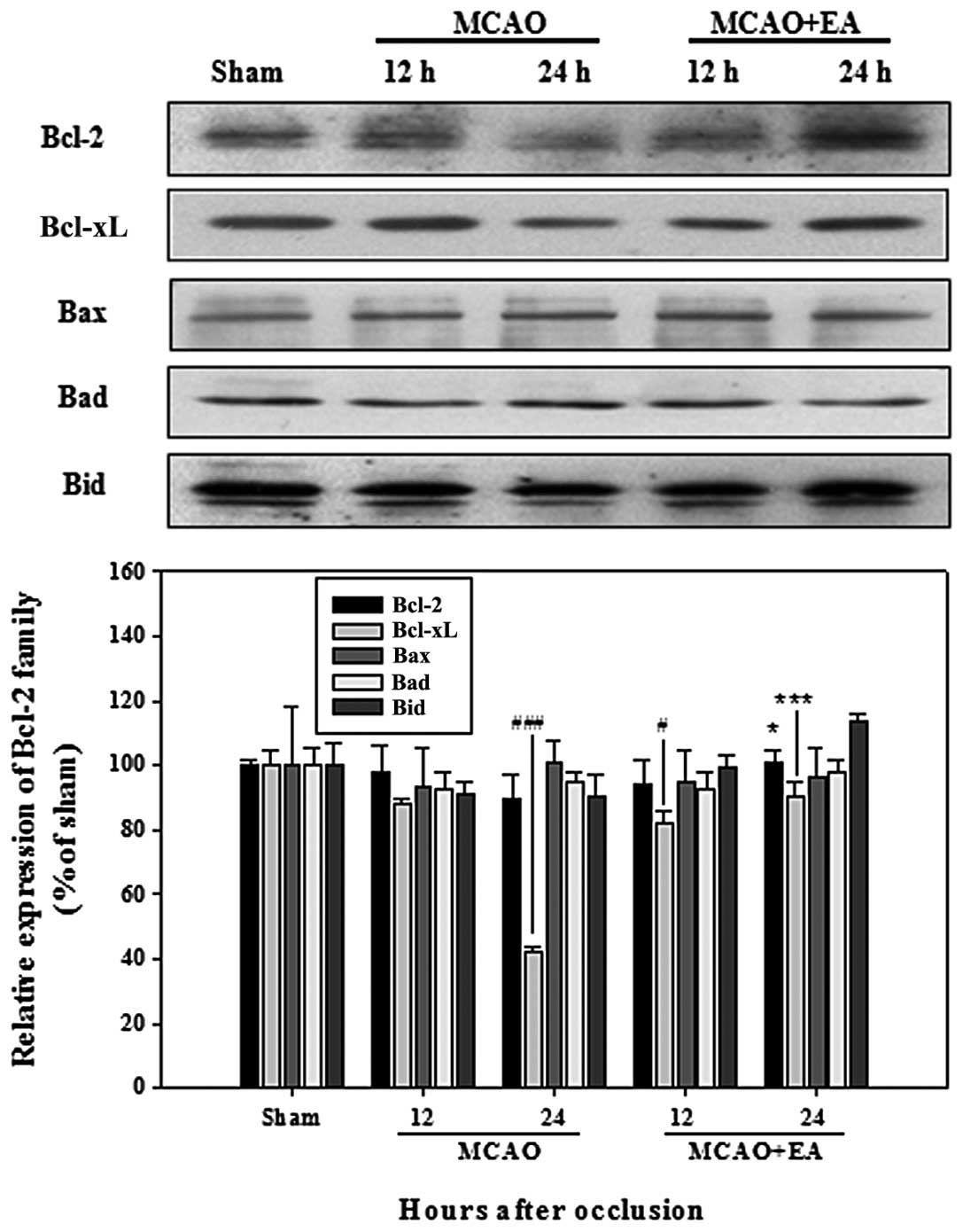
with EA (Fig. 3). Among the Bcl-2
family members, only the expression of Bcl-xL showed a significant
decrease in the rats with MCAO; however, a higher expression of
Bcl-2 and Bcl-xL was observed in the rats in the MCAO + EA group,
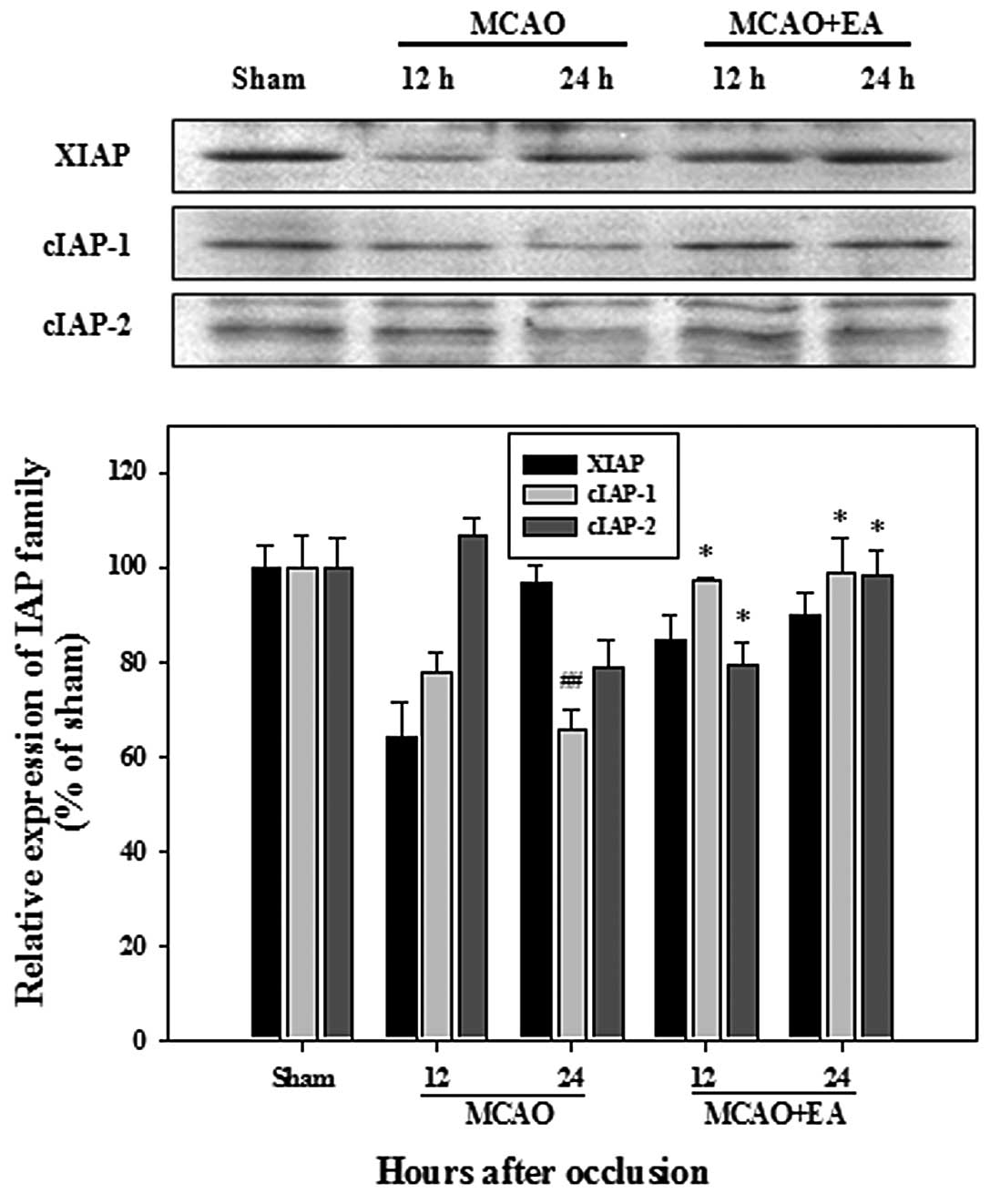
compared with the MCAO group rats at 24 h after occlusion (Fig. 4). The IAP family in the rats with
MCAO exhibited a lower expression compared with the sham-operated
rats; however, a significantly higher expression of cIAP-1 and
cIAP-2 was observed in the rats in the MCAO + EA group, when
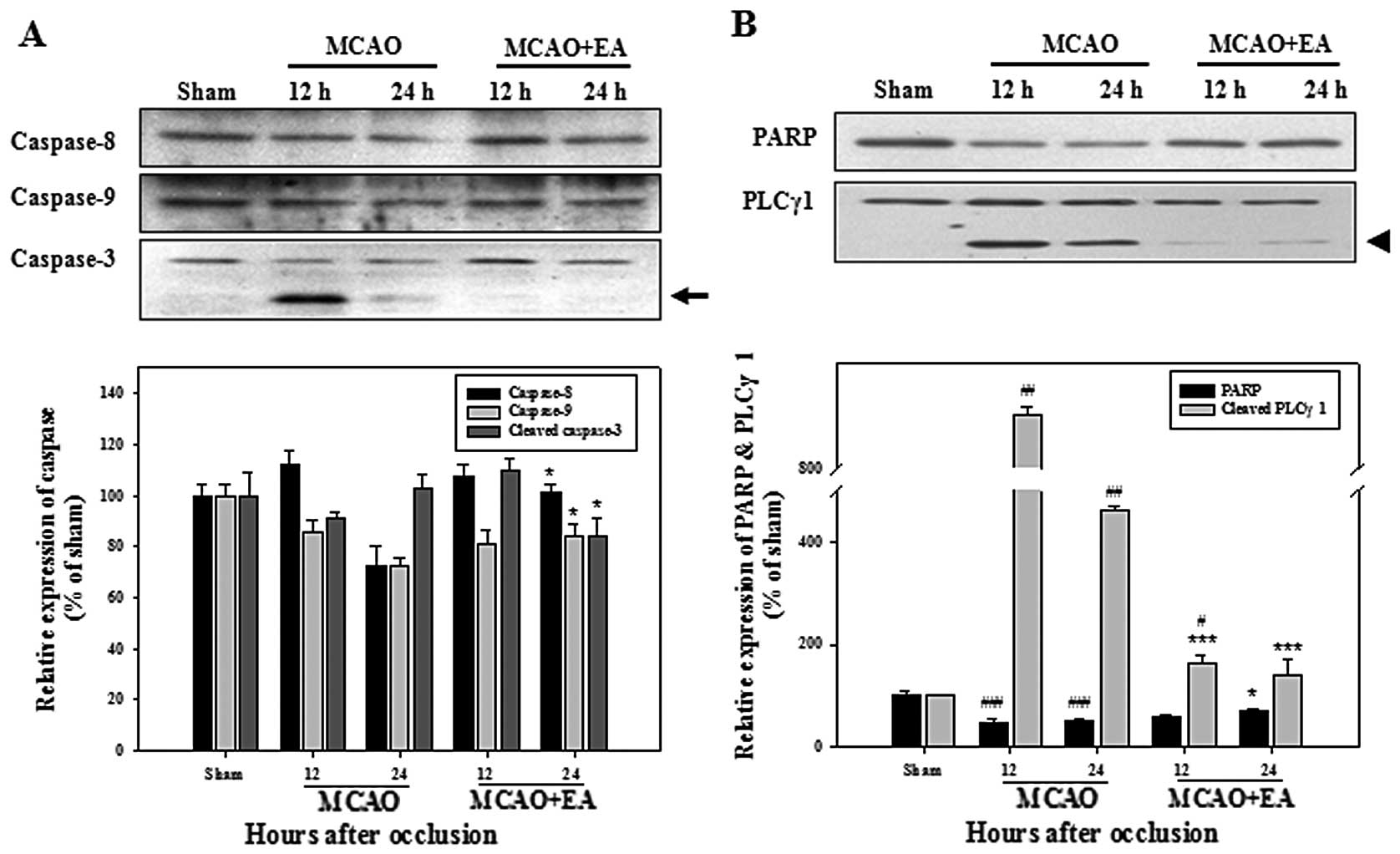
compared with the rats in the MCAO group (Fig. 5). We observed only one instance of
activated caspase-3 expression, which was significantly arrested by
treatment with EA in the rats with MCAO (Fig. 6A). Cleaved PLCγ1 in the rats with
MCAO was markedly inhibited by treatment with EA and a slightly
lower total PARP expression was observed in the rats with MCAO,
compared with the rats in the MCAO + EA group (Fig. 6B). These results suggest that EA
may protect cortical cells against ischemic-induced apoptosis
through the inhibition of the DR and mitochondrial apoptotic
pathways.
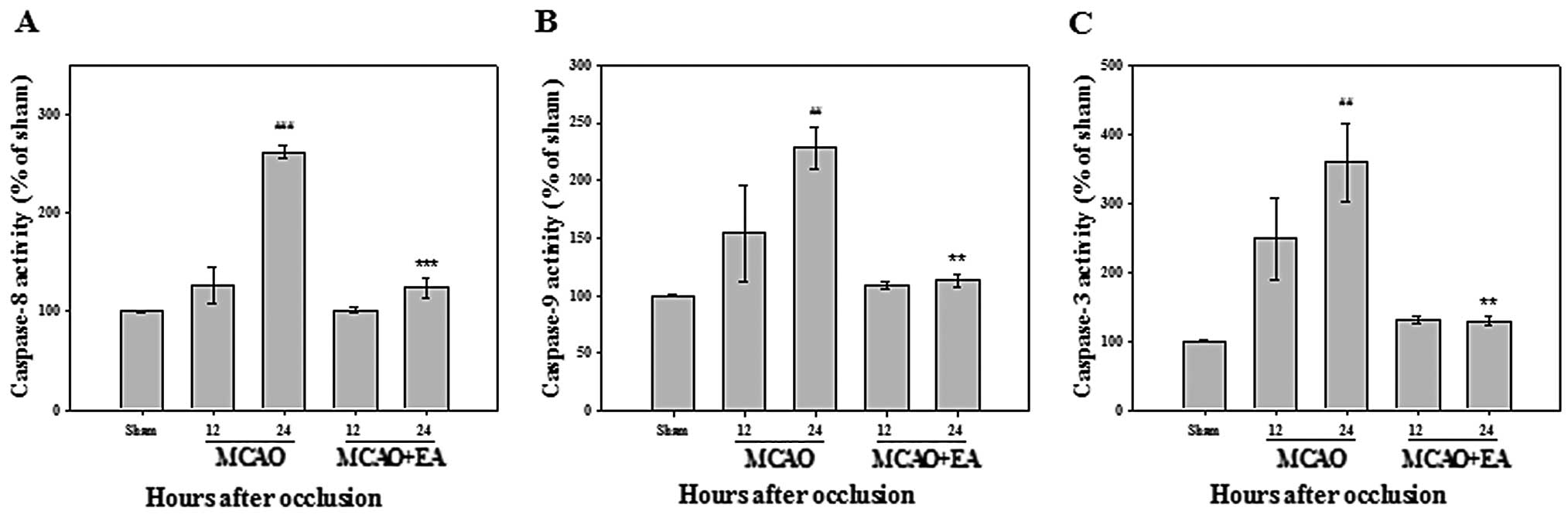
Effects of EA on caspase activity
To further confirm the underlying signaling pathways
mediating apoptosis, caspase activity assay was performed to assess
the activation of caspase-3, -8 and -9. The activities of
caspase-3, -8 and -9 showed a significant increase in the rats with
MCAO at 24 h after occlusion, compared with the sham-operated rats;
these were markedly inhibited following treatment with EA (Fig. 7). These results confirm that EA
may protect cortical cells against ischemic-induced apoptosis by
inhibition the activity of caspases.
Discussion
EA has been widely used as a potential therapy for
cerebral ischemia in clinical practice and basic research in Korea;
however, the detailed mechanisms responsible for its beneficial
effects remain unclear. In order to achieve clinical acceptance as
an effective treatment for cerebral ischemia, an understanding of
the mechanisms underlying the potent neuroprotective effects of EA
is important. In this study, we first attempted to determine which
region of the brain showed the most favorable neuroprotective
effects by EA in a rat model of transient focal cerebral ischemia.
We found that treatment with EA resulted in a significantly reduced
infarct area, particularly in the middle region of the brain
(Fig. 1).
We focused on the ischemic penumbra, which may be
able to tolerate longer ischemic states compared with any other
region; the effects of ischemia in this region may be reversible by
a novel therapeutic strategy (5,6).
As opposed to the classical ‘fried egg pattern’, the ischemic
penumbra is frequently embedded within the core (23); however, our morphological results
revealed a similar apoptotic pattern in the middle cortical region
of the brain. Chromatin condensation and TUNEL-positive cells can
be used as evidence of apoptosis, and we confirmed that the
ischemic penumbra contains apoptotic cells in the process of
undergoing apoptosis; thus, it has the capacity for recovery.
Apoptotic cells confirmed by Hoechst 33342 staining and TUNEL assay
showed a significant increase after occlusion in the MCAO group
rats, compared with the sham-operated rats; these apoptotic cells
were markedly arrested by EA stimulation (Fig. 2).
In order to determine which factors can protect
against apoptotic cell death of ischemic neurons, we analyzed
related proteins and their activities occurring through DR and
mitochondrial apoptosis. Death signals are initially induced by
various pro-apoptotic stimuli and eventually converge into a
mitochondrial-dependent mechanism activating death-execution
caspases (24).
The analysis of the expression of death ligands and
receptors revealed a significant upregulation of DR5, but not of
TRAIL. The upregulated expression of DR5 and TRAIL, which occurs
following transient ischemia-reperfusion, plays a deleterious role
in the pathogenesis of delayed neuronal damage after global
cerebral ischemia (25). However,
in the present study, the upregulation of DR5 was markedly arrested
by EA in the rats with MCAO (Fig.
3). This result suggests that the DR5-mediated death pathway
may underlie hypoxic-ischemic neuronal apoptosis and that treatment
with EA may result in the inhibition of DR5-induced apoptosis in
cerebral ischemia.
The upregulation of DRs mediates caspase-8,
initiator caspase-dependent neuronal death, and subsequently
induces neuronal cell death in focal cerebral ischemia (26,27). Activated caspase-8 leads to the
cleavage of Bid to the activated form, tBid, followed by the
triggering of mitochondrial cytochrome c release, which may
eventually lead to the activation of caspase-3 (28,29). In our study, we did not detect
cleaved (activated) caspase-8 and caspase-8 cleaved Bid, as shown
by western blot analysis. However, caspase-8 activity was
significantly inhibited by treatment with EA in the rats with MCAO
(Figs. 6A and 7A).
The IAP family plays a major role in preventing
apoptosis through the negative regulation of caspases. Members of
the IAP family include cIAP-1, cIAP-2 and XIAP (30). All IAPs can potentially bind to
caspases, but XIAP is the only member capable of blocking active
caspases (31). cIAPs can
regulate DR-mediated apoptosis, upstream of the mitochondria
(32). DR-mediated apoptosis
occurs through caspase 8-dependent cIAP-1 degradation in
vivo (33). Although other
caspases activated downstream of caspase-8 may be involved in this
pathway, as shown by our results, a significantly higher expression
of cIAP-1 was observed in the rats in the MCAO + EA group, compared
with the rats in the MCAO group (Fig.
5). These results suggest that treatment with EA may inhibit
neuronal cell death through a DR-mediated apoptotic pathway,
particularly by inhibiting DR5.
The involvement of the mitochondrial apoptotic
pathway is supported by the release of cytochrome c from the
mitochondria to the cytosol in cerebral ischemia (34). Cytochrome c in the cytosol
catalyzes the oligomerization of apoptotic protease activating
factor-1 (Apaf-1), which in turn promotes the activation of
pro-caspase-9, leading to the activation of downstream
pro-caspase-3, followed by apoptosis (35). An active role of the
mitochondrial-mediated apoptotic pathway involving caspases after
cerebral ischemia has been strongly suggested (11).
Caspases and Bcl-2 family members, along with
members of the IAP family, appear to play critical roles in the
regulation of multiple apoptotic cell death pathways during
cerebral ischemia (4). The Bcl-2
gene family includes both anti-apoptotic Bcl-2 and Bcl-xL, as well
as the pro-apoptotic proteins, Bax, Bak, Bad and Bid (4,36).
Bcl-2 and Bcl-xL play a critical role in determining cell survival
by inhibiting pro-apoptotic Bax (37,38). In our study, the higher expression
of the Bcl-2 family members, Bcl-2 and Bcl-xL, was observed in the
rats in the MCAO + EA group (Fig.
4).
Activated caspase-9 can induce the activation of
terminal caspase-3, which results in the mediation of the
mitochondrial-dependent apoptotic pathway following cerebral
ischemia (39,40). Caspase-3 is a major effector
protease in cell death and cleaved caspase presents activation
leading to cell death during ischemia (15,41). XIAP plays a major role in the
apoptotic process by directly inhibiting caspase-9, -3 and -7
(31); however, in this study,
only a slight increase in XIAP expression was observed in the rats
in the MCAO + EA group compared with the MCAO group rats (Fig. 5).
To further explore the underlying signaling pathways
in mitochondrial apoptosis, we performed assays for the assessment
of caspase activation and the cleavage of PARP and PLCγ1. Neuronal
cell death in stroke is associated with activated caspase-3 and
with cleaved PARP and PLCγ1 (4).
The activities of caspase-9 and -3 and activated caspase-3 were
markedly inhibited following treatment with EA in the MCAO group
rats (Figs. 6A and 7). The inhibited activation of caspase-3
was accompanied by the reduction of cleaved PLCγ1, a biochemical
characteristic of apoptosis (Fig.
6B). Our results suggest that treatment with EA may also result
in the inhibition of neuronal cell death through mitochondrial
apoptotic pathways, with a higher expression of anti-apoptotic
Bcl-2 and IAP family members in rats with MCAO.
We hypothesize that various signaling pathways may
be involved in EA-induced neuronal cell survival. The
phosphatidylinositol-3 kinase (PI3K)/Akt signaling pathway plays an
important role in neuronal survival by protecting cells from
caspase-mediated apoptosis and inhibiting pro-apoptotic proteins
(42,43). Our study demonstrated that EA
possibly activates the PI3K/Akt pathway to initiate the cell
survival pathway (44). EA may
induce the activation of the PI3K/Akt survival pathway, possibly
involving the anti-apoptotic form of the Bcl-2 and IAP families.
Consequently, we conclude that the EA-induced neuroprotective
effects against cerebral ischemia may be associated with the
inhibition of two major DR and mitochondrial apoptotic pathways.
Considering these results, treatment with EA may be an appropriate
therapy against cerebral ischemia by controlling two main routes of
the apoptotic pathway.
Acknowledgements
This study was supported by the R&D program of
MKE/KEIT (10040391, Development of Functional Food Materials and
Device for Prevention of Aging-associated Muscle Function
Decrease).
References
|
1
|
Shichita T, Hasegawa E, Kimura A, et al:
Peroxiredoxin family proteins are key initiators of post-ischemic
inflammation in the brain. Nat Med. 18:911–917. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lansberg MG, Bluhmki E and Thijs VN:
Efficacy and safety of tissue plasminogen activator 3 to 4.5 hours
after acute ischemic stroke: a metaanalysis. Stroke. 40:2438–2441.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Li X, Luo P, Wang Q and Xiong L:
Electroacupuncture pretreatment as a novel avenue to protect brain
against ischemia and reperfusion injury. Evid Based Complement
Alternat Med. 2012:1953972012.PubMed/NCBI
|
|
4
|
Nakka VP, Gusain A, Mehta SL and Raghubir
R: Molecular mechanisms of apoptosis in cerebral ischemia: multiple
neuroprotective opportunities. Mol Neurobiol. 37:7–38. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ginsberg MD: The new language of cerebral
ischemia. AJNR Am J Neuroradiol. 18:1435–1445. 1997.PubMed/NCBI
|
|
6
|
Love S: Apoptosis and brain ischaemia.
Prog Neuropsychopharmacol Biol Psychiatry. 27:267–282. 2003.
View Article : Google Scholar
|
|
7
|
Kato H and Kogure K: Biochemical and
molecular characteristics of the brain with developing cerebral
infarction. Cell Mol Neurobiol. 19:93–108. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kerr JF, Wyllie AH and Currie AR:
Apoptosis: a basic biological phenomenon with wide-ranging
implications in tissue kinetics. Br J Cancer. 26:239–257. 1972.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Choi DW: Glutamate neurotoxicity and
diseases of the nervous system. Neuron. 1:623–634. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lee JM, Zipfel GJ and Choi DW: The
changing landscape of ischaemic brain injury mechanisms. Nature.
399:A7–A14. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Krajewski S, Krajewska M, Ellerby LM, et
al: Release of caspase-9 from mitochondria during neuronal
apoptosis and cerebral ischemia. Proc Natl Acad Sci USA.
96:5752–5757. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lam TT, Abler AS and Tso MO: Apoptosis and
caspases after ischemia-reperfusion injury in rat retina. Invest
Ophthalmol Vis Sci. 40:967–975. 1999.PubMed/NCBI
|
|
13
|
Xiong ZQ and McNamara JO: Fleeting
activation of ionotropic glutamate receptors sensitizes cortical
neurons to complement attack. Neuron. 36:363–374. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Neumar RW, Hagle SM, DeGracia DJ, Krause
GS and White BC: Brain mu-calpain autolysis during global cerebral
ischemia. J Neurochem. 66:421–424. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Namura S, Zhu J, Fink K, et al: Activation
and cleavage of caspase-3 in apoptosis induced by experimental
cerebral ischemia. J Neurosci. 18:3659–3668. 1998.PubMed/NCBI
|
|
16
|
Broughton BR, Reutens DC and Sobey CG:
Apoptotic mechanisms after cerebral ischemia. Stroke. 40:e331–e339.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Campbell MT, Dagher P, Hile KL, et al:
Tumor necrosis factor-alpha induces intrinsic apoptotic signaling
during renal obstruction through truncated bid activation. J Urol.
180:2694–2700. 2008. View Article : Google Scholar
|
|
18
|
Wang SJ, Omori N, Li F, et al:
Potentiation of Akt and suppression of caspase-9 activations by
electroacupuncture after transient middle cerebral artery occlusion
in rats. Neurosci Lett. 331:115–118. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Du Y, Shi L, Li J, Xiong J, Li B and Fan
X: Angiogenesis and improved cerebral blood flow in the ischemic
boundary area were detected after electroacupuncture treatment to
rats with ischemic stroke. Neurol Res. 33:101–107. 2011. View Article : Google Scholar
|
|
20
|
Wang Q, Li X, Chen Y, et al: Activation of
epsilon protein kinase C-mediated anti-apoptosis is involved in
rapid tolerance induced by electroacupuncture pretreatment through
cannabinoid receptor type 1. Stroke. 42:389–396. 2011. View Article : Google Scholar
|
|
21
|
Sze FK, Wong E, Yi X and Woo J: Does
acupuncture have additional value to standard poststroke motor
rehabilitation? Stroke. 33:186–194. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Fang Z, Ning J, Xiong C and Shulin Y:
Effects of electroacupuncture at head points on the function of
cerebral motor areas in stroke patients: a PET study. Evid Based
Complement Alternat Med. 2012:9024132012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Foley LM, Hitchens TK, Barbe B, et al:
Quantitative temporal profiles of penumbra and infarction during
permanent middle cerebral artery occlusion in rats. Transl Stroke
Res. 1:220–229. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li P, Nijhawan D, Budihardjo I, et al:
Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9
complex initiates an apoptotic protease cascade. Cell. 91:479–489.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cui M, Wang L, Liang X, et al: Blocking
TRAIL-DR5 signaling with soluble DR5 reduces delayed neuronal
damage after transient global cerebral ischemia. Neurobiol Dis.
39:138–147. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Velier JJ, Ellison JA, Kikly KK, Spera PA,
Barone FC and Feuerstein GZ: Caspase-8 and caspase-3 are expressed
by different populations of cortical neurons undergoing delayed
cell death after focal stroke in the rat. J Neurosci. 19:5932–5941.
1999.PubMed/NCBI
|
|
27
|
Krupinski J, Lopez E, Marti E and Ferrer
I: Expression of caspases and their substrates in the rat model of
focal cerebral ischemia. Neurobiol Dis. 7:332–342. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li H, Zhu H, Xu CJ and Yuan J: Cleavage of
BID by caspase 8 mediates the mitochondrial damage in the Fas
pathway of apoptosis. Cell. 94:491–501. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Luo X, Budihardjo I, Zou H, Slaughter C
and Wang X: Bid, a Bcl2 interacting protein, mediates cytochrome c
release from mitochondria in response to activation of cell surface
death receptors. Cell. 94:481–490. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wilkinson JC, Wilkinson AS, Scott FL,
Csomos RA, Salvesen GS and Duckett CS: Neutralization of
Smac/Diablo by inhibitors of apoptosis (IAPs). A
caspase-independent mechanism for apoptotic inhibition. J Biol
Chem. 279:51082–51090. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Eckelman BP, Salvesen GS and Scott FL:
Human inhibitor of apoptosis proteins: why XIAP is the black sheep
of the family. EMBO Rep. 7:988–994. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Rothe M, Pan MG, Henzel WJ, Ayres TM and
Goeddel DV: The TNFR2-TRAF signaling complex contains two novel
proteins related to baculoviral inhibitor of apoptosis proteins.
Cell. 83:1243–1252. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Guicciardi ME, Mott JL, Bronk SF, Kurita
S, Fingas CD and Gores GJ: Cellular inhibitor of apoptosis 1
(cIAP-1) degradation by caspase 8 during TNF-related
apoptosis-inducing ligand (TRAIL)-induced apoptosis. Exp Cell Res.
317:107–116. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sugawara T, Fujimura M, Morita-Fujimura Y,
Kawase M and Chan PH: Mitochondrial release of cytochrome c
corresponds to the selective vulnerability of hippocampal CA1
neurons in rats after transient global cerebral ischemia. J
Neurosci. 19:RC391999.
|
|
35
|
Earnshaw WC, Martins LM and Kaufmann SH:
Mammalian caspases: structure, activation, substrates, and
functions during apoptosis. Annu Rev Biochem. 68:383–424. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yuan J and Yankner BA: Apoptosis in the
nervous system. Nature. 407:802–809. 2000. View Article : Google Scholar
|
|
37
|
Oltvai ZN, Milliman CL and Korsmeyer SJ:
Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that
accelerates programmed cell death. Cell. 74:609–619. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sedlak TW, Oltvai ZN, Yang E, et al:
Multiple Bcl-2 family members demonstrate selective dimerizations
with Bax. Proc Natl Acad Sci USA. 92:7834–7838. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kuida K, Haydar TF, Kuan CY, et al:
Reduced apoptosis and cytochrome c-mediated caspase activation in
mice lacking caspase 9. Cell. 94:325–337. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Noshita N, Sugawara T, Fujimura M,
Morita-Fujimura Y and Chan PH: Manganese superoxide dismutase
affects cytochrome c release and caspase-9 activation after
transient focal cerebral ischemia in mice. J Cereb Blood Flow
Metab. 21:557–567. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Shehadah A, Chen J, Zacharek A, et al:
Niaspan treatment induces neuroprotection after stroke. Neurobiol
Dis. 40:277–283. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hsu AL, Ching TT, Wang DS, Song X,
Rangnekar VM and Chen CS: The cyclooxygenase-2 inhibitor celecoxib
induces apoptosis by blocking Akt activation in human prostate
cancer cells independently of Bcl-2. J Biol Chem. 275:11397–11403.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Rau TF, Kothiwal A, Zhang L, et al: Low
dose methamphetamine mediates neuroprotection through a PI3K-AKT
pathway. Neuropharmacology. 61:677–686. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kim YR, Kim HN, Jang JY, et al:
Electroacupuncture confers beneficial effects through ionotropic
glutamate receptors involving phosphatidylinositol-3 kinase/Akt
signaling pathway in focal cerebral ischemia in rats. Eur J Integr
Med. 4:e413–420. 2012. View Article : Google Scholar
|