Introduction
Acute leukemia is the most common hematological
malignancy that is characterized by an uncontrolled expansion of
clonal leukemia cells in the bone marrow and/or peripheral blood
often accompanied with anemia, bleeding and frequent infectious
complications (1–4). Acute leukemia is generally
classified as either acute myelogenous leukemia (AML) or acute
lymphoblastic leukemia (ALL) and the primary treatments are
remission induction therapy and post-remission chemotherapy
(1,2). In recent years, with the
introduction of high-dose chemotherapy followed by allogeneic
hematopoietic stem cell transplantation (allo-HSCT) and the
discovery of novel therapeutic agents, considerable progress has
been made in the outcomes of patients with acute leukemia (3–5).
However, acute leukemia currently remains an incurable malignancy
due to the complex molecular mechanisms behind its tumorigenesis
(6,7). Thus, the identification of novel
molecular targets is mandatory for the early diagnosis and
long-term management of acute leukemia.
The human cervical cancer oncogene (HCCR) was first
identified from human cervical tissue through the differential
display RT-PCR method by Ko et al (8). The HCCR gene maps to the long arm of
the chromosome 12 and is classified into two types, HCCR1 and HCCR2
according to their molecular characteristics. A comparison of both
sequences has revealed that HCCR2 lacks the exon 1 of HCCR1
(8,9). Previous studies have demonstrated
that HCCR2 is not only overexpressed in human cervical cancer
tissues, but is expressed at high levels in a variety of human
malignancies, including breast, kidney, stomach, colon, liver and
ovarian cancer (8–13). The functional role of HCCR2 in
tumorigenesis may involve the negative regulation of the p53 tumor
suppressor gene (8,13,14). Animal experiments have
demonstrated that HCCR2-transgenic nude mice form spontaneous
breast tumors, which further confirms the crucial role of HCCR2 in
tumorigenesis (10). In our
previous study, we observed that HCCR2 mRNA was overexpressed in
patients with acute leukemia and leukemia cell lines, such as K562
and HL-60 cells, but not in normal leukocytes (15). Therefore, HCCR2 shows great
promise as an important molecular target for the diagnosis and
treatment of acute leukemia. Although accumulating data have shown
that HCCR2 expression in many solid tumors correlates with clinical
outcome has been confirmed as a good biomarker for monitoring
disease progression, its specific mechanisms of action in acute
leukemia remain elusive (10–13).
In the present study, we examined whether silencing
HCCR2 expression using small interfering RNA (siRNA) exerts
significant anti-proliferative effects in K562 cells. Furthermore,
we also wished to investigate the potential molecular mechanisms
behind these effects, which may help identify HCCR2 as a novel
therapeutic target for acute leukemia.
Materials and methods
Cell line and cell culture
The K562 cell line, established from human chronic
myelogenous leukemia (CML) cells in blastic crisis, was purchased
from the Cell Culture Center in Chinese Academy of Medical Sciences
(Shanghai, China). The cells were cultured in RPMI-1640 medium
(Invitrogen Life Technologies, Carlsbad, CA, USA) supplemented with
10% fetal bovine serum (HyClone, Logan, UT, USA), 100 μg/ml
penicillin, 10 μg/ml streptomycin and 2 mmol/l L-glutamine. The
cells were maintained in log phase growth at 37ºC in a humidified
atmosphere containing 5% CO2.
Silencing HCCR2 expression by siRNAs
We used the K562 cells to perform the HCCR2
silencing experiment, as a high expression level of HCCR2 in K562
cells was demonstrated in our previous study (15). The siRNAs used for the silencing
of HCCR2 were designed and synthesized from Invitrogen Life
Technologies according to previous literature (13). The oligonucleotide sequences used
to generate the three synthesized HCCR2-targeting siRNA are listed
in Table I. siRNA-H1, targeting
HCCR2 mRNA coding sequence 475–493 bp; siRNA-H2, targeting HCCR2
mRNA coding sequence 611–629 bp; and siRNA-H3, targeting HCCR2 mRNA
coding sequence 854–872 bp. A scrambled sequence
ACTACCGTTGTATAGGTGT was used as the negative control siRNA that is
absent in human, mouse, and rat genomes. Each oligonucleotide pair
was annealed by incubation at 95ºC for 5 min and cooled slowly, and
was ligated separately into plasmid vector pcDNA3 (Invitrogen Life
Technologies), which had been digested with BamHI and
EcoRI. These HCCR2-siRNA vectors were transfected into the
K562 cells using Lipofectamine 2000 (Invitrogen Life Technologies)
according to the manufacturer’s instructions. The ability of the
three transfected HCCR2-siRNAs to silence HCCR2 expression was
investigated by quantitative RT-PCR (qRT-PCR) and western blot
analysis.
 | Table ISequences used to generate the three
synthesized HCCR2-targeting siRNA. |
Table I
Sequences used to generate the three
synthesized HCCR2-targeting siRNA.
| No. | Primer sequences |
|---|
| siRNA-H1 |
5′-GATCCGACAGATCTGTGCACCAAGATCAAGACGTCTTGGTGCACAGATCTGTTTTTTTA-3′
3′-GCTGTCTAGACACGTGGTTCTAGTTCTGCAGAACCACGTGTCTAGACAAAAAAATTCGA-5′ |
| siRNA-H2 |
5′-GATCCGTAAGATGTGAGAAGCATGGTCAAGACGCCATGCTTCTCACATCTTATTTTTTA-3′
3′-GCATTCTACACTCTTCGTACCAGTTCTGCGGTACGAAGAGTGTAGAATAAAAAATTCGA-5′ |
| siRNA-H3 |
5′-GATCCGTTGTGCAGCAAGAGAGACATCAAGACGTGTCTCTCTTGCTGCACAATTTTTTA-3′
3′-GCAACACGTCGTTCTCTCTGTAGTTCTGCACAGAGAGAACGACGTGTTAAAAAATTCGA-5′ |
Cell proliferation assay
Cell proliferation was determined in cells in
96-well plates using the
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium
(MTS)/phenazine ethosulfate (PES) assay (Promega Corp., Madison,
WI, USA) according to manufacturer’s instructions. In brief, the
blank control cells, and the negative control- and
HCCR2-siRNA-transfected K562 cells were seeded in 96-well plates at
densities of 5×103 cells/well. Cultures were set up in
triplicate and maintained at 37ºC in a humidified atmosphere with
5% CO2. At 24, 48, 72, 96 and 120 h after transfection,
10 μl MTS-PES (10 mg/ml) (Promega Corp.) were added into each well
for an additional 6 h of incubation. A microplate reader was then
used to measure the absorbance value at 490 nm for each well, which
represented K562 cell proliferation.
Morphological observation of K562
cells
After transfection with HCCR2-siRNA, the K562 cells
were smeared on microscope glass plates and stained with
Wright-Giemsa. The morphological changes in the K562 cells was
observed under a light microscope.
Cell cycle analysis and assessment of
apoptosis
Cell cycle distribution was determined using DNA
staining with propidium iodide (PI) (BioLegend, San Diego, CA, USA)
followed by flow cytometry. Apoptosis assay was performed according
to the directions contained in the manual provided with the Annexin
V/PI apoptosis assay kit (BioLegend). In brief, the cells were
harvested and washed twice with cold phosphate-buffered saline
(PBS), and then the cells were resuspended in 1X binding buffer at
a concentration of 1×106 cells/ml. A total of 100 μl of
the solution was then transferred to a 5-ml culture tube with 5 μl
of Annexin V-FITC and 10 μl of PI (20 mg/ml). The cells were gently
vortexed and incubated for 15 min at room temperature in the dark.
After the addition of 400 ml of 1X binding buffer, the cells were
analyzed by flow cytometry. Annexin V− and
PI− cells were considered as viable cells. Annexin
V+ and PI− cells were considered as
early-stage apoptotic cells. Annexin V+ and
PI+ cells were considered as late-stage apoptotic
cells.
qRT-PCR
Total RNA was isolated from the K562 cells using the
TRIzol reagent (Invitrogen Life Technologies) according to the
manufacturer’s instructions, and then cDNA was synthesized from 2
μg total RNA using a first-strand cDNA synthesis kit (Invitrogen
Life Technologies). The PCR amplification protocol was as follows:
denaturation at 94ºC for 10 min, 40 cycles of 94ºC for 15 sec, 58ºC
30 sec, and 72ºC for 40 sec. The mRNA expression levels of HCCR2,
Bcl-2, Bax, p21, p27 and β-actin were detected using ABI 7900
(Applied Biosystems, Foster City, CA, USA) and SYBR-Green
chemistry. The relative quantification of the target genes was
calculated using the 2−ΔΔCt formula by the comparative
cycle threshold (Ct) value method (16). The PCR primers were designed based
on the corresponding gene structure, and the sequences are listed
in Table II. qRT-PCR assays were
performed in triplicate.
| Table IIPrimers used for quantitative
RT-PCR. |
Table II
Primers used for quantitative
RT-PCR.
| Gene | | Primer sequences |
|---|
| HCCR2 | Sense |
5′-GGAGGCAGAGAGAGGAGCAG-3′ |
| Antisense |
5′-AGCAAGAGGGTTTGTTTCAGTTCT-3′ |
| Bcl-2 | Sense | 5′-ATCGCCCTG
TGGATGACTGAG-3′ |
| Antisense |
5′-CAGCCAGGAGAAATCAAACAGAGG-3′ |
| Bax | Sense |
5′-GGACGAACTGGACAGTAACATGG-3′ |
| Antisense |
5′-GCAAAGTAGAAAAGGGCGACAAC-3′ |
| p21 | Sense |
5′-GCAGACCAGCATGACAGATTT-3′ |
| Antisense |
5′-GGATTAGGGCTTCCTCTTGGA-3′ |
| p27 | Sense |
5′-CCTCTTCGGCCCGGTGGAC-3′ |
| Antisense |
5′-TTTGGGGAACCGTCTGAAAC-3′ |
| p53 | Sense |
5′-TGCTCAGATAGCGATGGTC-3′ |
| Antisense |
5′-TAGGGCACCACCACACTAT-3′ |
| β-actin | Sense |
5′-TCTGGCACCACACCTTCTACAATG-3′ |
| Antisense |
5′-AGCACAGCCTGGATAGCAACG-3′ |
Western blot analysis
The K562 cells were harvested and subsequently
protein was separated by 15% SDS- polyacrylamide gel
electrophoresis (SDS-PAGE) before being transferred onto a
polyvinylidene difluoride (PVDF) membrane. The blots were blocked
with 5% non-fat dry milk in Tris-buffered saline containing 0.1%
Tween-20 (TBST) at 37ºC for 2 h, followed by incubation with
specific primary antibodies against HCCR2, Bcl-2, Bax, p21, p27 or
β-actin (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA) at 4ºC
overnight. The blots were washed with TBST and incubated with a
horseradish peroxidase-conjugated secondary antibody at 37ºC for 2
h. After several washes with TBST, the immunoreactive bands were
visualized on X-ray film and scanned. All experiments were
performed in triplicate. The quantitative data from bands were
expressed as the target protein/β-actin ratio using BandScan
analysis software.
Statistical analysis
All statistical data are presented as the means ±
standard deviation (SD). The effects of treatment among the
different groups were compared using a one-way analysis of
variance. All analyses were performed using GraphPad Prism software
version 5.0 (GraphPad Software Inc., San Diego, CA, USA) and a
value of P<0.05 was considered to indicate a statistically
significant difference.
Results
Inhibition of HCCR2 expression by siRNAs
in K562 cells
We analyzed the ability of the three HCCR2-siRNAs to
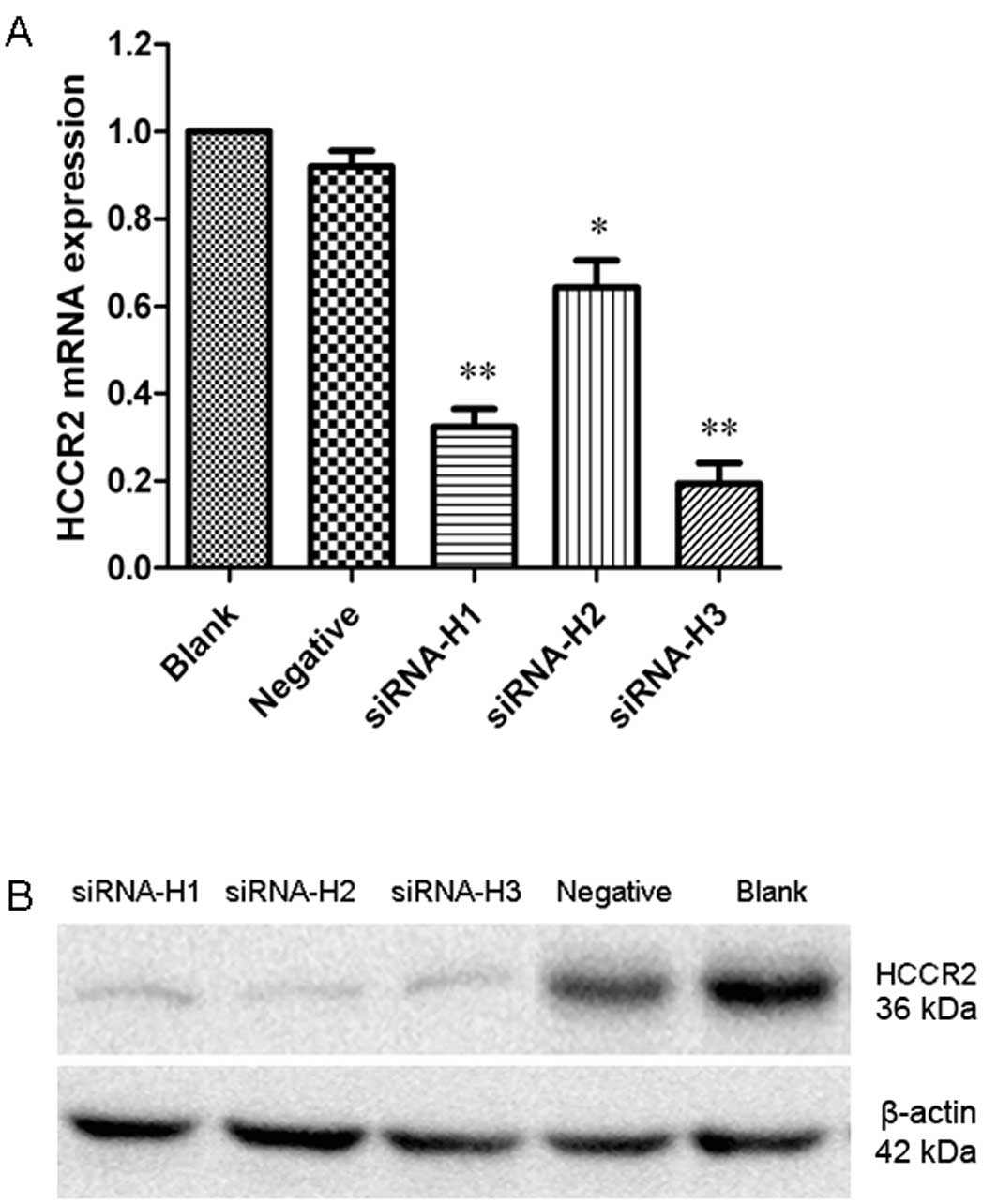
suppress HCCR2 mRNA expression by qRT-PCR. The results revealed
that transfection with the three HCCR2-siRNAs resulted in varying
degrees of reduced HCCR2 mRNA expression in the K562 cells. The
quantification analysis revealed that siRNA-H1 (P<0.01),
siRNA-H2 (P<0.05) and siRNA-H3 (P<0.01) significantly
inhibited HCCR2 expression at the mRNA level compared with the
negative control cells (Fig. 1A).
The western blot analysis results also indicated that HCCR2 protein
expression was significantly suppressed following transfection with
the three HCCR2-siRNAs (Fig. 1B).
The HCCR2 mRNA and protein levels were not altered in the blank
group cells or in the negative control siRNA-transfected cells. As
siRNA-H3 showed the highest efficiency in silencing HCCR2
expression, we thus selected siRNA-H3 for the subsequent
experiments.
HCCR2 knockdown suppresses the
proliferation of K562 cells
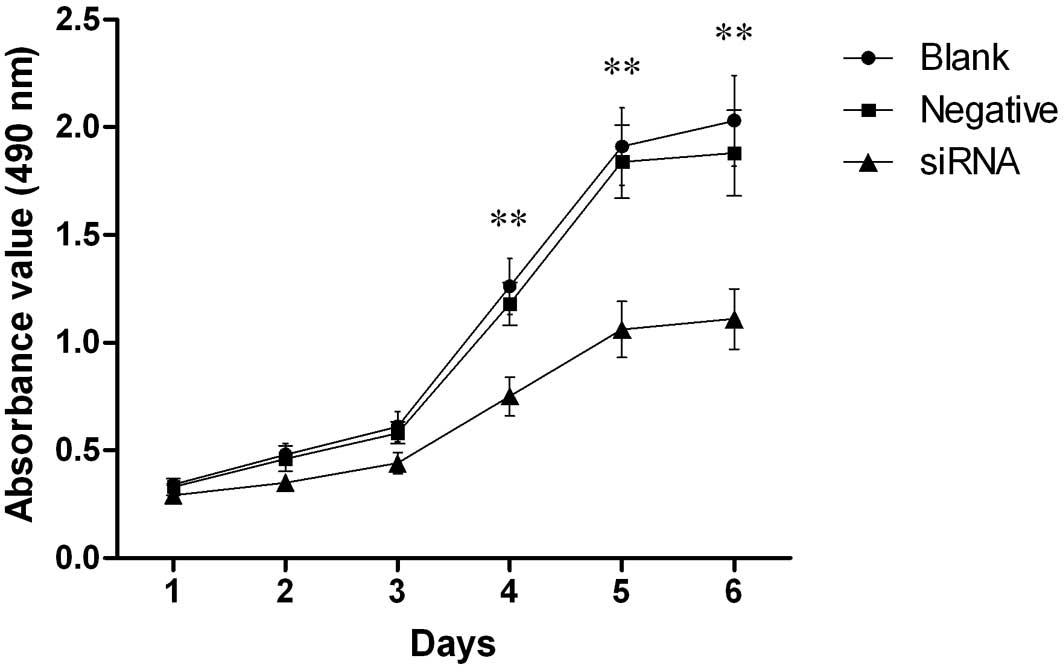
Our results revealed that silencing the HCCR2 gene
affected the proliferation of K562 cells. As shown in Fig. 2, cell proliferation was markedly
inhibited in the HCCR2-siRNA-transfected cells from day 4 to 6
after transfection when compared with the blank control and the
negative control cells (P<0.01). However, there was no
statistically significant difference in cell proliferation during
the first three days among the three groups of cells.
Morphological changes in K562 cells
following transfection with HCCR2-siRNA
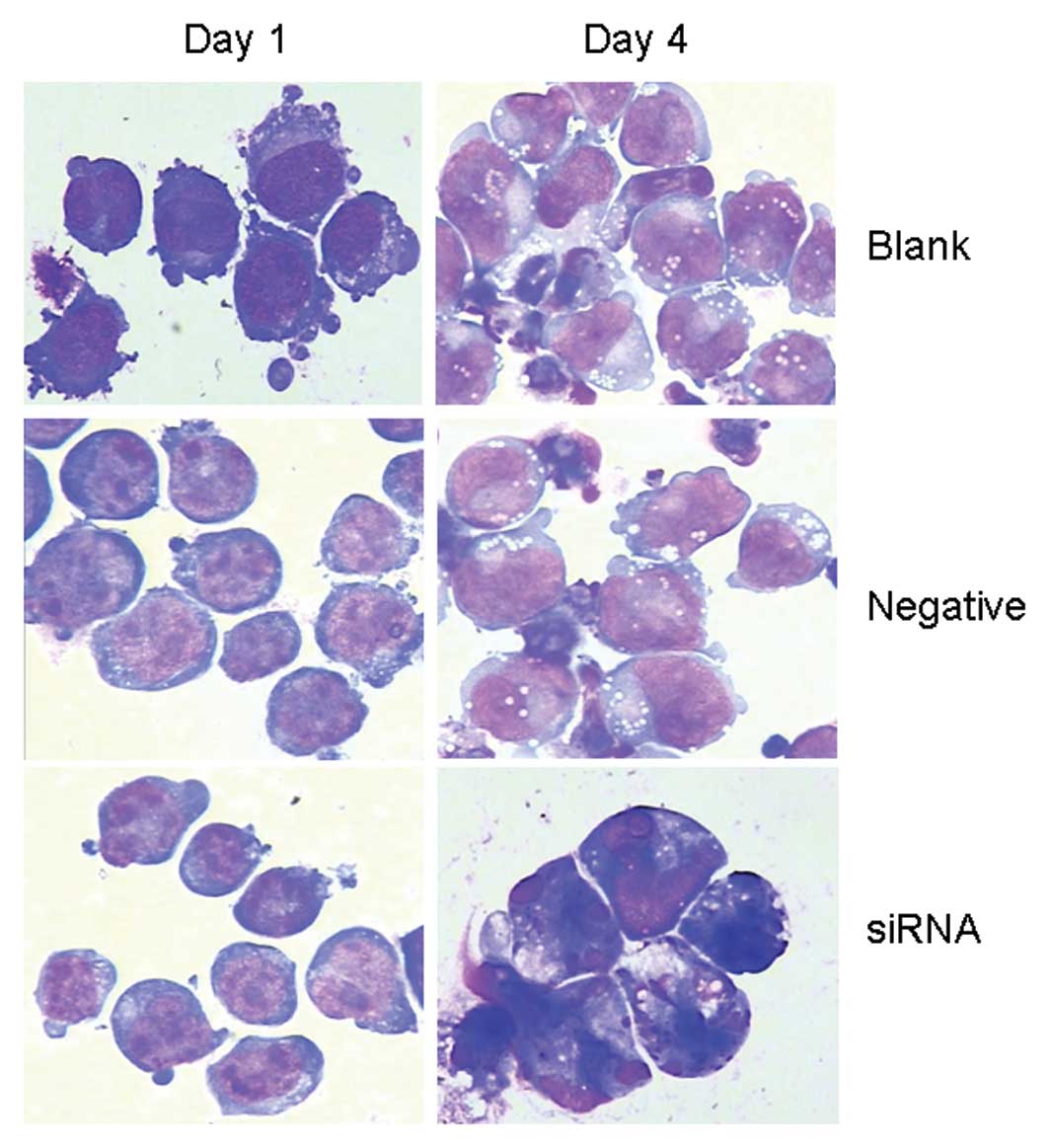
After transfection for 96 h, a morphological
examination of the HCCR2-siRNA-transfected K562 cells revealed the
typical appearance of apoptotic cells, which included the
appearance of condensed nuclear chromatin, nuclei shrinkage,
nuclear fragmentation and the formation of apoptotic bodies.
However, these morphological changes were not observed among the
blank control and negative control cells (Fig. 3).
HCCR2 knockdown inhibits cell cycle
progression and induces apoptosis
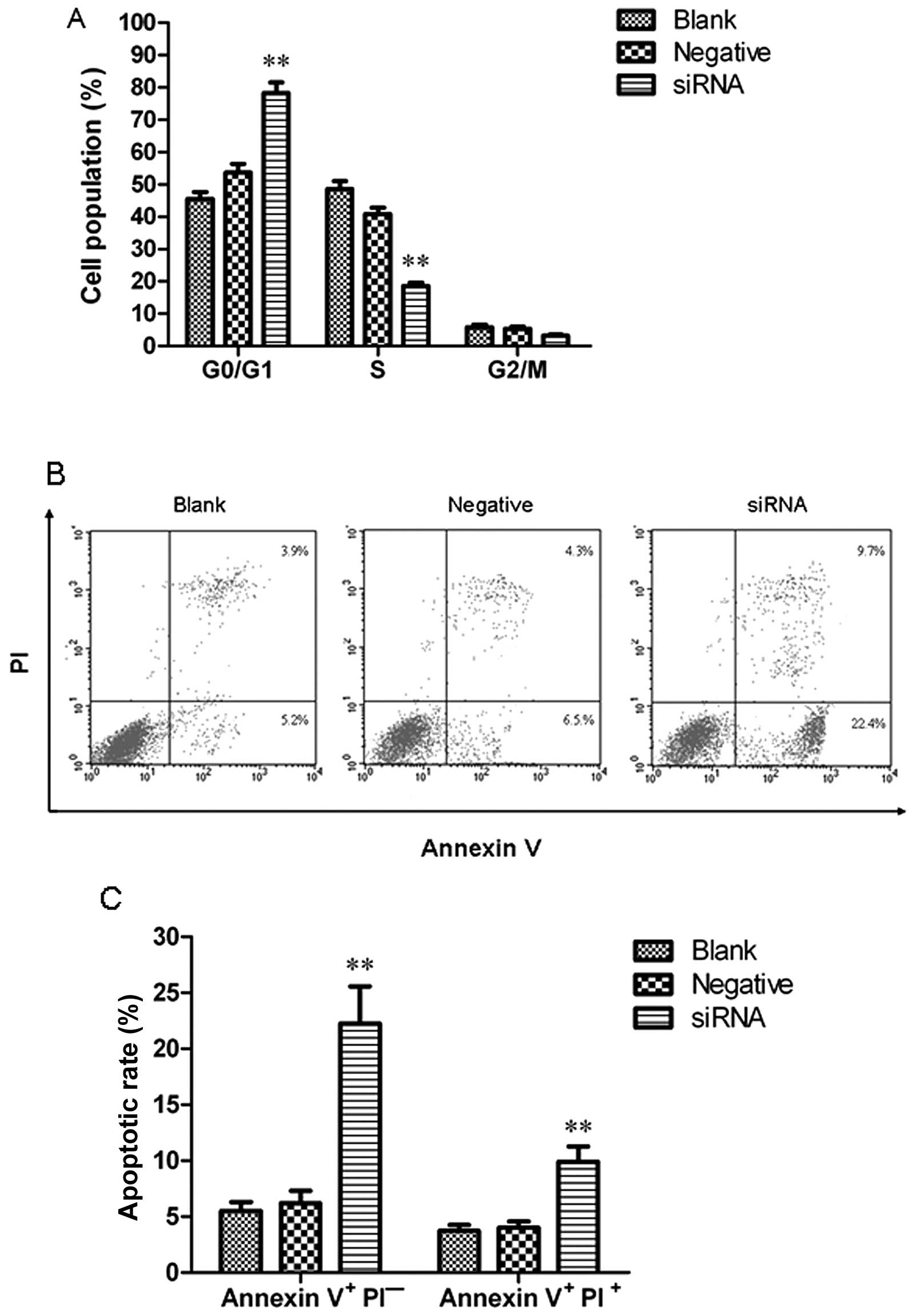
To determine whether HCCR2 knockdown results in cell
cycle changes, the cellular DNA content was measured by flow
cytometry. The results revealed that the percentage of K562 cells
in the G0/G1 phase was significantly increased when compared with
the blank control (P<0.01). By contrast, the percentage of K562
cells in the S phase was significantly decreased as compared with
the blank control (P<0.01). However, the number of K562 cells in
the G2/M phase was relatively unaffected. These data demonstrated
that HCCR2 knockdown by siRNA induced a significant arrest of cell
cycle progression in the G1 phase, resulting in a decreased number
of cells in the S phase with a corresponding accumulation of cells
in the G0–G1 phase (Fig. 4A).
Fig. 4B presents a representative
example of apoptotic cells in the blank control, and the negative
control- and HCCR2-siRNA-transfected group. The results revealed
that the ratio of early-stage and late-stage apoptotic cells in the
HCCR2-siRNA-transfected cells was significantly higher than that in
the blank control and negative control cells (P<0.01) (Fig. 4C).
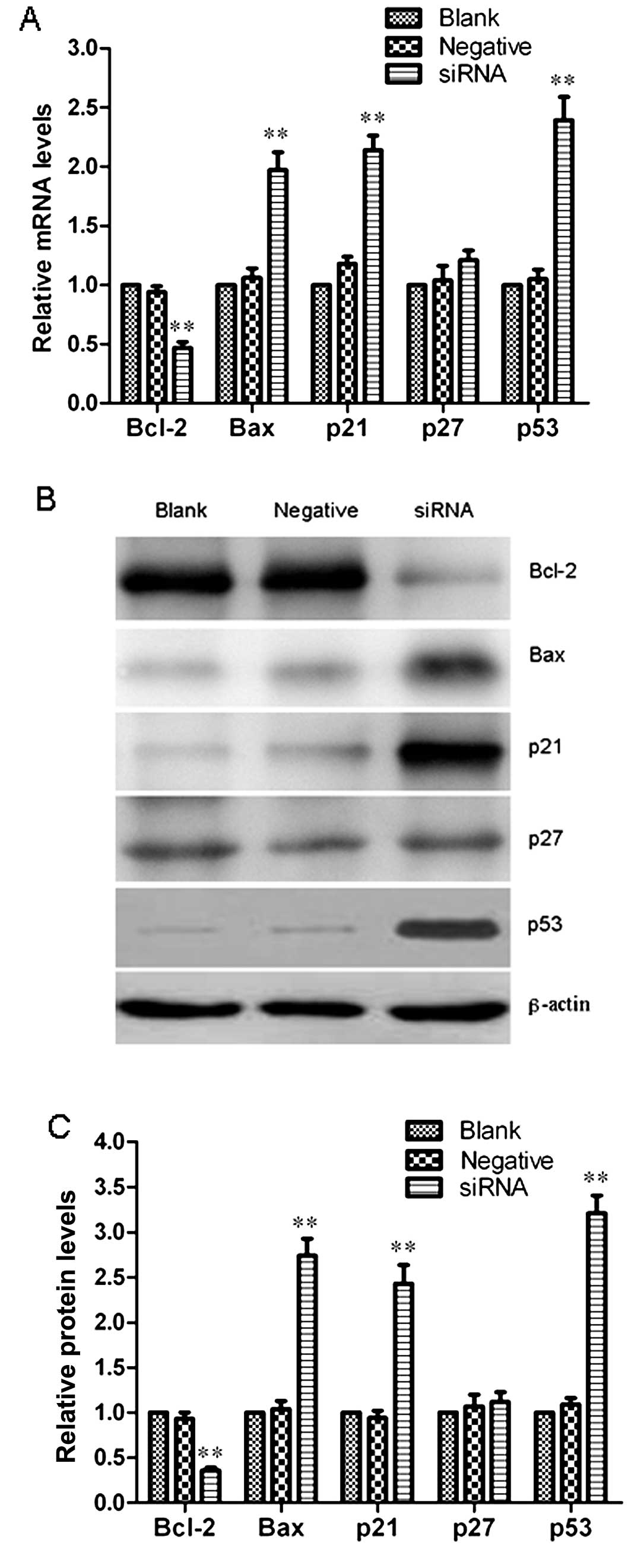
Effect of HCCR2 knockdown on the
expression of apoptosis- and cell cycle-related genes
To further investigate the molecular mechanisms
behind the suppression of cell proliferation and G0/G1 cell cycle
arrest induced by transfection with HCCR2-siRNA, we analyzed the
mRNA and protein expression levels of Bcl-2, Bax, p21, p27 and p53
in the K562 cells at 96 h after transfection by qRT-PCR and western
blot analysis. Fig. 5A represents
the relative mRNA quantification of these genes in the blank
control group, and negative control- and HCCR2-siRNA-transfected
K562 cells. The results indicated that Bcl-2 mRNA expression
significantly decreased (P<0.01), while Bax mRNA expression
markedly increased (P<0.01) when compared with the blank
control. Furthermore, the Bax/Bcl-2 ratio was also significantly
increased in the HCCR2-siRNA-transfected cells. In addition, we
also observed that the mRNA expression levels of p21 and p53 were
markedly elevated in the HCCR2-siRNA-transfected K562 cells as
compared with the blank control and negative control (P<0.01);
however, p27 mRNA expression was not markedly altered in the three
groups of cells. Representative immunoblots are shown in Fig. 5B. The western blot analysis
results confirmed a marked decrease in Bcl-2 protein expression
(P<0.01), and a significant increase in Bax, p21 and p53 protein
expression following transfection with HCCR2-siRNA (P<0.01). No
significant difference was observed in the p27 protein expression
level among the three groups (Fig.
5C).
Discussion
siRNA is a powerful tool for gene knockdown, in
which double-stranded RNA (dsRNA) is processed into siRNA, inducing
the activation of cellular pathways, leading to selective
sequence-specific silencing of target genes with homology to dsRNA
(17). A large number of studies
have indicated that siRNA is an ideal method in molecular biology
research and a very attractive therapeutic alternative to chemical
drugs (17–19). In the present study, in order to
investigate the function role of the HCCR2 gene in the
tumorigenesis of acute leukemia, we silenced the expression of
HCCR2 in K562 cells using siRNA. It is well known that K562 cells
are derived from human CML cells in blastic crisis and are often
used as an in vitro model of leukemia cells. Our results
demonstrated that the mRNA and protein expression levels of HCCR2
were markedly decreased following transfection with HCCR2-targeting
siRNA in K562 cells.
A previous report by Yoon et al (11) identified that HCCR2 was
overexpressed in patients with hepatocellular carcinoma and thus
may be recognized as a novel biomarker for hepatocarcinogenesis.
The same study indicated that the HCCR2 assay has an advantage over
the alpha-fetoprotein (AFP) assay in the early diagnosis of
hepatocellular carcinoma. In our previous study, we found that the
HCCR2 gene was abnormally overexpressed in newly diagnosed AML and
ALL patients when compared with the normal control group.
Furthermore, the patients who achieved complete remission after
chemotherapy presented a significant decrease in HCCR2 expression;
however, the patients with no remission showed not decrease in
expression. In addition, the moment a patient relapsed, the level
of HCCR2 expression once again increased (15). These results indicate that HCCR2
may be used as biomarker for monitoring minimal residual disease in
patients with acute leukemia.
In this study, we examined whether silencing HCCR2
expression by siRNA affects cell proliferation, cell cycle
progression and apoptosis in K562 cells. We observed that the cell
growth of HCCR2-knockdown clones was significantly attenuated
compared with the blank or negative control clones, while the cell
numbers were comparable among all the groups at 4 days after
transfection. Additionally, we measured the apoptosis of K562 cells
using Annexin V/PI staining and found that the rate of early-stage
and late-stage apoptotic cells was markedly increased in the
HCCR2-siRNA-transfected cells. In the cell cycle analysis, a
decreased number of cells in the S phase and an increased number of
cells in the G0/G1 phase were observed in the
HCCR2-siRNA-transfected cells; however, the number of cells in the
G2/M phase was relatively unaffected. Taken together, our results
demonstrated that silencing HCCR2 expression effectively suppressed
the proliferation and survival of K562 cells, inducing marked
antitumor effects, which were associated with the induction of
apoptosis and the inhibition of cell cycle G1/S phase
transition.
p53 is a putative tumor suppressor gene, which plays
a crucial role in controlling cell proliferation, and suppressing
the growth and transformation of cells primarily through the
induction of cell cycle arrest or apoptosis (20). The function of the p53 protein is
to maintain the integrity of the genome and to repair damaged DNA.
Before the initiation of damaged DNA repair, the p53 protein first
induces the cells into G1 phase arrest. If DNA repair is not
completed, p53 may induce the damaged cells into programmed cell
death (21). In addition, the p53
protein is well known as the master regulator of the p21 gene
(22,23). In agreement with previous reports
demonstrating the function of HCCR2 as a negative regulator of p53
in tumorigenesis (8,13,14), we found that HCCR2 was
overexpressed and accompanied with an almost null expression of p53
in K562 cells. Furthermore, p53 expression was markedly upregulated
following transfection with HCCR2-siRNA, which was consistent with
the increased number of apoptotic K562 cells. However, there have
been conflicting reports, in which it was found that siRNA
targeting HCCR2 did not alter the p53 protein expression level in
HepG2 cells (24). This
difference in the role of p53 may be explained by the fact that
cells derived from various tissues may have different genetic
backgrounds.
Cell cycle progression is regulated by
cyclin-dependent kinases (CDKs), which are activated by cyclins
binding or inhibited by CDK inhibitors (CKIs) (25). The p21 proteins belong to the
first family of CKIs, which inhibit cell cycle progression mainly
through the suppression of CDK2 activity and block the transition
from the G1 phase into the S phase after DNA damage (26–28). Although this activity is shared by
other CKIs, such as p27 and p57, a number of clinical and
experimental studies have suggested that p21 plays a crucial role
in tumorigenesis (29–31). In this study, our data
demonstrated that silencing HCCR2 expression upregulated the
expression of p21, but did not affect p27 expression in K562
cells.
Bcl-2, as an anti-apoptotic protein, inhibits
apoptotic cell death and its overexpression is considered to
promote survival in tumor cells (32). On the contrary, Bax is a
pro-apoptotic protein, whose overexpression can inhibit the
malignant progression of a tumor (33). The upregulation of the Bax/Bcl-2
ratio directs the cells more towards apoptosis than survival
(34). In this study, we found
that the knockdown of HCCR2 decreased Bcl-2 expression and
increased Bax expression. Furthermore, the Bax/Bcl-2 ratio was
significantly increased in the K562 cells following the knockdown
of HCCR2 using siRNA. These results demonstrated that silencing
HCCR2 expression induced a greater number of K562 cells into
irreversible apoptosis by regulating the expression of Bcl-2 and
Bax.
In conclusion, the results from the present study
suggest that silencing HCCR2 gene expression by siRNA suppresses
cell proliferation, promotes cell cycle G1 phase arrest and induces
apoptosis in K562 cells. The data presented in this study also
provide a mechanistic explanation for the anti-proliferative and
anti-apoptotic effects induced by silencing HCCR2 expression in
K562 cells. Although the details of the functional mechanisms of
HCCR2 in vivo require further investigation, HCCR2 may serve
as an attractive molecular target for the treatment of
leukemia.
References
|
1
|
Ferrara F and Schiffer CA: Acute myeloid
leukaemia in adults. Lancet. 381:484–495. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Inaba H, Greaves M and Mullighan CG: Acute
lymphoblastic leukaemia. Lancet. 381:1943–1955. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hamilton BK and Copelan EA: Concise
review: the role of hematopoietic stem cell transplantation in the
treatment of acute myeloid leukemia. Stem Cells. 30:1581–1586.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Khaled SK, Thomas SH and Forman SJ:
Allogeneic hematopoietic cell transplantation for acute
lymphoblastic leukemia in adults. Curr Opin Oncol. 24:182–190.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ungewickell A and Medeiros BC: Novel
agents in acute myeloid leukemia. Int J Hematol. 96:178–185. 2012.
View Article : Google Scholar
|
|
6
|
Roboz GJ: Current treatment of acute
myeloid leukemia. Curr Opin Oncol. 24:711–719. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kimby E, Nygren P and Glimelius B: A
systematic overview of chemotherapy effects in acute myeloid
leukaemia. Acta Oncol. 40:231–252. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ko J, Lee YH, Hwang SY, et al:
Identification and differential expression of novel human cervical
cancer oncogene HCCR-2 in human cancers and its involvement in p53
stabilization. Oncogene. 22:4679–4689. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chung YJ and Kim JW: Novel oncogene HCCR:
its diagnostic and therapeutic implications for cancer. Histol
Histopathol. 20:999–1003. 2005.PubMed/NCBI
|
|
10
|
Ko J, Shin SM, Oh YM, Lee YS, Ryoo ZY, Lee
YH, Na DS and Kim JW: Transgenic mouse model for breast cancer:
induction of breast cancer in novel oncogene HCCR-2 transgenic
mice. Oncogene. 23:1950–1953. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yoon SK, Lim NK, Ha SA, et al: The human
cervical cancer oncogene protein is a biomarker for human
hepatocellular carcinoma. Cancer Res. 64:5434–5441. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jung SS, Park HS, Lee IJ, et al: The HCCR
oncoprotein as a biomarker for human breast cancer. Clin Cancer
Res. 11:7700–7708. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ha SA, Lee YS, Shin SM, et al: Oncoprotein
HCCR-1 expression in breast cancer is well correlated with known
breast cancer prognostic factors including the HER2 overexpression,
p53 mutation, and ER/PR status. BMC Cancer. 9:512009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ha SA, Shin SM, Lee YJ, et al: HCCRBP-1
directly interacting with HCCR-1 induces tumorigenesis through P53
stabilization. Int J Cancer. 122:501–508. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Qiao SK, Guo XN, Xu SR and Wang Y: The
expression and clinical prognostic value of HCCR genes in patients
with acute leukemia. Chin Journal Hematol. 7:481–483. 2010.(In
Chinese).
|
|
16
|
Schmittgen TD and Livak KJ: Analyzing
real-time PCR data by the comparative C (T) method. Nat Protoc.
3:1101–1108. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ichim TE, Li M, Qian H, Popov IA, Rycerz
K, Zheng X, White D, Zhong R and Min WP: RNA interference: a potent
tool for gene-specific therapeutics. Am J Transplant. 4:1227–1236.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Leung RK and Whittaker PA: RNA
interference: from gene silencing to gene- specific therapeutics.
Pharmacol Ther. 107:222–239. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Takeshita F and Ochiya T: Therapeutic
potential of RNA interference against cancer. Cancer Sci.
97:689–696. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Olivier M, Petitjean A, Marcel V, Pétré A,
Mounawar M, Plymoth A, de Fromentel CC and Hainaut P: Recent
advances in p53 research: an interdisciplinary perspective. Cancer
Gene Ther. 16:1–12. 2009. View Article : Google Scholar
|
|
21
|
Wang B, Xiao Z and Ren EC: Redefining the
p53 response element. Proc Natl Acad Sci USA. 106:14373–14378.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Peller S and Rotter V: TP53 in
hematological cancer: low incidence of mutations with significant
clinical relevance. Hum Mutat. 21:277–284. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Janssen A, Schiffmann S, Birod K, Maier
TJ, Wobst I, Geisslinger G and Grösch S: p53 is important for the
anti-proliferative effect of ibuprofen in colon carcinoman cells.
Biochem Biophys Res Commun. 365:698–703. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Guo J, Yang L, Zhang Y, Wang J, Wan S, Xia
S, Yang S, Wang R and Fang D: Silencing of the HCCR2 gene induces
apoptosis and suppresses the aggressive phenotype of hepatocellular
carcinoma cells in culture. J Gastrointest Surg. 15:1807–1813.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Vermeulen K, Van Bockstaele DR and
Berneman ZN: The cell cycle: a review of regulation, deregulation
and therapeutic targets in cancer. Cell Prolif. 36:131–149. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Warfel NA and El-Deiry WS: p21WAF1 and
tumourigenesis: 20 years after. Curr Opin Oncol. 25:52–58.
2013.PubMed/NCBI
|
|
27
|
Stivala LA, Cazzalini O and Prosperi E:
The cyclin-dependent kinase inhibitor p21CDKN1A as a target of
anti-cancer drugs. Curr Cancer Drug Targets. 12:85–96. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kizildag S, Ates H and Kizildag S:
Treatment of K562 cells with 1,25-dihydroxyvitamin D3 induces
distinct alterations in the expression of apoptosis-related genes
BCL2, BAX, BCLXL, and p21. Ann Hematol. 89:1–7. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cmielová J and Rezáčová M: p21Cip1/Waf1
protein and its function based on a subcellular localization
[corrected]. J Cell Biochem. 112:3502–3506. 2011.PubMed/NCBI
|
|
30
|
Wang X, Gao P, Long M, Lin F, Wei JX, Ren
JH, Yan L, He T, Han Y and Zhang HZ: Essential role of cell cycle
regulatory genes p21 and p27 expression in inhibition of breast
cancer cells by arsenic trioxide. Med Oncol. 28:1225–1254. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Coqueret O: New roles for p21 and p27
cell-cycle inhibitors: a function for each cell compartment? Trends
Cell Biol. 13:65–70. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Tomek M, Akiyama T and Dass CR: Role of
Bcl-2 in tumour cell survival and implications for pharmacotherapy.
J Pharm Pharmacol. 64:1695–1702. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yin C, Knudson CM, Korsmeyer SJ and Van
Dyke T: Bax suppresses tumorigenesis and stimulates apoptosis in
vivo. Nature. 385:637–640. 1997. View
Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chiu TL and Su CC: Curcumin inhibits
proliferation and migration by increasing the Bax to Bcl-2 ratio
and decreasing NF-κBp65 expression in breast cancer MDA-MB-231
cells. Int J Mol Med. 23:469–475. 2009.PubMed/NCBI
|