Introduction
Osteoarthritis (OA), the most common chronic
disability in older adults, is characterized by the degeneration of
articular cartilage. With the aging of the population, it is
estimated that the number of OA cases will double over the next 3
decades (1). Recent studies have
suggested that aging plays an important role in the pathogenesis of
OA (2,3). However, whether the aging process is
responsible for the development of OA has not yet been fully
elucidated. Considering that chondrocytes are the only type of cell
type present in articular cartilage, the OA process is
characterized by the changes that occur in these cells. Therefore,
the understanding of the cellular processes that regulate
aging-associated changes in chondrocytes is essential to the
development of novel therapeutic interventions for progressive
joint diseases.
Autophagy is a cellular response to various types of
stress, whereby cellular organelles and macromolecules are engulfed
and recycled to sustain cellular metabolism. Evidence indicates
that autophagy is required for lifespan extension in various
organisms, and that numerous autophagy-related proteins are
directly regulated by longevity pathways (4). Autophagic dysfunction may contribute
to the pathogenesis of numerous neurodegenerative diseases,
including Parkinson’s disease (5), Alzheimer’s disease (6), Frontoteporal dementia (7), Lafora disease (8) and Huntington’s disease (9). Constitutive autophagy is important
for maintaining the quality of organelles and maintaining cell
function. Autophagy degrades damaged organelles, cell membranes and
proteins, and the failure of autophagy is thought to be one of the
main causes of cell aging. Therefore, the level of autophagic
activity decreases with aging, whereas the stimulation of autophagy
may have potent anti-aging effects (10).
However, to date, little is known about the role of
autophagy in articular cartilage. Roach et al (11) firstly described a peculiar variant
of apoptotic cell death in articular cartilage, termed
chondroptosis, a term that also included an autophagy component.
Previous studies have suggested that autophagy is associated with
OA. Caramés et al (12)
demonstrated that aged and OA articular cartilage were associated
with the reduced expression of Unc-51-like kinase 1 (ULK1),
Beclin-1 and microtubule-associated protein 1 light chain 3 (LC3),
and speculated that autophagy may play a protective role against
chondrocyte death. In addition, hypoxia-inducible factor (HIF)-2α
has been suggested to suppress autophagy in chondrocytes (13). Certainly, autophagy plays a dual
role, and is involved in both cell survival and death (4,14).
However, the potential role of autophagy in the initiation or the
development of OA remains unknown. Therefore, further studies are
required to elucidate the possible role of autophagy in OA and to
understand the association between autophagy, cell survival and
cell death.
In present study, we investigated the role of
autophagy in chondrocyte responses in the pathogenesis of articular
cartilage degeneration in OA. By intervening in autophagy, we
assessed the differences among the chondrocytes and ultrastructural
changes. Our results demonstrated that Beclin-1 and LC3 expression
in response to hypoxic conditions differed in the young and aging
cartilage, respectively, and confirmed that autophagic levels were
decreased in the aging compared with the young cartilage. Of note,
although decreased autophagy was observed in aging cartilage, the
levels of autophagic markers in patients with OA did not differ
from those in the aging cartilage. We also detected cell death in
the presence of autophagy, and reduced cell death by inhibitors of
autophagy in OA chondrocytes. Accordingly, we suggest that during
the cartilage pathological process, autophagy may play a
cytoprotective and death-promoting role in the pathogenesis of
OA.
Materials and methods
Human cartilage sampling
All cartilage samples were collected from human
donors who were undergoing surgery at the Department of
Orthopaedics, the First Affiliated Hospital of Anhui Medical
University, Hefei, China. The specimens were divided into 3 groups:
group 1, young cartilage specimens were obtained from 9 patients
undergoing lower limb amputation in severe trauma; group 2, 13
aging cartilage specimens, also having no history of joint disease,
were harvested from patients with femoral neck fracture who were
undergoing femoral head replacement and used as age-matched
non-arthritic articular cartilage samples; and group 3, OA
cartilage tissues were obtained from 12 patients with early-stage
osteoarthritis undergoing total knee arthroplasty. Human tissues
were obtained under the approval of the Hospital Ethics Committee
of Anhui Medical University. The characteristics of the patients
participating in this study are shown in Table I.
 | Table ICharacteristics of the patients
participating in this study. |
Table I
Characteristics of the patients
participating in this study.
|
Characteristics | OA cartilage
(n=12) | Aged cartilage
(n=13) | Young cartilage
(n=9) |
|---|
| Age (mean ±
SD) | 65.4±8.6 years | 63.4±6.2 years | 22.4 ±6.2
years |
| Gender
(female/male) | 7/5 | 6/7 | 4/5 |
| Diagnosis | Osteoarthritis | Femoral neck
fracture | Destructive limb
injury |
| Medications | Total knee
arthroplasty | Total hip
replacement | Amputation |
| OA grade | II–III | I–II | I |
| Mankin score | 4–7 | 0–2 | 0 |
Isolation and culture of primary
chondrocytes
The cartilage tissue was incubated with trypsin (0.5
mg/ml; Sigma-Aldrich, St. Louis, MO, USA) at 37°C for 10 min. After
the trypsin solution was removed, the tissue slices were treated
for 12–16 h with type II collagenase (2 mg/ml; Sigma-Aldrich, St.
Louis, MO, USA) in Dulbecco’s modified Eagle’s medium (DMEM;
Gibco-Life Technologies) with 5% fetal calf serum. The isolated
chondrocytes were recovered in DMEM supplemented with 10% fetal
calf serum, L-glutamine and antibiotics and were allowed to attach
to the culture flasks. The cells were incubated at 37°C in a
humidified gas mixture containing 5% CO2 balanced with
air. The chondrocytes were used in the experiments at confluency
(2–3 weeks in primary culture).
Histological and immunohistochemical
analysis
The serial sections were stained with hematoxylin
and eosin (H&E) in order to observe histological changes in the
articular cartilage. Immunohistochemistry (IHC) was performed
according to the indirect immunoperoxidase method. In brief,
following deparaffinization, hydration and blockage of endogenous
peroxidase, the specimens were incubated for 20 min with 10%
non-fat milk in phosphate-buffered saline (PBS) in order to block
specific sites and then individually incubated at 4°C overnight
with the following primary antibodies: rabbit polyclonal LC3
antibody (NB100-2220, 1:200; Novus Biologicals, LLC, Littleton, CO,
USA) and rabbit polyclonal Beclin-1 antibody (ab55878, 1:400;
Abcam, Cambridge, MA, USA). After rinsing, the slides were washed,
and the sections were incubated with biotinylated goat anti-rabbit
secondary antibody for 30 min at room temperature, and then
incubated using the Vectastain ABC-AP kit (Vector Laboratories,
Burlingame, CA, USA) for 30 min. Finally, the sections were washed
and incubated with 3,3-diaminobenzidine tetrahydrochloride (DAB)
substrate for 2–8 min. Visual impressions of the staining intensity
in the cytoplasm and the percentage of immunopositive chondrocytes
were recorded by analyzing the digital photomicrographs using
ImageJ software.
Transient transfection and confocal
microscopy
After the chondrocytes reached 60–70% confluency,
they were transfected with green fluorescent protein-light chain 3
(GFP-LC3) plasmid (OriGene Technologies, Inc., USA), using the
Lipofectamine 2000 reagent (no. 11668–27; Invitrogen, Carlsbad, CA,
USA) according to the manufacturer’s instructions, and 24 h after
transfection, they were incubated in an O2 tension of 2%
overnight in an Invivo2 Hypoxia Workstation (Ruskinn
Technology Ltd., Bridgend, UK) or under normoxic conditions for 24
h, and examined under a confocal microscope. For quantification
purposes, the proportion of chondrocytes that contain GFP-LC3
puncta (number of cells with green fluorescent puncta/total number
of transfected cells) (15,16), at least 100 green fluorescent
chondrocytes should be counted for each sample to obtain
statistically significant results.
Western blot analysis
The chondrocytes were isolated from confluent
monolayer cultures. Before immunoblotting, protein was quantified
using the Bradford method with a BCA detection kit (Thermo
Scientific-Pierce, Rockford, IL, USA) and adjusted to equal
concentrations across different samples. Equal amounts of protein
were separated by SDS-PAGE on precise 10% polyacrylamide gels.
Following electrophoresis, the proteins were transferred onto a
polyvinylidene fluoride (PVDF) membranes (Millipore, Billerica, MA,
USA). The membranes were then blocked with 5% non-fat milk in
Tris-buffered saline containing 0.1% Tween-20 (TBST) at room
temperature for 1 h, and then incubated overnight at 4°C with
primary antibodies: rabbit polyclonal LC3 antibody (NB100-2220,
1:2,000; Novus) and rabbit polyclonal Beclin-1 antibody (ab55878,
1:2,000; Abcam). Following incubation, the membranes were washed in
TBST buffer for 5 min and probed with anti-rabbit secondary
antibody (401393 -2ML, 1:50,000; Millipore) for 1 h at room
temperature. They were then washed with TBST 3 times for 5 min.
Protein bands of interest were detected using the ECL dectection
kit (no. 34094; Thermo Scientific-Pierce). The expression of
glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (ab9485, 1:2,000;
Abcam) was used as the loading control.
Transmission electron microscopy
(TEM)
TEM analysis was performed as previously described
(17). The chondrocytes were
harvested, and fixed in 2.5% glutaraldehyde in phosphate buffer,
post-fixed in 2% osmium tetroxide and embedded in Luveak-812
(Nacalai Tesque, Inc., Kyoto, Japan). Ultrathin sections were
stained with uranyl acetate for 10 min, then with lead citrate for
10 min, and evaluated under a JEM-1230 electron microscope (Jeol
Ltd., Tokyo, Japan).
Monodansylcadaverine (MDC) staining
Three groups of chondrocytes were seeded on
coverslips overnight and then treated with rapamycin or
3-methyladenine (3-MA) (both from Sigma-Aldrich, Shanghai, China).
After autophagy activation or inhibition, the cells were incubated
with 0.05 mM MDC (Sigma-Aldrich, Shanghai, China) in PBS at 37°C
for 10 min, then washed 3 times with PBS and fixed with a solution
of 4% paraformaldehyde for 30 min. The coverslips were examined
under a fluorescence microscope (XSZ-D2; Olympus, Tokyo, Japan). To
quantify autophagic cells after treatment, we counted the number of
autophagic cells demonstrating MDC-labeled particles among 200
cells.
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay
Cells (2×104) were plated into a 96-well
plate and cultured for >12 h. The chondrocytes were then
subjected to autophagy activation or inhibition using
pharmacological tools for 24 h at various concentrations, and then
incubated with MTT (0.5 g/l) for 4 h. The formazan precipitate was
dissolved in 200 μl dimethyl sulfoxide and the absorbance at 570 nm
was measured using a microplate reader (Bio-Rad, Hercules, CA,
USA).
Acridine orange (AO) staining and
quantification by flow cytometry
AO is a widely used to visualize acidic vesicular
organelles (AVOs) (18). AO is a
fluorescent dye that stains DNA and the cytoplasm bright green. At
low pH, AO emits red fluorescence with intensity proportional to
the degree of acidity. To detect the cell death rate, following the
pharmacological activation or inhibition of autophagy, the cells
were washed twice with PBS, fixed with 4% paraformaldehyde and
stained with AO (Molecular Probes, Eugene, OR, USA) at 1 μg/ml for
15 min. They were then washed with PBS to remove the unbound dye.
AVO formation in the AO-stained cells was measured by flow
cytometry, and the cells were analyzed on a flow cytometer using
CellQuest software.
Quantification of DNA hypoploidy and cell
cycle phase analysis by flow cytometry
When the chondrocytes reached 60–70% confluency, we
treated the cells with 3-MA or rapamycin (Sigma-Aldrich, Shanghai,
China). Following treatment for the desired amount of time, the
cells were harvested. The digested cells were then washed 3 times
in PBS, fixed with ice cold 70% ethanol and then incubated at −20°C
overnight. Subsequently, the fixed cells were centrifuged and
washed 3 times with PBS. The cells were resuspended in 250 μl
propidium iodide (50 mg/ml) and incubated in the dark at room
temperature for 3 h. The cells were then analyzed on a flow
cytometer using CellQuest software.
Statistical analyses
All data and results presented in this study were
derived from at least 3 independent experiments. The data are
expressed as the means ± SD. The significance of the data was
evaluated by one-way ANOVA. Values of P<0.05 were considered to
indicate statistically significant differences.
Results
Differential expression of Beclin-1 and
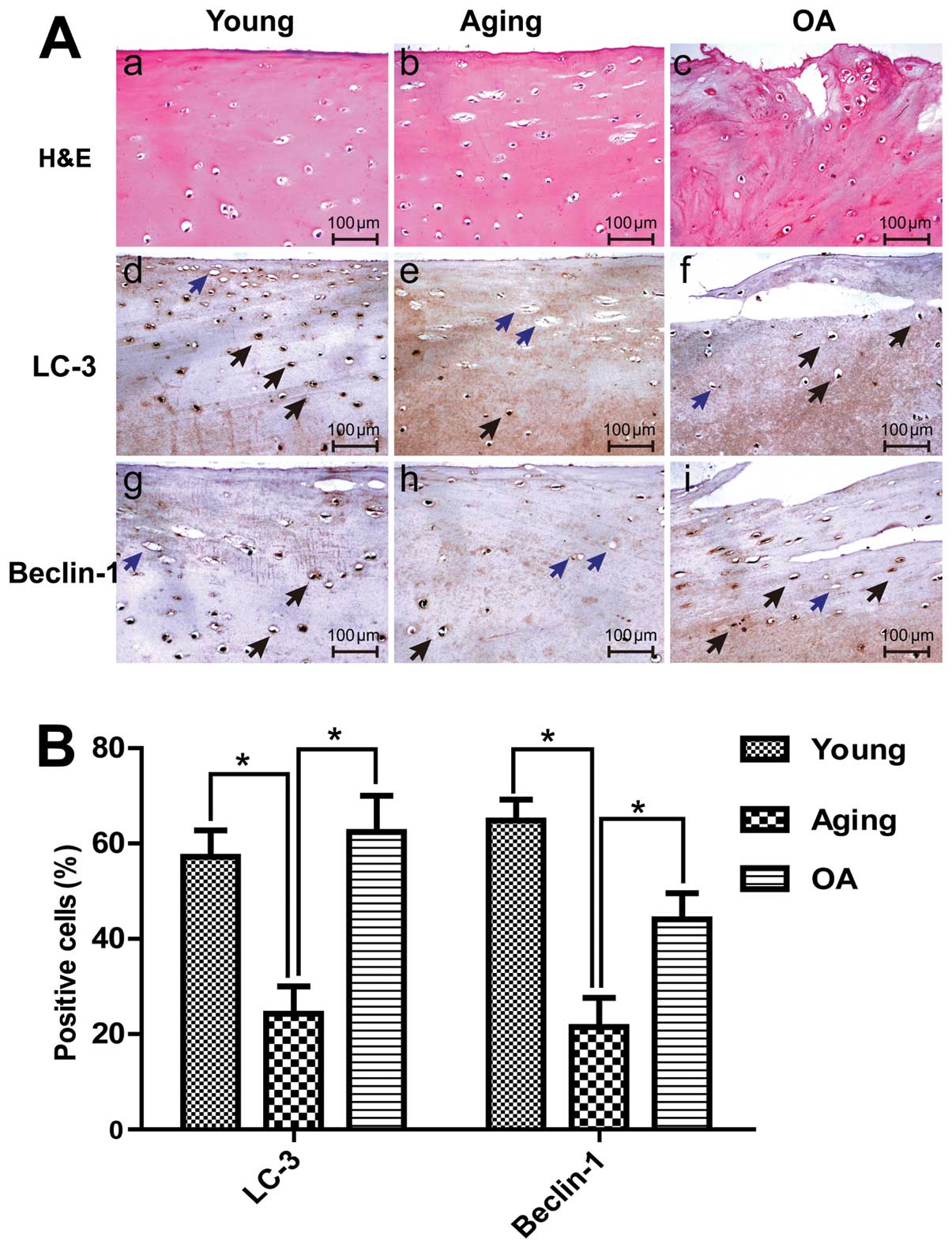
LC3 in human articular cartilage tissues
To determine the role of autophagy in the
development of OA, we examined Beclin-1 and LC3 expression in
young, aging and OA cartilage by IHC in the upper zone of the
cartilage (including the superficial and middle zones). IHC
analysis indicated that Beclin-1 and LC3 were ubiquitously
expressed in the young cartilage group, but lower expression levels
were observed in the aging cartilage. Cartilage samples were
evaluated by staining with H&E (Fig. 1A-a–c) and Beclin-1 and LC3
expression levels were determine by IHC (Fig. 1A-d–g). The percentage of
chondrocytes positive for Beclin-1 was substantially decreased in
the aging group (21.5±6.1%) compared with the young group
(64.8±4.4%), while the percentage of LC3-positive cells in the
young and aging groups (57.2±5.6 and 24.3±5.7%, respectively)
indicated a statistically significant decrease in aging cartilage
(P<0.05) (Fig. 1B). These
results indirectly suggest a decline in autophagic activity in
aging tissue. Of note, we found that the expression of autophagic
proteins was not reduced in patients with OA. In the OA group, the
percentages of Beclin-1- and LC3-positive cells were 44.1±5.6 and
62.4±7.6%, respectively (Fig.
1B).
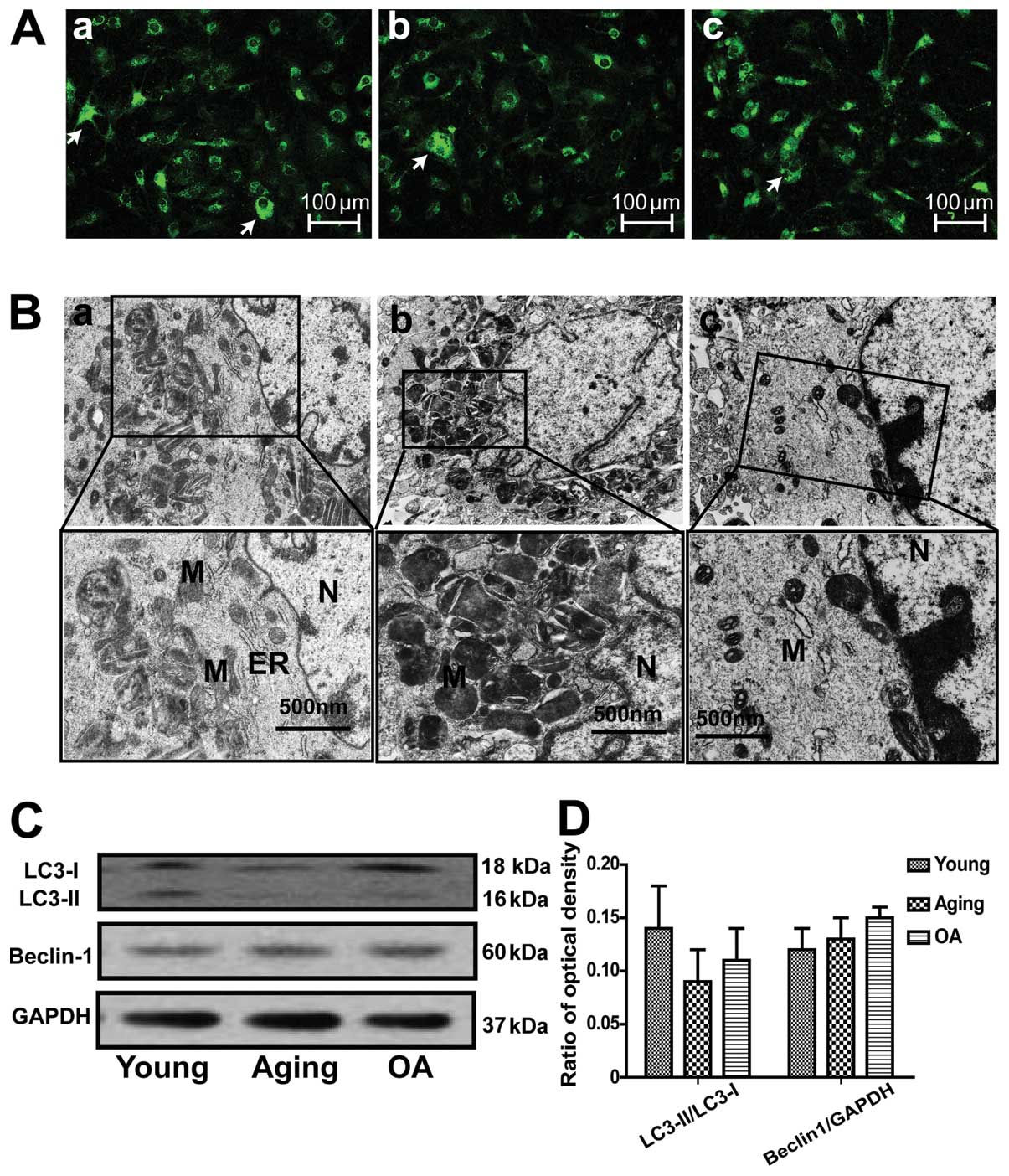
Differences in the autophagic response of
cultured articular chondrocytes
We used human primary chondrocytes transiently
expressing GFP-LC3 to quantify autophagosome formation. The results
revealed that the percentage of chondrocytes with GFP-LC3 puncta
structures was low (Fig. 2A).
When comparing aging and OA chondrocytes, the percentage of
chondrocytes with GFP-LC3 puncta structures did not differ. The
results of electron microscopy also revealed few autophagosomes in
these 3 groups of chondrocytes. Western blot analysis also revealed
no significant differences in the expression levels of autophagic
proteins in the chondrocytes (Fig. 2C
and D). Taken together, these results indicate that autophagic
activity was low and did not differ between the 3 groups of
cultured chondrocytes in vitro.
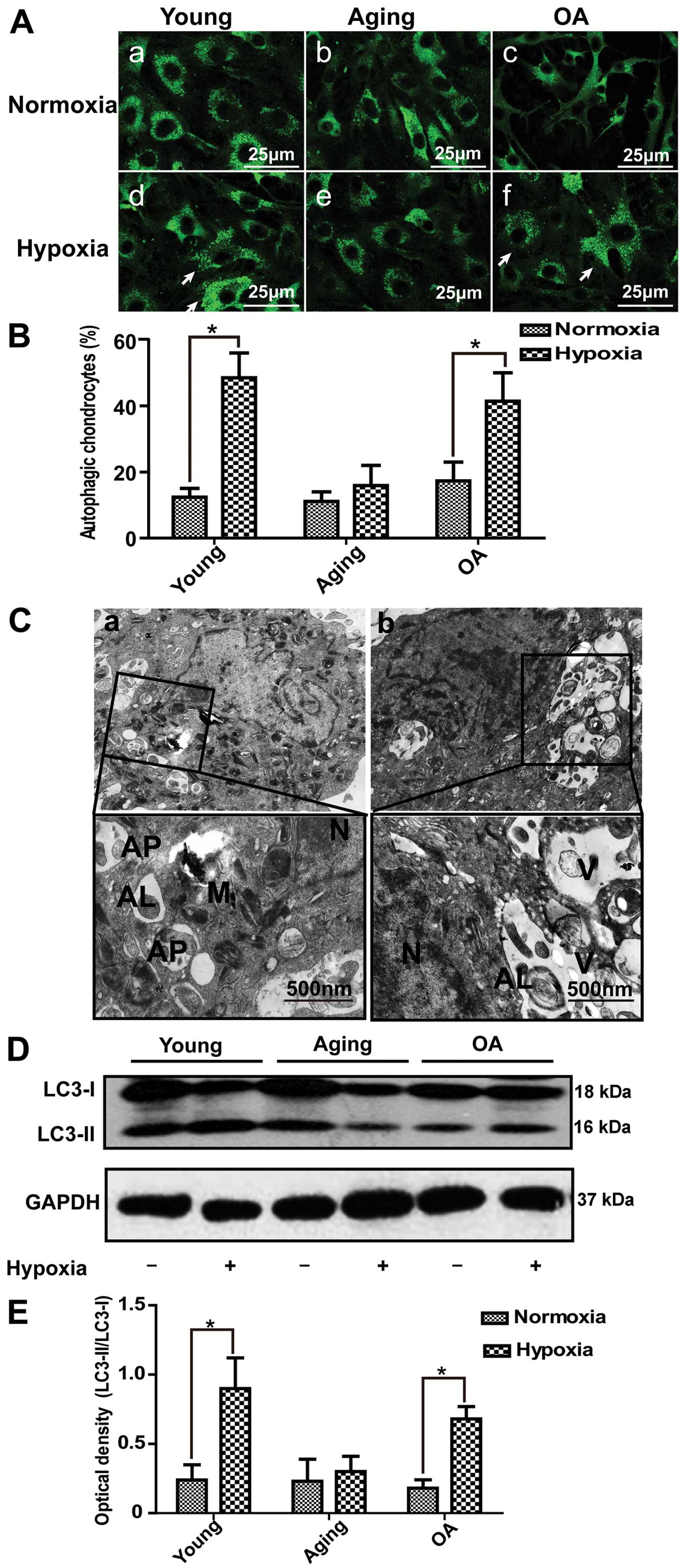
Hypoxia plays an essential role in
autophagy in chondrocytes
Chondrocytes grow in a hypoxic environment in
vivo (19). Thus, in this
study, we further examined the role of hypoxia in autophagy in
cultured chondrocytes. In the young and OA chondrocytes, the
numbers of GFP-LC3 puncta structures were markedly increased, but
in the aging group, no significant change was observed (Fig. 3A). Compared with the groups
exposed to normoxic conditions, the percentage of cells with
GFP-LC3 puncta structures in the groups exposed to hypoxic
conditions was significantly increased. We subsequently used
electron microscopy to observe the changes in the ultrastructure of
chondrocytes following hypoxia-induced autophagy. A large number of
autophagosomes with degraded organelles was observed in the young
chondrocytes (Fig. 3C-a). By
contrast, the aging chondrocytes contained few autophagosomes. Of
note, in the OA chondrocytes, we found characteristic structures of
autophagic cell death (Fig.
3C-b); these cells with autophagic vacuoles contained cell
fragments, autolysosomes and few or no chromatin was condensed. In
addition, our results revealed a significant increase in the
expression of LC3-II protein in the young and OA groups; however,
no significant changes were observed in the aging group (Fig. 3D and E).
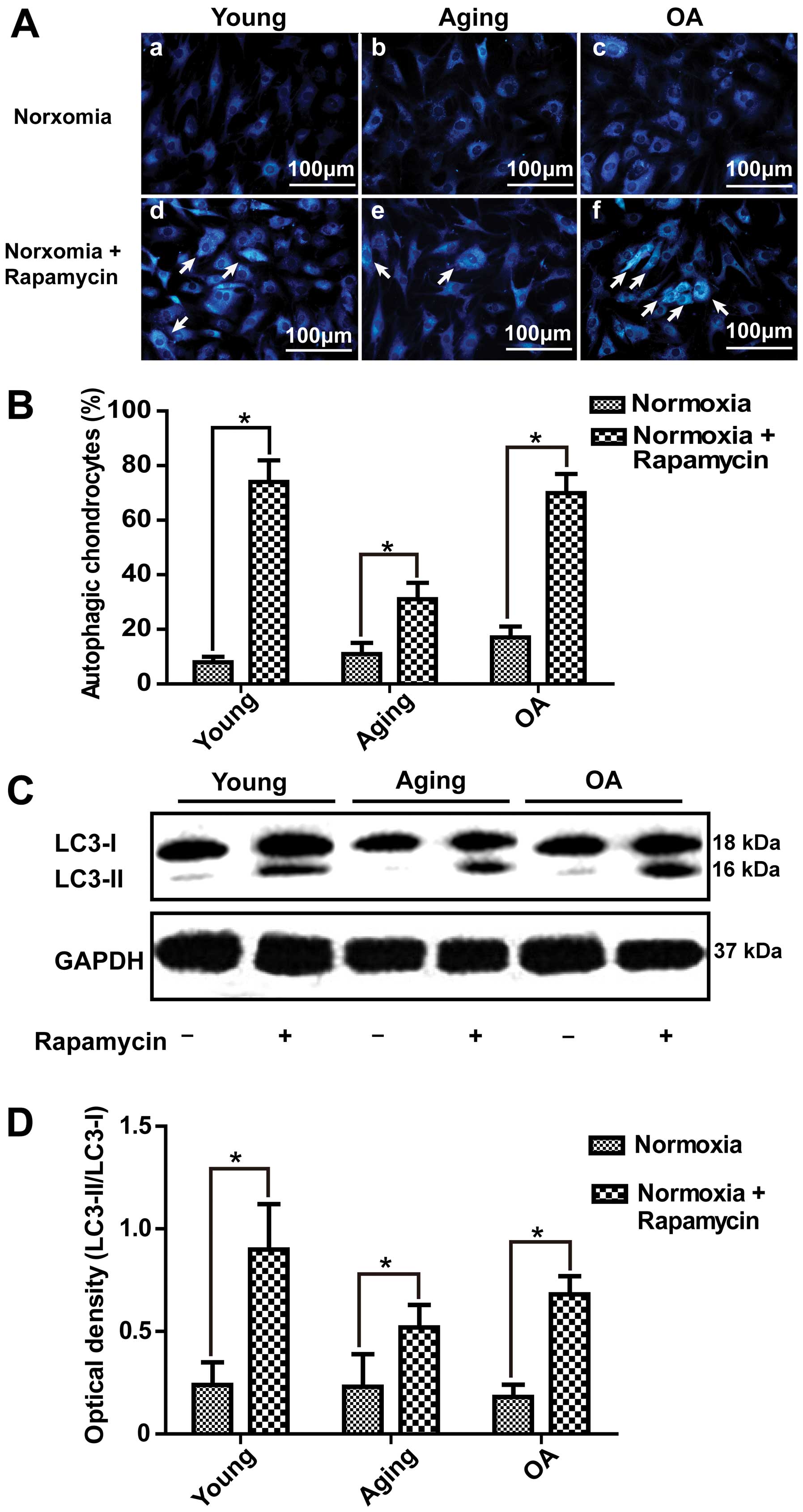
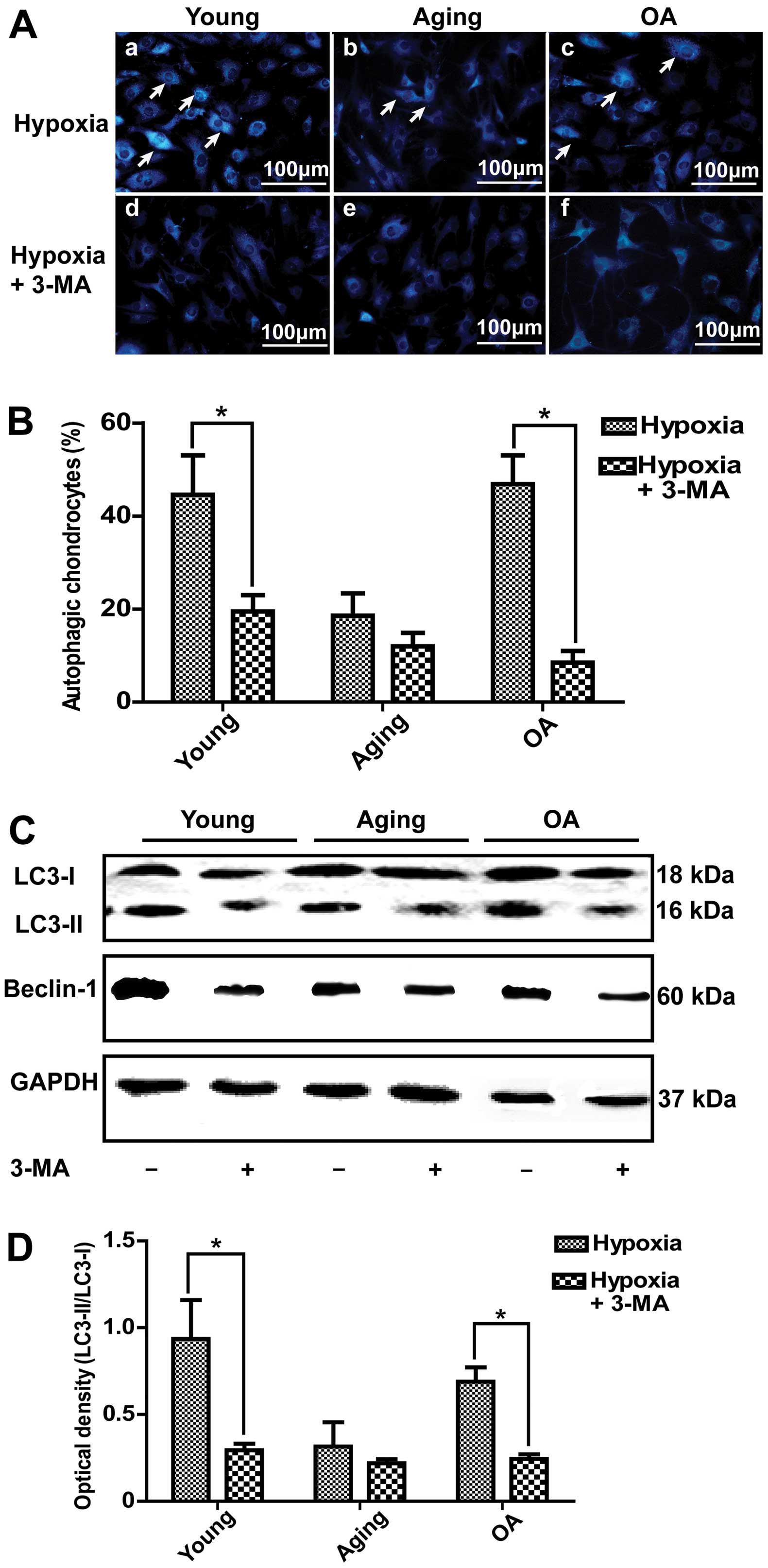
Effect of rapamycin and 3-MA on
autophagic of chondrocytes
In this study, we demonstrated that autophagic
activity was increased under hypoxic conditions compared with
normoxic conditions. Thus, we induced autophagy under normoxic
conditions and inhibited autophagy under hypoxic conditions. As
illustrated in Fig. 4A-d–f,
following the treatment of the chondrocytes with 10 μM rapamycin,
which has been reported to be a potent inducer of autophagy
(13,20,21), under normoxic conditions, the
number of vesicles was increased by MDC staining (Fig. 4B). Western blot analysis also
indicated that the expression of LC3-II was significantly increased
in the young and OA chondrocytes (Fig. 4C). This result indicated that
autophagy in young and OA chondrocytes was more easily induced than
in aging chondrocytes (Fig. 4D).
Under hypoxic conditions, following the pre-treatment of the
chondrocytes with 3-MA, which is one of the most commonly used
inhibitors of autophagy (22,23), the number of autophagic vacuoles
stained by MDC was much lower (Fig.
5A and B). Moreover, LC3-II and Beclin-1 expression
significantly decreased in the young and OA chondrocytes (Fig. 5C and D).
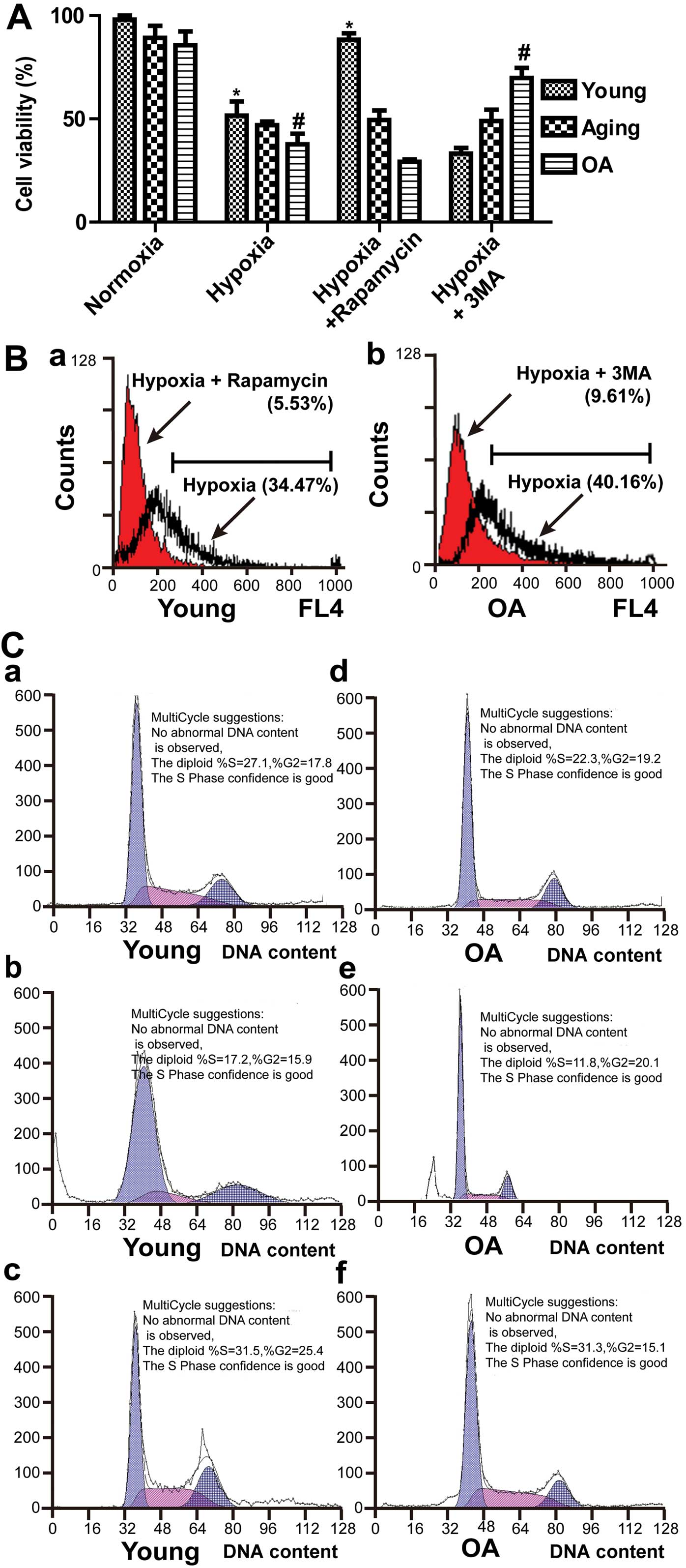
Effects of autophagy on cell viability
and cell cycle of chondrocytes
To extend our analysis, we intervened in the
autophagic pathway to examine the effects on cell viability. In
particular, we examined whether autophagy contributes to the cell
survival or cell death process. Cell viability was determined by
MTT assay. The cell survival rate increased significantly in the
young chondrocytes after the induction of autophagy (P<0.05). In
addition, the cell survival rate changed from 37.69±5.17 to
69.86±4.81% in the OA chondrocytes after the inhibition of
autophagy (Fig. 6A). Flow
cytometry was performed to detect cell death by AO staining. As
shown in Fig. 6B-a, the cell
death rate changed from 34.47 to 5.53% after the induction of
autophagy in young chondrocytes. In the OA chondrocytes, by
contrast, it changed from 40.16 to 9.61% after the inhibition of
autophagy (Fig. 6B-b). These
results demonstrated that autophagy was beneficial to cell survival
in young chondrocytes but led to cell death in OA chondrocytes. To
further clarify the possible mechanisms by which autophagy affected
the viability of chondrocytes, we used flow cytometry for the
quantification of DNA hypoploidy, analysis of the cell cycle and
the measurement of cell sub-G1 peaks. In the young chondrocytes,
the majority of cells were in the G0/G1 phase of the cell cycle
(Fig. 6C-a). Flow cytometry
demonstrated that rapamycin prevented the accumulation of
subdiploid cells (Fig. 6C-b and
c). These findings suggest that increased autophagosome
formation is required for chondrocyte survival in young cartilage.
As shown in Fig. 6C-e, autophagy
was critical for the OA chondrocyte cycle progression from the G2/M
to the G1 phase under hypoxic conditions. The sub-G1 population of
the cell cycle is represented as total cell death, and we found
that the sub-G1 population in the OA chondrocytes decreased from
16.24% to 4.47% following treatment with 3-MA under hypoxic
conditions (Fig. 6C-e and f).
Discussion
The aim of this study was to confirm that autophagy
plays a dual role in articular cartilage tissue. Firstly, we
confirmed that autophagic activity was downregulated in the
chondrocytes of aged articular cartilage. Surprisingly, we found
that the expression of autophagic-related proteins was not
decreased in the tissues of patients with OA. In addition, we also
found that hypoxia may induce the autophagy of chondrocytes.
Another observation was that rapamycin may activate autophagy,
which plays a protective role in young chondrocytes. However, the
excessive activation of autophagy led to autophagic cell death in
OA chondrocytes in vitro.
The present study demonstrated that autophagic
activity declined in the aging articular cartilage. We found that
LC3-II conversion occurred more easily in young chondrocytes than
in aging chondrocytes. In early autophagy, LC3-I is converted to
LC3-II through lipidation by the ubiquitin-like system, which
correlates with the extent of autophagosome formation (24,25). Therefore, decreased autophagic
activity may participate in the pathological process of cartilage
aging. Aging is one of the main factors in the pathogenesis of OA,
and older patients who experience a joint injury develop OA much
more rapidly than young patients with a similar knee injury
(26). During aging, the
efficiency of autophagic degradation is declined and intracellular
waste products are accumulated. Furthermore, there is a general
consensus that the function of autophagy is decreased during aging
(27–29). This observation is consistent with
the notion that basal autophagic activity is decreased with age,
thus contributing to the accumulation of damaged macromolecules and
susceptibility to aging-related diseases (30).
To our surprise, the autophagic markers were found
to be increased in patients with OA. Beclin-1 and LC3 expression
levels in OA cartilage were significantly increased in the upper
zone compared with those in aging articular cartilage. These
results are not completely consistent with those of Caramés et
al (12). They reported that
the expression of ULK1, Beclin-1 and LC3 in OA cartilage was
significantly decreased in the superficial zone. We speculated that
their results may be due to the wear and tear with the loss of
chondrocytes in the superficial zone. However, we found the cell
clusters which localized in the middle zone in OA cartilage showed
a strong expression of Beclin-1 and LC3. In addition, the results
of GFP-LC3 transfection indicated that the chondrocytes in OA were
more easily susceptible to autophagy than those in the aging group,
which further demonstrated that autophagic activity in OA was
higher than the aging group.
We found that autophagy may play a protective role
in young cartilage. In our experiments, rapamycin-induced autophagy
protected chondrocytes in the young group from death under hypoxic
conditions, to a certain extent. Autophagy is a mechanism for the
turnover of proteins and elimination of damaged organelles to
maintain cell homeostasis. The induction of autophagy under
pathological conditions is generally considered to play a
cytoprotective role in young cartilage. Recent studies provide
compelling evidence that autophagy protects against diverse
pathologies, such as neurodegenerative diseases (31), cancer (32), aging (33) and heart diseases (34). Therefore, we suggested that in
young articular cartilage, autophagy was activated as an adaptive
response to hypoxic conditions. These results indicate that
compromised autophagy represents a novel mechanism in the
development of cartilage degeneration.
The most intriguing finding of our study was that
autophagic chondrocyte death occured in OA. Roach et al
(11) described a morphological
change of cell death in articular cartilage which was similar to
autophagic cell death. The present study first reported that this
type of death is classic autophagy in chondrocytes. We detected
cell death in the presence of autophagy, and demonstrated the
reduction in cell death by the inhibition of autophagy. Such
findings are consistent with most basic characteristics of
autophagic cell death (35).
Autophagic cell death is morphologically defined as a type of cell
death. This autophagy-dependent non-apoptotic cell death is defined
as autophagic cell death, or type II programmed cell death (PCD)
(36). Therefore, our results
provided a starting point to study autophagic chondrocyte death in
OA.
In addition, although our results demonstrated that
autophagy induced chondrocyte survival and death in different
stages of OA progression. Certain studies have found that autophagy
can promote cell survival or cell death, depending on the type of
cellular stress (35). Autophagy
has also been reported to play a role in cell survival under other
types of stress, such as exposure to DNA-damaging reagents
(37), endoplasmic reticulum
stress (38) and radiation
(39). Exposure to treatment
conditions, such as hypoxia (40)
and arsenic trioxide (41) have
been reported to induce autophagic cell death.
In conclusion, the present study demonstrates that
rapamycin-induced autophagy does not lead to chondrocyte death in
young patients, while it can induce cell death by autophagy in the
patients with OA. We described a novel and paradoxical role for
autophagic cellular degradative pathways in OA cartilage. These
results suggest that autophagy may play both a cytoprotective and
death-promoting role in chondrocytes. Therefore, the dual role of
autophagy in cytoprotection and cell death, as well as its impact
on longevity is one of the most fascinating features of this
process, which clearly has a direct impact on the age-related
development of cartilage degeneration. Understanding the role of
autophagy in chondrocyte responses should provide new targets for
attenuating and preventing the development of OA.
Acknowledgements
The authors thank Wen Hu for helping with electron
microscopy. This study was supported by the Doctoral Fund of the
Ministry of Education of China (no. 20123420110002).
References
|
1
|
Zhang Y and Jordan JM: Epidemiology of
osteoarthritis. Clin Geriatr Med. 26:355–369. 2010. View Article : Google Scholar
|
|
2
|
Loeser RF: Aging and osteoarthritis. Curr
Opin Rheumatol. 23:492–496. 2011. View Article : Google Scholar
|
|
3
|
Bijlsma JW, Berenbaum F and Lafeber FP:
Osteoarthritis: an update with relevance for clinical practice.
Lancet. 377:2115–2126. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Levine B and Kroemer G: Autophagy in the
pathogenesis of disease. Cell. 132:27–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Matsuda N, Sato S, Shiba K, et al: PINK1
stabilized by mitochondrial depolarization recruits Parkin to
damaged mitochondria and activates latent Parkin for mitophagy. J
Cell Biol. 189:211–221. 2010. View Article : Google Scholar
|
|
6
|
Lee JH, Yu WH, Kumar A, et al: Lysosomal
proteolysis and autophagy require presenilin 1 and are disrupted by
Alzheimer-related PS1 mutations. Cell. 141:1146–1158. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Parkinson N, Ince PG, Smith MO, et al: ALS
phenotypes with mutations in CHMP2B (charged multivesicular body
protein 2B). Neurology. 67:1074–1077. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Aguado C, Sarkar S, Korolchuk VI, et al:
Laforin, the most common protein mutated in Lafora disease,
regulates autophagy. Hum Mol Genet. 19:2867–2876. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Martinez-Vicente M, Talloczy Z, Wong E, et
al: Cargo recognition failure is responsible for inefficient
autophagy in Huntington’s disease. Nat Neurosci. 13:567–576.
2010.PubMed/NCBI
|
|
10
|
Cuervo AM, Bergamini E, Brunk UT, Droge W,
Ffrench M and Terman A: Autophagy and aging: the importance of
maintaining ‘clean’ cells. Autophagy. 1:131–140. 2005.
|
|
11
|
Roach HI, Aigner T and Kouri JB:
Chondroptosis: a variant of apoptotic cell death in chondrocytes?
Apoptosis. 9:265–277. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Caramés B, Taniguchi N, Otsuki S, Blanco
FJ and Lotz M: Autophagy is a protective mechanism in normal
cartilage, and its aging-related loss is linked with cell death and
osteoarthritis. Arthritis Rheum. 62:791–801. 2010.PubMed/NCBI
|
|
13
|
Bohensky J, Terkhorn SP, Freeman TA, et
al: Regulation of autophagy in human and murine cartilage:
hypoxia-inducible factor 2 suppresses chondrocyte autophagy.
Arthritis Rheum. 60:1406–1415. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Baehrecke EH: Autophagy: dual roles in
life and death? Nat Rev Mol Cell Biol. 6:505–510. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Klionsky DJ, Cuervo AM and Seglen PO:
Methods for monitoring autophagy from yeast to human. Autophagy.
3:181–206. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Klionsky DJ, Abdalla FC, Abeliovich H, et
al: Guidelines for the use and interpretation of assays for
monitoring autophagy. Autophagy. 8:445–544. 2012. View Article : Google Scholar
|
|
17
|
Chang J, Wang W, Zhang H, Hu Y and Yin Z:
Bisphosphonates regulate cell proliferation, apoptosis and
pro-osteoclastic expression in MG-63 human osteosarcoma cells.
Oncol Lett. 4:299–304. 2012.PubMed/NCBI
|
|
18
|
Paglin S, Hollister T, Delohery T, et al:
A novel response of cancer cells to radiation involves autophagy
and formation of acidic vesicles. Cancer Res. 61:439–444.
2001.PubMed/NCBI
|
|
19
|
Murphy CL, Thoms BL, Vaghjiani RJ and
Lafont JE: Hypoxia. HIF-mediated articular chondrocyte function:
prospects for cartilage repair. Arthritis Res Ther. 11:2132009.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sasaki H, Takayama K, Matsushita T, et al:
Autophagy modulates osteoarthritis-related gene expression in human
chondrocytes. Arthritis Rheum. 64:1920–1928. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Carames B, Hasegawa A, Taniguchi N, Miyaki
S, Blanco FJ and Lotz M: Autophagy activation by rapamycin reduces
severity of experimental osteoarthritis. Ann Rheum Dis. 71:575–581.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lin YC, Kuo HC, Wang JS and Lin WW:
Regulation of inflammatory response by 3-methyladenine involves the
coordinative actions on Akt and glycogen synthase kinase 3β rather
than autophagy. J Immunol. 189:4154–4164. 2012.PubMed/NCBI
|
|
23
|
Petiot A, Ogier-Denis E, Blommaart EF,
Meijer AJ and Codogno P: Distinct classes of phosphatidylinositol
3′-kinases are involved in signaling pathways that control
macroautophagy in HT-29 cells. J Biol Chem. 275:992–998. 2000.
|
|
24
|
Tanida I, Ueno T and Kominami E: LC3
conjugation system in mammalian autophagy. Int J Biochem Cell Biol.
36:2503–2518. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ohsumi Y and Mizushima N: Two
ubiquitin-like conjugation systems essential for autophagy. Semin
Cell Dev Biol. 15:231–236. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Roos H, Adalberth T, Dahlberg L and
Lohmander LS: Osteoarthritis of the knee after injury to the
anterior cruciate ligament or meniscus: the influence of time and
age. Osteoarthritis Cartilage. 3:261–267. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bergamini E: Autophagy: a cell repair
mechanism that retards ageing and age-associated diseases and can
be intensified pharmacologically. Mol Aspects Med. 27:403–410.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cuervo AM: Autophagy and aging: keeping
that old broom working. Trends Genet. 24:604–612. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Salminen A and Kaarniranta K: Regulation
of the aging process by autophagy. Trends Mol Med. 15:217–224.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Cuervo AM and Dice JF: Age-related decline
in chaperone-mediated autophagy. J Biol Chem. 275:31505–31513.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Mariño G, Madeo F and Kroemer G: Autophagy
for tissue homeostasis and neuroprotection. Curr Opin Cell Biol.
23:198–206. 2011.PubMed/NCBI
|
|
32
|
Janku F, McConkey DJ, Hong DS and Kurzrock
R: Autophagy as a target for anticancer therapy. Nat Rev Clin
Oncol. 8:528–539. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Rubinsztein DC, Mariño G and Kroemer G:
Autophagy and Aging. Cell. 146:682–695. 2011. View Article : Google Scholar
|
|
34
|
Iglewski M, Hill JA, Lavandero S and
Rothermel BA: Mitochondrial fission and autophagy in the normal and
diseased heart. Curr Hypertens Rep. 12:418–425. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chen Y, Azad MB and Gibson SB: Methods for
detecting autophagy and determining autophagy-induced cell death.
Can J Physiol Pharmacol. 88:285–295. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kroemer G and Levine B: Autophagic cell
death: the story of a misnomer. Nat Rev Mol Cell Biol. 9:1004–1010.
2008. View
Article : Google Scholar : PubMed/NCBI
|
|
37
|
Katayama M, Kawaguchi T, Berger MS and
Pieper RO: DNA damaging agent-induced autophagy produces a
cytoprotective adenosine triphosphate surge in malignant glioma
cells. Cell Death Differ. 14:548–558. 2007. View Article : Google Scholar
|
|
38
|
Ogata M, Hino S, Saito A, et al: Autophagy
is activated for cell survival after endoplasmic reticulum stress.
Mol Cell Biol. 26:9220–9231. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lomonaco SL, Finniss S, Xiang C, et al:
The induction of autophagy by gamma-radiation contributes to the
radioresistance of glioma stem cells. Int J Cancer. 125:717–722.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Azad MB, Chen Y, Henson ES, et al: Hypoxia
induces autophagic cell death in apoptosis-competent cells through
a mechanism involving BNIP3. Autophagy. 4:195–204. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kanzawa T, Kondo Y, Ito H, Kondo S and
Germano I: Induction of autophagic cell death in malignant glioma
cells by arsenic trioxide. Cancer Res. 63:2103–2108.
2003.PubMed/NCBI
|