Introduction
Naringin
(4′,5,7-trihydroxyflavonone-7-rhamnoglucoside), is a flavonone
found in citrus fruit, such as grapefruit. Accumulating evidence
has indicated that naringin has a broad spectrum of pharmacological
and therapeutic properties, including antioxidant (1–5),
antihypercholesterolemic (5–7),
anti-inflammatory (1,4,5,8)
and anti-apoptotic effects (9–12).
Notably, naringin has antihyperglycemic properties (13,14) and cardioprotective effects
(12,15,16). A recent study demonstrated that
naringin protects H9c2 cardiac cells against high glucose
(HG)-induced apoptosis by inhibiting the activation of the p38
mitogen-activated protein kinase (MAPK) pathway (12). Accordingly, we hypothesized that
the molecules which activate the p38 MAPK pathway may contribute to
HG-induced cardiomyocyte injury and to the cardioprotective effects
of naringin. Thus, in this study, we investigated one ot these
molecules, leptin.
Leptin, a 16-kDa peptide released from adipocytes
and other cells, has been implicated in the regulation of appetite
and energy metabolism (17,18). Leptin was initially thought to
mainly regulate obesity or body weight. However, increasing
evidence has demonstrated the effects of leptin in the regulation
of inflammation (19), blood
pressure homeostasis and cardiovascular disease (20–23). Leptin receptors have a widespread
tissue distribution, including the kidneys, pancreas, lungs and
heart (22,24–26), through which leptin exerts its
physiological effects, primarily by activating Janus tyrosine
kinase (JAK), signal transducers and signal transducers and
activators of transcription (STATs) (27). In addition, previous studies have
demonstrated that leptin can also activate other signaling
cascades, such as extracellular signal-regulated kinase 1/2
(ERK1/2) (28), c-Jun N-terminal
kinase/stress-activated protein kinase (JNK) (29) and p38 MAPK (30–33) of the MAPK family in a variety of
cell types. A previous study demonstrated that leptin induces the
selective translocation of p38 MAPK from the cytoplasmic to the
nuclear fraction and is dependent on intact caveolae and on the
activity of the RhoA pathway (33). In rat vascular smooth muscle
cells, leptin has been shown to induce hypertrophy through the
activation of the p38 MAPK pathway (31).
Thus, there seems to be a link between leptin and
diabetes-induced phathophysiological processes. Leptin has been
shown to regulate the diabetes-induced increase in extracellular
matrix (ECM) protein production in renal mesangial cells (34). Leptin is also involved in the
increased production of fibronectin (FN) induced by diabetes and in
cardiomyocyte hypertrophy (22).
However, evidence that the leptin-induced activation of the p38
MAPK pathway may be involved in hyperglycemia-induced cardiomyocyte
injury, including cytotoxicity, apoptosis, generation of reactive
oxygen species (ROS) and the dissipation of mitochondrial membrane
potential (MMP), is lacking. Thus, the aim purpose of the present
study was to determine whether the leptin-induced activation of the
p38 MAPK pathway contributes to hyperglycemia-induced cardiomyocyte
injury. We also aimed to determine whether the inhibition of the
leptin-induced activation of the-p38 MAPK pathway is involved in
the protective effects of naringin against HG-induced injury in
H9c2 cardiac cells.
Materials and methods
Materials
Naringin with a purity of ≥98%, was obtained from
Sigma-Aldrich, St. Louis, MO, USA, stored at 2–4°C and protected
from sunlight. Dichlorofluorescein diacetate (DCFH-DH), rhodamine
(Rh123) and Hoechst 33258 were also purchased from Sigma-Aldrich.
Leptin antagonist (LA) was supplied by Prospec (East Brunswick, NJ,
USA). The Cell Counting kit-8 (CCK-8) was purchased from Dojindo
Laboratories (Kumamoto, Japan). Fetal bovine serum (FBS) and
DMEM-F12 medium were obtained from Gibco-BRL (Grand Island, NY,
USA). Anti-p38 MAPK, anti-phospho-p38 MAPK, anti-leptin and
anti-leptin receptor antibodies were procured from Cell Signaling
Technology (Boston, MA, USA). HRP-conjugated secondary antibody and
the BCA protein assay kit were obtained from KangChen Bio-tech,
Inc. (Shanghai, China). Enhanced chemiluminescence (ECL) solution
was purchased from Nanjing KeyGen Biotech Co., Ltd. (Nanjing,
China).
Cell culture and treatments
H9c2 cardiac cells, a rat cardiomyoblast cell line,
were supplied by the Sun Yat-sen University Experimental Animal
Center (Guangzhou, China). The cells were grown in DMEM-F12 medium
supplemented with 10% FBS under an atmosphere of 5% CO2
at 37°C and 95% air.
The H9c2 cells were preconditioned with 80 μmol/l
naringin for 2 h prior to exposure to 35 mmol/l glucose (HG) for 24
h. To further determine whether the protective effects of naringin
and the activation of the p38 MAPK pathway induced by HG are
associated with leptin, the H9c2 cells were preconditioned with 50
ng/ml LA for 24 h prior to exposure to 35 mmol/l glucose for 24
h.
Measurement of cell viability
The H9c2 cells were seeded in 96-well plates at a
concentration of 1×104cells/ml, and incubated at 37°C.
The CCK-8 assay was employed to assess the viability of the H9c2
cardiac cells. After the indicated treatments, 10 μl of CCK-8
solution were added to each well at a 1/10 dilution and then the
plate was incubated for 1.5 h in an incubator. With the use of a
microplate reader (Molecular Devices, Sunnyvale, CA, USA)
absorbance was assayed at 450 nm. The mean optical density (OD) of
3 wells in the indicated groups was used to calculate the
percentage of cell viability according to the following formula:
cell viability (%) = (ODtreatment group/ODcontrol
group) ×100%. The experiment was repeated 5 times.
Hoechst 33258 nuclear staining for the
detection of apoptosis
Apoptotic cell death was observed by Hoechst 33258
staining followed by photofluorography. In brief, the H9c2 cells
were plated in 35 mm dishes at a density of 1×106
cells/well. After the indicated treatments, the H9c2 cells were
fixed with 4% paraformaldehyde in 0.1 mol/l phosphate-buffered
saline (PBS, pH 7.4) for 10 min. The slides were then washed 5
times with PBS. After staining with 5 mg/ml Hoechst 33258 for 5
min, the H9c2 cells were washed 5 times with PBS and then
visualized under a fluorescence microscope (Bx50-FLA; Olympus,
Tokyo, Japan). Viable H9c2 cells displayed a uniform blue
fluorescence throughout the nucleus and normal nuclear size.
However, apoptotic H9c2 cells showed condensed, distorted or
fragmented nuclei. The experiment was carried out 3 times.
Measurement of intracellular ROS
generation
The generation of intracellular ROS was determined
by the oxidative conversion of the cell-permeable oxidation of
DCFH-DA into fluorescent DCF. The H9c2 cells were cultured on a
slide with DMEM-F12 medium. After the different treatments, the
slides were washed twice with PBS. DCFH-DA (10 μmol/l) solution in
serum-free medium was then added to the slides, and the H9c2 cells
were then incubated in an incubator at 37°C for a further 30 min.
The slides were washed 5 times with PBS, and DCF fluorescence was
measured over the entire field of vision with the use of a
fluorescence microscope connected to an imaging system (BX50-FLA;
Olympus). The mean fluorescence intensity (MFI) from 5 random
fields was measured with the use of ImageJ 1.47i software and the
MFI was used as an index of the amount of ROS. The experiment was
carried out 5 times.
Measurement of MMP
MMP was assessed using a fluorescent dye, Rh123, a
cell-permeable carionic dye that preferentially enters the
mitochondria based on the highly negative MMP. The depolarization
of MMP results in a decrease in green fluorescence. The H9c2 cells
were cultured on a slide with DMEM-F12. After the indicated
treatments, the slides were washed 3 times with PBS. The cells were
incubated with 1 mg/l Rh123 at 37°C for 30 min in an incubator and
briefly washed 3 times with PBS and air dried. Fluorescence was
then measured over the whole field of vision with the use of a
fluorescence microscope connected to an imaging system (BX50-FLA;
Olympus). The MFI of Rh123 from 5 random fields was analyzed using
ImageJ 1.47i software The MFI was taken as an index of the MMP
levels. The experiment was carried out 5 times.
Western blot analysis
After the indicated treatments, the H9c2 cells were
harvested and lysed with cell lysis solution at 4°C for 30 min.
Total protein was quantified using the BCA protein assay kit.
Loading buffer was added to the cytosolic extracts, and boiled for
5 min; the same amounts of supernatant from each sample were
subjected to 10% sodium dodecyl sulphate-polyacrylamide gel
electrophoresis (SDS-PAGE), and then the total number of proteins
was transferred onto polyvinylidene difluoride (PVDF) membranes.
The membranes were blocked for 60 min in 5% fat-free milk in fresh
blocking buffer [0.1% Tween-20 in Tris-buffered saline (TBS-T)] at
room temperature, and incubated with either anti-p38 (1:1,000
dilution), anti-phosphorylated (p)-p38 (1:1,000 dilution),
anti-leptin (1:1,000 dilution) or anti-leptin receptor (1:1,000
dilution) antibodies in freshly prepared TBS-T with 3% fat-free
milk overnight with gentle agitation at 4°C. The membranes were
washed for 15 min with TBS-T and incubated with horseradish
peroxidase (HRP)-conjugated goat anti-rabbit secondary antibody
(1:2,500 dilution; KangChen Biotech, Inc.) in TBS-T with 3%
fat-free milk for 1.5 h at room temperature. The membranes were
then washed 3 times with TBS-T for 15 min. The immunoreactive
signals were visualized by using an ECL detection. In order to
quantify the protein expression levels, the X-ray films were
scanned and analyzed using ImageJ 1.47i software. The experiment
was carried out 3 times.
Statistical analysis
All data are presented as the means ± SEM.
Differences between groups were analyzed by one-way analysis of
variance (ANOVA) followed by the LSD post hoc comparison test.
Statistical analyses were performed using SPSS 13.0 statistical
software (SPSS, Chicago, IL, USA). Values of P<0.05 were
considered to indicate statistically significant differences.
Results
Naringin and LA attenuate the HG-induced
increase in leptin expression levels in H9c2 cardiac cells
In order to determine the effects of HG (35 mmol/l
glucose) on leptin expression levels in H9c2 cardiac cells, a
time-response experiment on leptin expression levels was performed.
Following exposure of the H9c2 cardiac cells to 35 mmol/l glucose
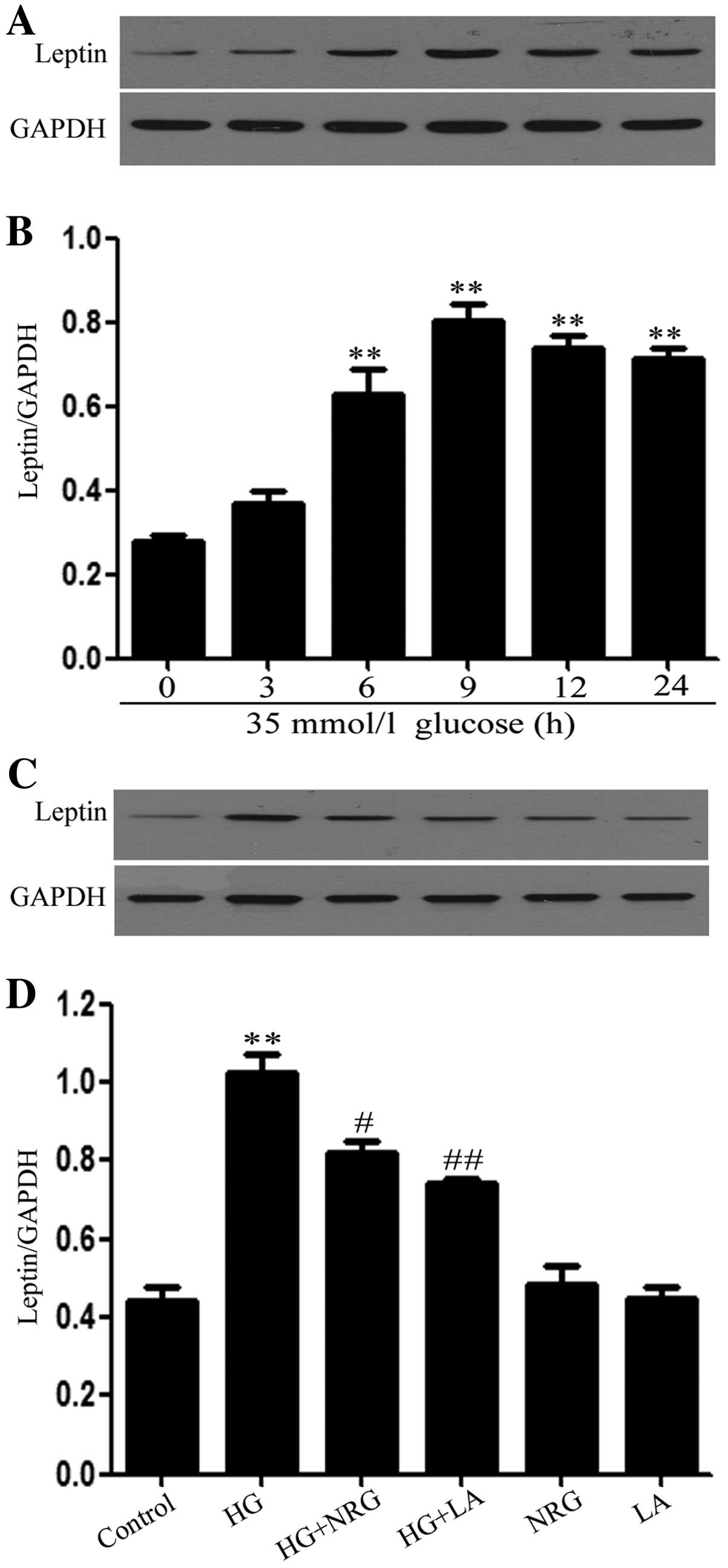
for 3, 6, 9, 12 and 24 h, the leptin expression levels began to
markedly increase at 6 h, reaching a peak at 9 h (Fig. 1A and B). Thus, leptin expression
levels were examined 9 h post-exposure to HG in the following
experiments. Notably, prior to exposure to 35 mmol/l glucose, the
H9c2 cells were treated with 80 μmol/l naringin for 2 h. This
pre-treatment significantly reduced the expression levels of leptin
which were increased by HG (Fig. 1C
and D).
In addition, the administration of 50 ng/ml LA for
24 h prior to exposure to HG, led to marked decrease in leptin
expression levels (Fig. 1C and
D). The basal expression levels of leptin (control) were not
altered by the separate treatment with 80 μmol/l naringin or 50
ng/ml LA (Fig. 1C and D).
Naringin attenuates the HG-induced
increase in the expression levels of leptin receptors in H9c2
cardiac cells
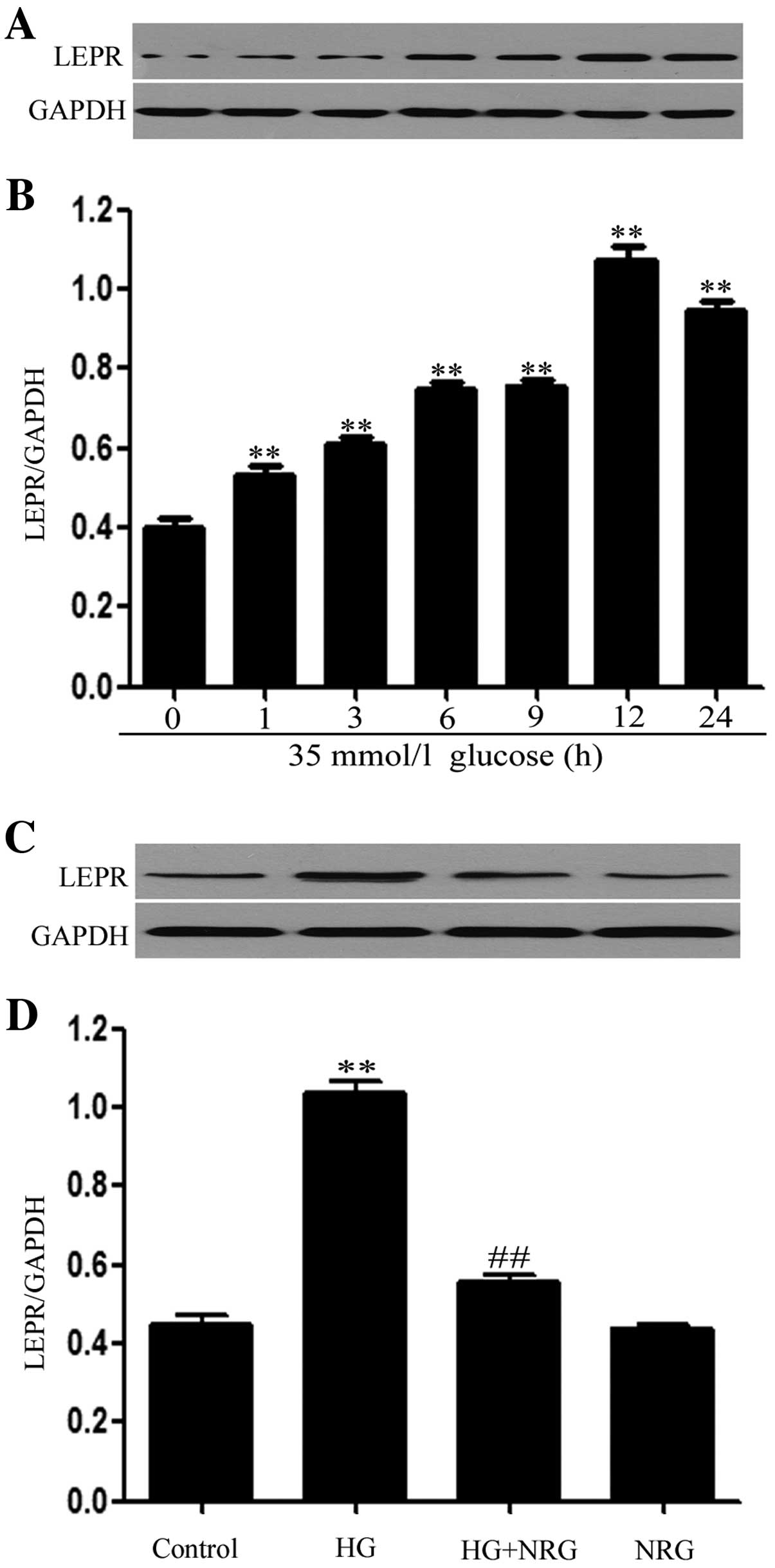
Western blot analysis (Fig. 2A and B) revealed that the exposure
of H9c2 cells to 35 mmol/l glucose for 1, 3, 6, 9, 12 and 24 h
induced the overexpression of leptin receptor (Ob-R) protein,
peaking at 12 h. The expression levels of Ob-R began to decrease at
24 h, but remained much higher than those observed at 9 h (Fig. 2A and B).
Additionally, treatment with 80 μmol/l naringin for
2 h prior to exposure to HG markedly alleviated the increased Ob-R
expression levels detected 12 h after exposure to HG (p<0.01)
compared with the HG-treated group (Fig. 2C and D). Naringin alone, at 80
μmol/l, did not alter the basal expression levels of leptin
receptors in H9c2 cells.
LA modulates the activation of p38 MAPK
induced by HG in H9c2 cardiac cells
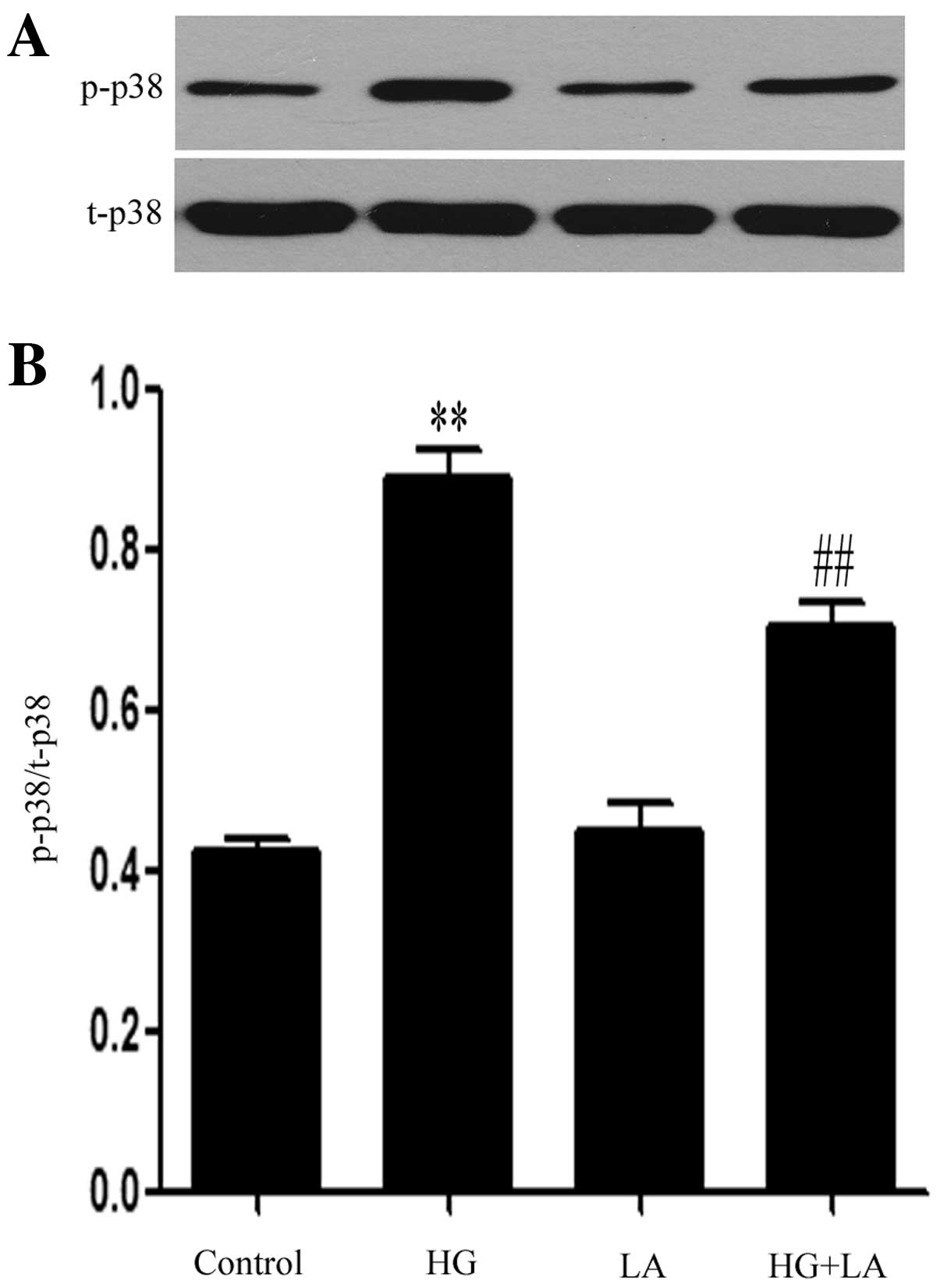
Western blot analysis revealed that exposure of the
H9c2 cells to HG markedly upregulated the expression levels of
p-p38 MAPK (Fig. 3). The
increased p-p38 MAPK expression levels were markedly suppressed by
treatment with LA (50 ng/ml) for 24 h prior to exposure to HG,
indicating the involvement of leptin in the HG-induced activation
of p38 MAPK in H9c2 cardiac cells.
Naringin and LA alleviate HG-induced
cytotoxicity in H9c2 cardiac cells
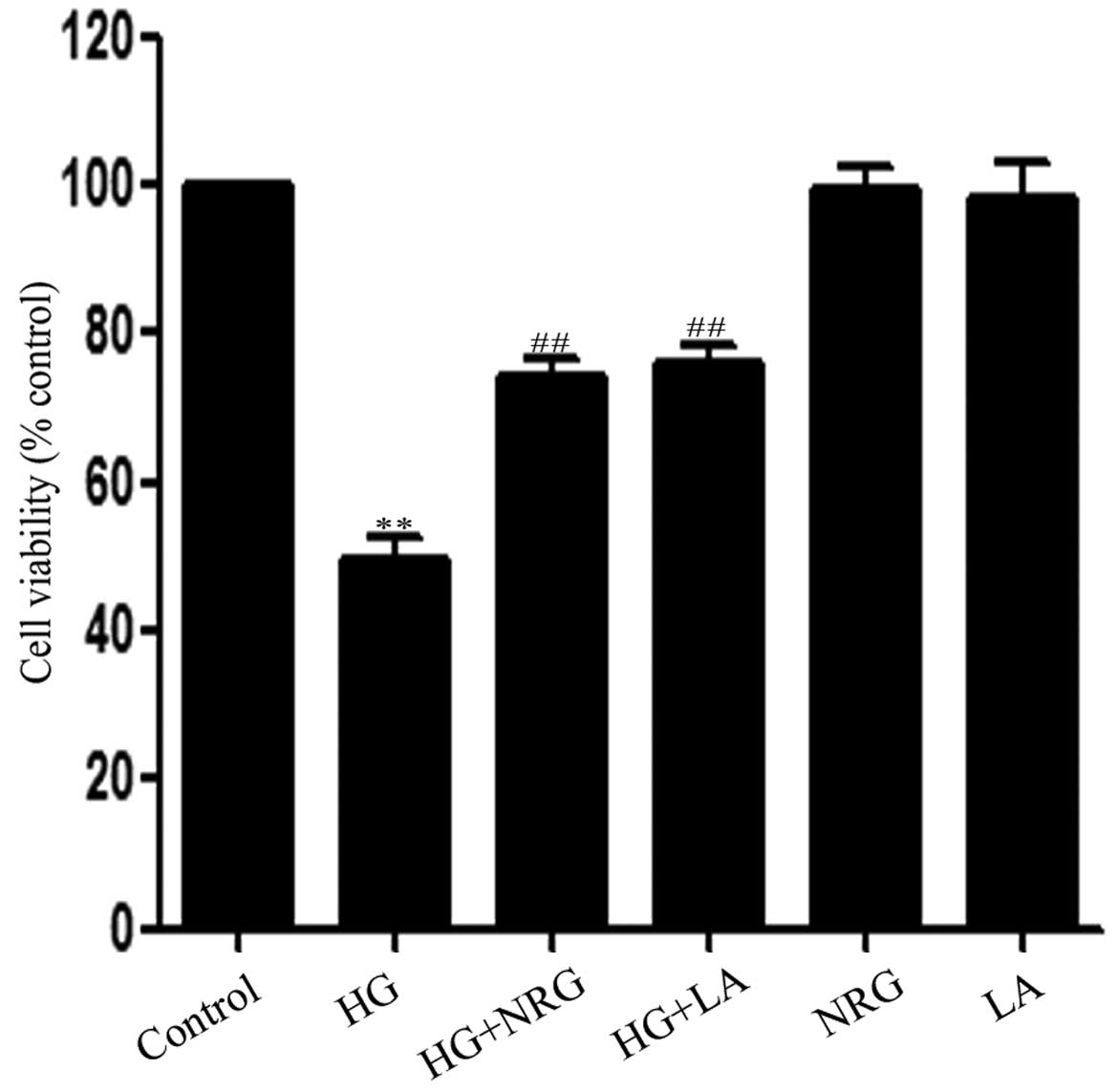
Exposure of the H9c2 cells to HG for 24 h induced
marked cytotoxic effects, leading to reduced cell viability
(Fig. 4). However,, the decreased
cell viability was attenuated by treatment with 80 μmol/l naringin
for 2 h or by treatment with 50 ng/ml LA for 24 h prior to exposure
to HG. cell viability levels still remained reduced compaired to
the control group. Naringin or LA alone did not affect the
viability of the H9c2 cardiac cells.
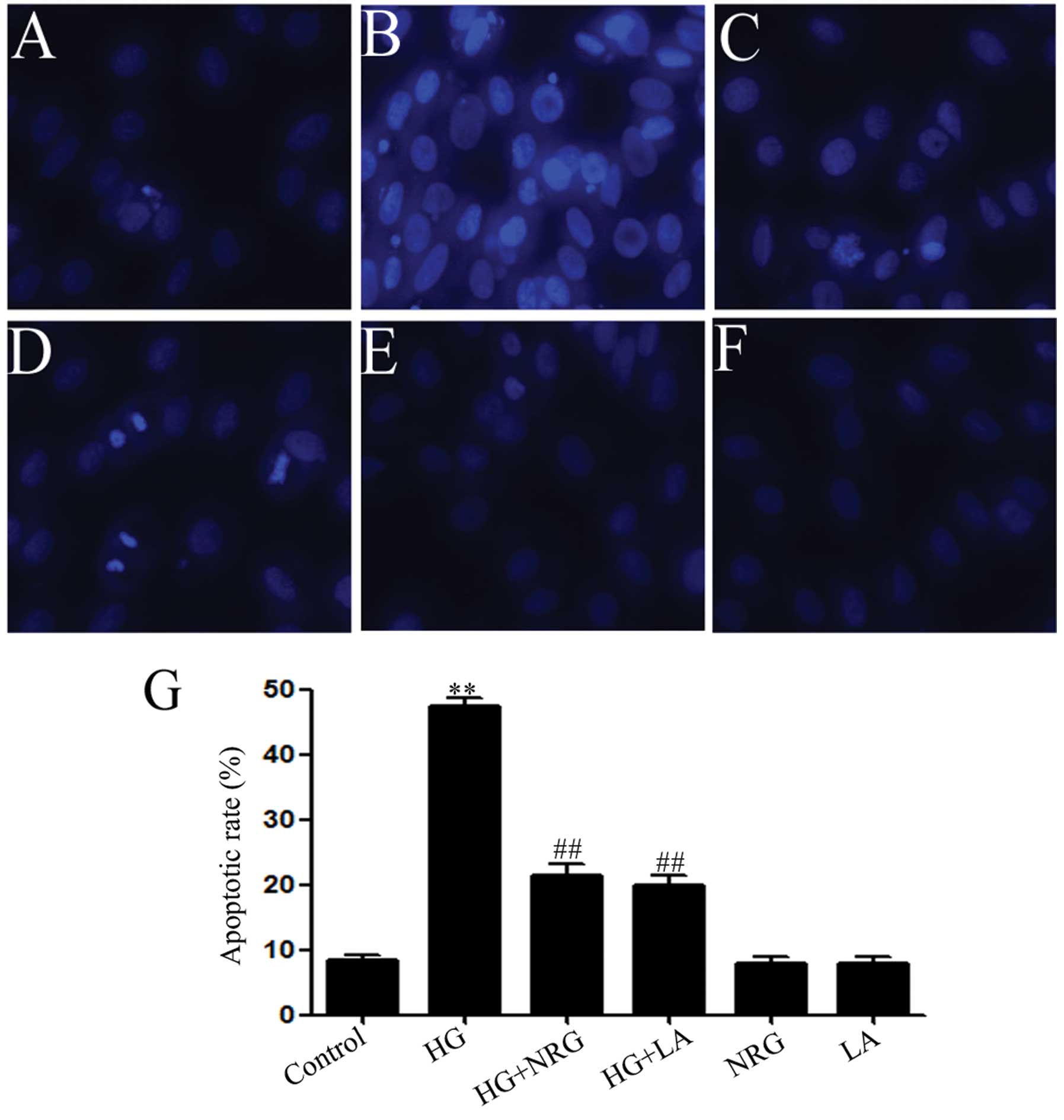
Naringin and LA attenuate HG-induced
apoptosis in H9c2 cardiac cells
As illustrated in Fig.
5B, exposure of the H9c2 cells to HG for 24 h induced typical
characteristics of apoptosis, as indicated by the condensation of
chromatin, the shrinkage of nuclei and the formation of apoptotic
bodies. However, treatment of the cells with 80 μmol/l naringin for
2 h prior to exposure to HG markedly ameliorated the increase in
the number of cells which presented nuclear condensation and
fragmentation (Fig. 5C). In
addition, treatment of the cells with 50 ng/ml LA for 24 h prior to
exposure to HG also alleviated HG-induced apoptosis (Fig. 5D). Naringin or LA alone did not
significantly alter the morphology or the percentage of apoptotic
H9c2 cells (Fig. 5E–G). These
findings suggest that naringin protects the H9c2 cells against
HG-induced apoptosis, which is related at least in part, with the
increase in leptin expression.
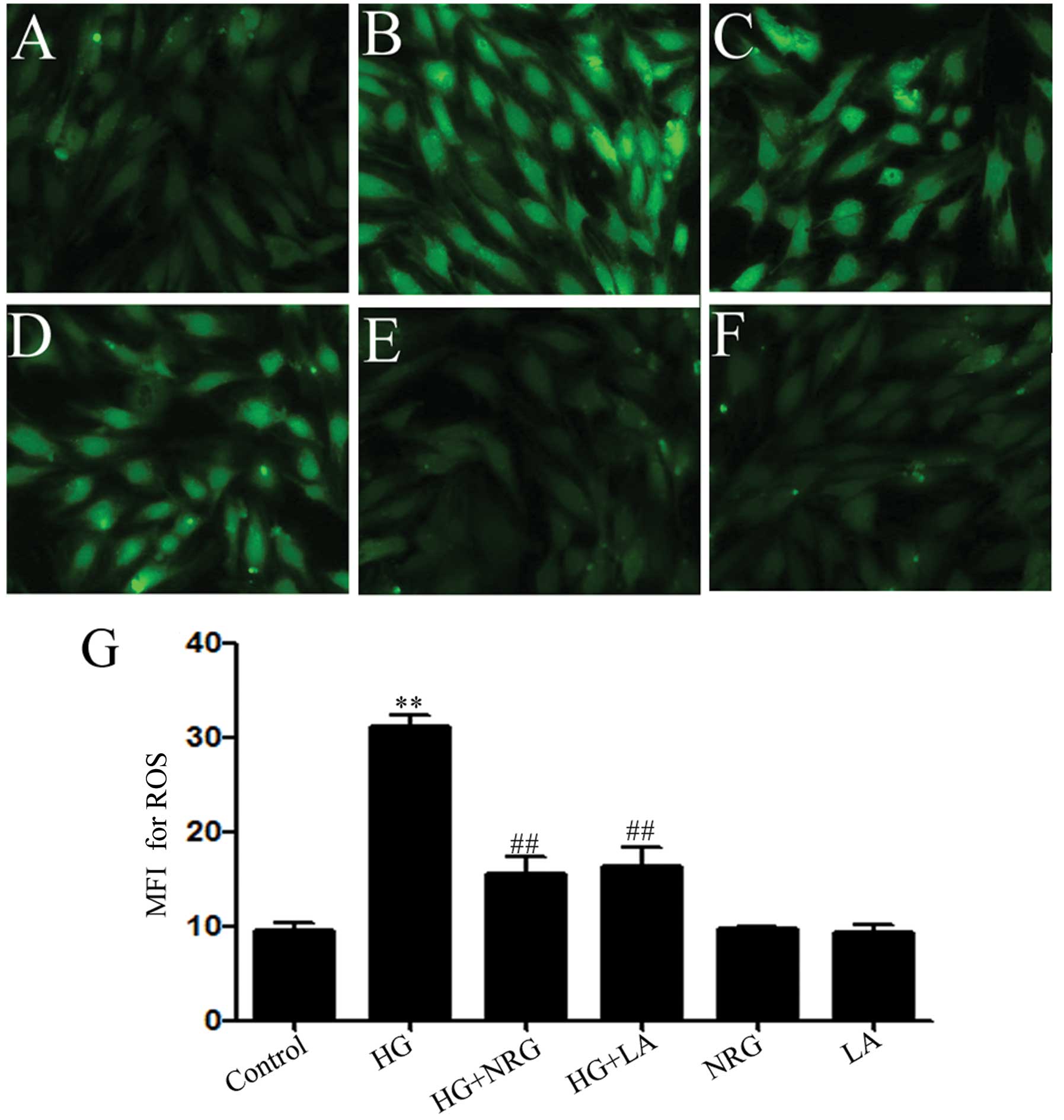
Naringin and LA alleviate the HG-induced
increase in ROS generation in H9c2 cells
Previous studies have demonstrated that ROS
generation is involved in HG-induced cardiomyocyte injury (35,36). Thus, in this study, we explored
the effects of naringin on HG-induced ROS generation in H9c2 cells.
The exposure of the H9c2 cells to HG for 24 h markedly enhanced ROS
generation (Fig. 6B and G). The
increase in ROS generation was suppressed by treatment of the cells
with 80 μmol/l naringin for 2 h prior to exposure to HG (Fig. 6C and G), revealing the inhibitory
effects of naringin on HG-induced oxidative stress. In order to
determine whether leptin is involved in the HG-induced
overproduction of ROS, the H9c2 cells were treated with 50 ng/ml LA
for 24 h prior to exposure to HG. Our results revealed that
treatment with LA considerably diminished the HG-induced increase
in ROS generation (Fig. 6D and
G), indicating the involvement of leptin in HG-induced ROS
overproduction.
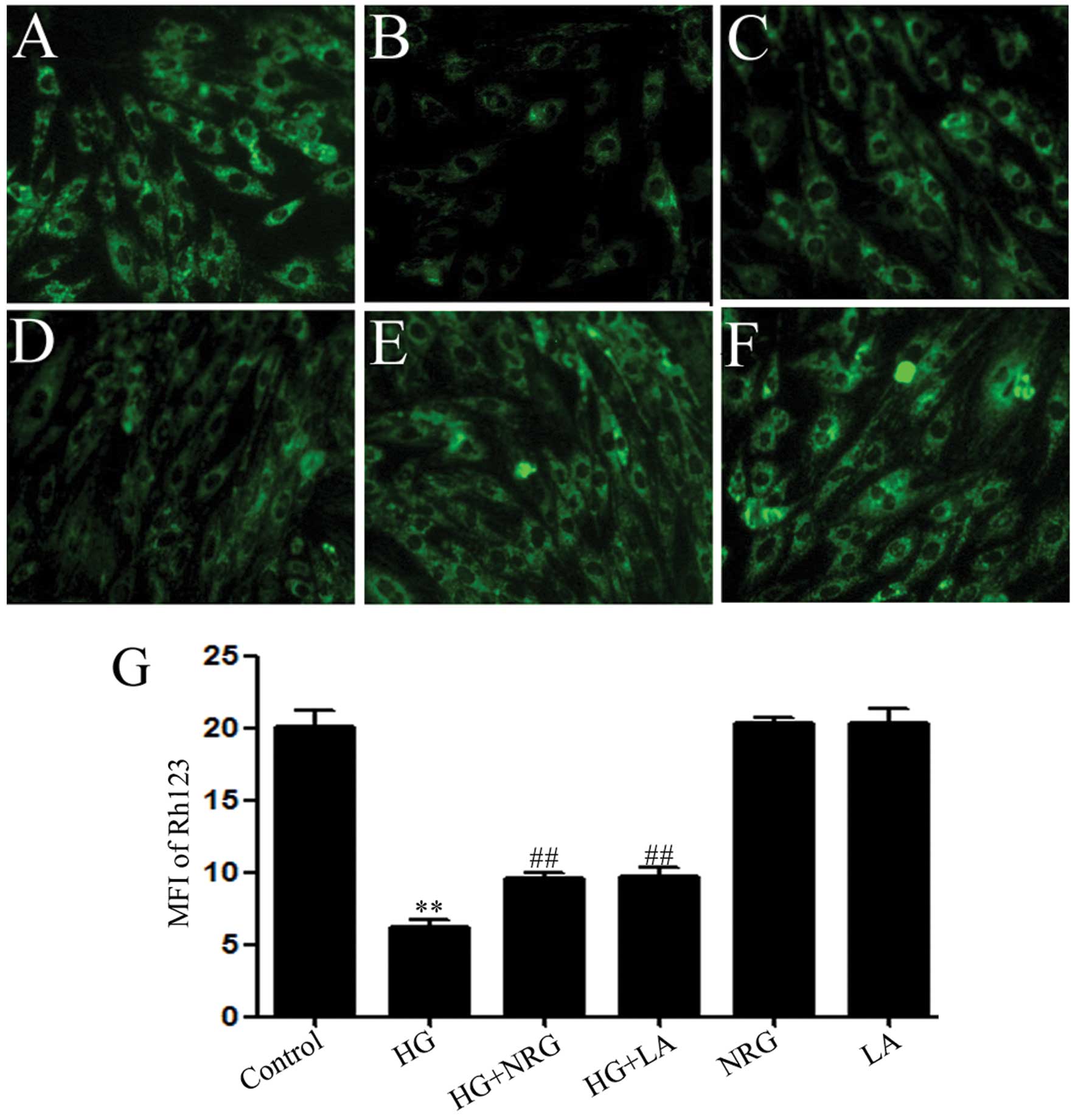
Naringin and LA block the HG-induced
dissipation of MMP in H9c2 cells
Since mitochondrial damage has been shown to
contribute to HG-induced cardiomyocyte injury (37,38), we investigated whether naringin
can prevent the loss of MMP in HG-treated H9c2 cells. Our results
revealed that the exposure of the cells to HG for 24 h induced
mitochondrial damage, as indicated by the dissipation of MMP
(Fig. 7B and G). Of note, the
dissipation of MMP was attenuated by treatment of the cells with 80
μmol/l naringin for 2 h prior to exposure to HG (Fig. 7C and G), indicating that naringin
protected the H9c2 cells against HG-induced mitochondrial damage.
Similarly, treatment of the cells with 50 ng/ml LA for 24 h prior
to exposure to HG, attenuated the HG-induced dissipation of MMP,
suggesting the involvement of leptin in the HG-induced dissipation
of MMP (Fig. 7D and G).
Discussion
Hyperglycemia, an important feature of diabetes
mellitus (DM), is considered to be the most important factor
leading to almost all complications associated with chronic
diabetes, including diabetic cardiomyopathy (39,40). However, the mechanisms underlying
the deteriorative effects of hyperglycemia on cardiomyocytes are
not yet fully understood. A number of factors, such as ROS
generation (35,36) mitochondrial insult (37,38) and the activation of MAPKs
(12,41–43), have been shown to participate in
hyperglycemia-induced injury. Majumdar et al (22) reported that the exposure of
neonatal rat cardiomyocytes to 25 mmol/l glucose induced increased
mRNA and protein expression levels of both leptin and endothelin-l
(ET-l), which are involved in HG-induced cardiomyocyte hypertrophy.
Consistent with these findings (22), our results demonstrated that the
exposure of H9c2 cardiac cells to HG (35 mmol/l glucose) markedly
upregulated the expression levels of both leptin and leptin
receptor. Since leptin has been shown to participate in HG-induced
cardiomyocyte hypertrophy (22),
we investigated whether leptin conributes to other injuries induced
by HG. The findings of this study demonstrated that the exposure of
the cells to HG induced marked changes, as shown by the decrease in
cell viability, the increase in the apoptotic cell number and ROS
generation, as well as in the dissipation of MMP. However, these
injuries induced by HG were significantly attenuated by treatment
with LA prior to exposure to HG, suggesting that leptin is involved
in HG-induced cytotoxicity, apoptosis, ROS generation and
mitochondrial damage. Our data confirm the data from a previous
study (22), and provide novel
evidence of the role of leptin in hyperglycemia-induced
cardiomyocyte injury.
Additionally, we observed that the exposure of the
cells to HG, induced an increase in p-p38 MAPK expression levels,
which is in accordance with the findings of previous studies
(12,41–43). Importantly, the high expression
levels of p-p38 MAPK were decreased by treatment with LA prior to
exposure to HG, indicating that leptin acts upstream of p38 MAPK
and contributes to the HG-induced activation of the p38 MAPK
pathway in H9c2 cells. It is known that p38 MAPK is activated by a
diverse range of physical and chemical stresses, such as
hypoxia/ischemia (44,45), drugs (46,47) and oxidative stress (48). To the best of our knowledge, only
a few studies to date have addressed the role of p38 MAPK in leptin
signaling in different cell types (30–33,49); for example, leptin has been shown
to induce hypertrophy through he activation of p38 MAPK in rat
vascular smooth muscle cells (31). However, it remains unclear as to
whether the leptin-induced activation of the p38 MAPK pathway is
involved in HG-induced cardiomyocyte injury. Recently, we (12), as well as others [Xu et al
(42)] demonstrated the
contribution of p38 MAPK activation to HG-induced injury in H9c2
cardiac cells. Thus, the data from the current study (Figs. 3–7), as well as those from previous
studies (12,42), provide evidence that the
leptin-induced activation of the p38 MAPK pathway may be an
important mechanism responsible for HG-induced injury in H9c2
cardiac cells.
Naringin, a citrus flavonone, has been shown to
exert protective effects against hyperglycemia-induced cardiac
injury in DM and has attracted considerable attention due to its
broad spectrum of pharmacological and therapeutic properties
(1–16), including its antihyperglycemic
(13,14), anti-apoptotic (9–12)
and cardioprotective (12,15,16)
effects. In a recent study, we demonstrated that naringin protects
cardiomyocytes against HG-induced injury by modulating the
activation of the p38 MAPK pathway (12). However, the mechanisms underlying
the cardioprotective effects of naringin, including the effects of
naringin on the leptin-induced activation of the p38 MAPK pathway
remain unclear. Thus, in the present study, we explored the effects
of naringin on the leptin-induced activation of the p38 MAPK
pathway in H9c2 cardiac cells exposed to HG. Our results
demonstrated that naringin markedly attenuated the HG-induced
increase in the expression levels of both leptin and leptin
receptors, indicating the inhibitory effects of naringin on the
increase in leptin expression induced by HG. Since we have
previously demonstrated that naringin inhibits the HG-induced
activation of the p38 MAPK pathway (12), the findings of the current study
support the notion that naringin protects H9c2 cardiac cells
against HG-induced injury, at least in part through the inhibition
of the leptin-induced activation of the p38 MAPK pathway. This is
clearly supported by the following evidence: i) the inhibitory
effects of naringin on the increased expression of both leptin and
leptin receptor induced by HG; ii) the inhibitory effects of LA on
the HG-induced activation of p38 MAPK; iii) the inhibitory effects
of naringin on the HG-induced activation of p38 MAPK (12); iv) the protective effects of both
naringin and LA against HG-induced cardiomyocyte injury, leading to
an increase in cell viability, as well as a decrease in the number
of apoptotic cells, ROS generation, and in the attenuation of the
loss of MMP; v) the inhibitory effects of SB203580, an inhibitor of
p38 MAPK, on HG-induced injury (12,42).
In conclusion, this study provides the first
evidence that the leptin-induced activation of the p38 MAPK pathway
contributes to HG-induced cardiomyocyte injury, including
cytotoxicity, apoptosis, oxidative stress and mitochondrial damage.
The understanding of the role of such a pathway is important, as it
may lead to the development of novel treatment strategies for
diabetic cardiomyopathy. In addition, we provide novel evidence
that the inhibition of the leptin-induced activation of the p38
MAPK pathway is involved in the cardioprotective effects of
naringin against hyperglycemia-induced cardiomyocyte injury. These
findings provider a deeper understanding of the mechanisms
responsible for the cytoprotective effects of naringin against
cardiovascular complications associated with diabetes and its
pharmacological and therapeutic properties.
Acknowledgements
This study was supported by grants from the Science
and Technology Planning Project of Guangdong in China (no.
2012A080202020) and the Guangdong Natural Science Foundation (no.
S2011010002620).
References
|
1
|
Mahmoud AM, Ashour MB, Abdel-Moneim A and
Ahmed OM: Hesperidin and naringin attenuate hyperglycemia-mediated
oxidative stress and proinflammatory cytokine production in high
fat fed/streptozotocin-induced type 2 diabetic rats. J Diabetes
Complications. 26:483–490. 2012. View Article : Google Scholar
|
|
2
|
Rajadurai M and Prince PS: Preventive
effect of naringin on isoproterenol-induced cardiotoxicity in
Wistar rats: an in vivo and in vitro study. Toxicology.
232:216–225. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jeon SM, Park YB and Choi MS:
Antihypercholesterolemic property of naringin alters plasma and
tissue lipids, cholesterol-regulating enzymes, fecal sterol and
tissue morphology in rabbits. Clin Nutr. 23:1025–1034. 2004.
View Article : Google Scholar
|
|
4
|
Jain M and Parmar HS: Evaluation of
antioxidative and anti-inflammatory potential of hesperidin and
naringin on the rat air pouch model of inflammation. Inflamm Res.
60:483–491. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bodas R, Prieto N, López-Campos O,
Giráldez FJ and Andrés S: Naringin and vitamin E influence the
oxidative stability and lipid profile of plasma in lambs fed fish
oil. Res Vet Sci. 91:98–102. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jung UJ, Kim HJ, Lee JS, et al: Naringin
supplementation lowers plasma lipids and enhances erythrocyte
antioxidant enzyme activities in hypercholesterolemic subjects.
Clin Nutr. 22:561–568. 2003. View Article : Google Scholar
|
|
7
|
Kim HJ, Oh GT, Park YB, Lee MK, Seo HJ and
Choi MS: Naringin alters the cholesterol biosynthesis and
antioxidant enzyme activities in LDL receptor-knockout mice under
cholesterol fed condition. Life Sci. 74:1621–1634. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Nie YC, Wu H, Li PB, et al:
Anti-inflammatory effects of naringin in chronic pulmonary
neutrophilic inflammation in cigarette smoke-exposed rats. J Med
Food. 15:894–900. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jagetia GC, Venkatesha VA and Reddy TK:
Naringin, a citrus flavonone, protects against radiation-induced
chromosome damage in mouse bone marrow. Mutagenesis. 18:337–343.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kanno S, Shouji A, Asou K and Ishikawa M:
Effects of naringin on hydrogen peroxide-induced cytotoxicity and
apoptosis in P388 cells. J Pharmacol Sci. 92:166–170. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kanno S, Shouji A, Hirata R, Asou K and
Ishikawa M: Effects of naringin on cytosine arabinoside
(Ara-C)-induced cytotoxicity and apoptosis in P388 cells. Life Sci.
75:353–365. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Huang H and Wu K, You Q, Huang R, Li S and
Wu K: Naringin inhibits high glucose-induced cardiomyocyte
apoptosis by attenuating mitochondrial dysfunction and modulating
the activation of the p38 signaling pathway. Int J Mol Med.
32:396–402. 2013.
|
|
13
|
Adel AM, Mohamed BA, Ayman MM and Osama
MA: Insulin sensitizing effects of hesperidin and naringin in
experimental kmodel of induced type 2 diabetes in rats: focus on
tumor necrosis factor-alpha and resistin. Nat Sci. 7:134–141.
2011.
|
|
14
|
Osama MA, Ayman MM, Adel AM and Mohamed
BA: Antihyperglycemic and antihyperlipidemic effects of hesperidin
and naringin in high fat diet/streptozotocin type 2 diabetic rats.
Life Sci J. 8:91–101. 2011.
|
|
15
|
Rajadurai M and Prince PS: Preventive
effect of naringin on cardiac mitochondrial enzymes during
isoproterenol-induced myocardial infarction in rats: a transmission
electron microscopic study. J Biochem Mol Toxicol. 21:354–361.
2007. View Article : Google Scholar
|
|
16
|
Rajadurai M and Prince PS: Naringin
ameliorates mitochondrial lipid peroxides, antioxidants and lipids
in isoproterenol-induced myocardial infarction in Wistar rats.
Phytother Res. 23:358–362. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tartaglia LA: The leptin receptor. J Biol
Chem. 272:6093–6096. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Murad A, Nath AK, Cha ST, Demir E,
Flores-Riveros J and Sierra-Honigmann MR: Leptin is an
autocrine/paracrine regulator of wound healing. FASEB J.
17:1895–1897. 2003.PubMed/NCBI
|
|
19
|
La Cava A, Alviggi C and Matarese G:
Unraveling the multiple roles of leptin in inflammation and
autoimmunity. J Mol Med (Berl). 82:4–11. 2004.PubMed/NCBI
|
|
20
|
Ren J: Leptin and hyperleptinemia - from
friend to foe for cardiovascular function. J Endocrinol. 181:1–10.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Figenschau Y, Knutsen G, Shahazeydi S,
Johansen O and Sveinbjörnsson B: Human articular chondrocytes
express functional leptin receptors. Biochem Biophys Res Commun.
287:190–197. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Majumdar P, Chen S, George B, Sen S,
Karmazyn M and Chakrabarti S: Leptin and endothelin-1 mediated
increased extracellular matrix protein production and cardiomyocyte
hypertrophy in diabetic heart disease. Diabetes Metab Res Rev.
25:452–463. 2009. View
Article : Google Scholar
|
|
23
|
Wallace AM, McMahon AD, Packard CJ, et al:
Plasma leptin and the risk of cardiovascular disease in the west of
Scotland coronary prevention study (WOSCOPS). Circulation.
104:3052–3056. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hoggard N, Mercer JG, Rayner DV, Moar K,
Trayhurn P and Williams LM: Localization of leptin receptor mRNA
splice variants in murine peripheral tissues by RT-PCR and in situ
hybridization. Biochem Biophys Res Commun. 232:383–387. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kieffer TJ, Heller RS and Habener JF:
Leptin receptors expressed on pancreatic beta-cells. Biochem
Biophys Res Commun. 224:522–537. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lin J, Barb CR, Matteri RL, Kraeling RR,
Chen X, Meinersmann RJ and Rampacek GB: Long form leptin receptor
mRNA expression in the brain, pituitary, and other tissues in the
pig. Domest Anim Endocrinol. 19:53–61. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bjørbaek C, Uotani S, da Silva B and Flier
JS: Divergent signaling capacities of the long and short isoforms
of the leptin receptor. J Biol Chem. 272:32686–32695.
1997.PubMed/NCBI
|
|
28
|
Bjørbaek C, Buchholz RM, Davis SM, et al:
Divergent roles of SHP-2 in ERK activation by leptin receptors. J
Biol Chem. 276:4747–4755. 2001.PubMed/NCBI
|
|
29
|
Bouloumie A, Marumo T, Lafontan M and
Busse R: Leptin induces oxidative stress in human endothelial
cells. FASEB J. 13:1231–1238. 1999.PubMed/NCBI
|
|
30
|
van den Brink GR, O’Toole T, Hardwick JC,
van den Boogaardt DE, Versteeg HH, van Deventer SJ and
Peppelenbosch MP: Leptin signaling in human peripheral blood
mononuclear cells, activation of p38 and p42/44 mitogen-activated
protein (MAP) kinase and p70 S6 kinase. Mol Cell Biol Res Commun.
4:144–150. 2000.PubMed/NCBI
|
|
31
|
Shin HJ, Oh J, Kang SM, et al: Leptin
induces hypertrophy via p38 mitogen-activated protein kinase in rat
vascular smooth muscle cells. Biochem Biophys Res Commun.
329:18–24. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Rajapurohitam V, Gan XT, Kirshenbaum LA
and Karmazyn M: The obesity-associated peptide leptin induces
hypertrophy in neonatal rat ventricular myocytes. Circ Res.
93:277–279. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zeidan A, Javadov S, Chakrabarti S and
Karmazyn M: Leptin-induced cardiomyocyte hypertrophy involves
selective caveolae and RhoA/ROCK-dependent p38 MAPK translocation
to nuclei. Cardiovasc Res. 77:64–72. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Han DC, Isono M, Chen S, Casaretto A, Hong
SW, Wolf G and Ziyadeh FN: Leptin stimulates type I collagen
production in db/db mesangial cells: glucose uptake and TGF-beta
type II receptor expression. Kidney Int. 59:1315–1323. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Peake BF, Nicholson CK, Lambert JP, Hood
RL, Amin H, Amin S and Calvert JW: Hydrogen sulfide preconditions
the db/db diabetic mouse heart against ischemia-reperfusion injury
by activating Nrf2 signaling in an Erk-dependent manner. Am J
Physiol Heart Circ Physiol. 304:H1215–H1224. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ganesan K, Gani SB and Arunachalam GM:
Effect of Helicteres isora bark extracts on heart
antioxidant status and lipid peroxidation in streptozotocin
diabetic rats. J Appl Biomed. 6:89–95. 2008.
|
|
37
|
Boudina S, Sena S, Theobald H, et al:
Mitochondrial energetics in the heart in obesity-related diabetes:
direct evidence for increased uncoupled respiration and activation
of uncoupling proteins. Diabetes. 56:2457–2466. 2007. View Article : Google Scholar
|
|
38
|
Ceriello A: Cardiovascular effects of
acute hyperglycaemia: pathophysiological underpinnings. Diab Vasc
Dis Res. 5:260–268. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Brownlee M: Biochemistry and molecular
cell biology of diabetic complications. Nature. 414:813–820. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ren J and Davidoff AJ: Diabetes rapidly
induces contractile dysfunctions in isolated ventricular myocytes.
Am J Physiol. 272:H148–H158. 1997.PubMed/NCBI
|
|
41
|
Soetikno V, Sari FR, Sukumaran V, et al:
Curcumin prevents diabetic cardiomyopathy in streptozotocin-induced
diabetic rats: possible involvement of PKC-MAPK signaling pathway.
Eur J Pharm Sci. 47:604–614. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Xu W, Wu W, Chen J, Guo R, Lin J, Liao X
and Feng J: Exogenous hydrogen sulfide protects H9c2 cardiac cells
against high glucose-induced injury by inhibiting the activities of
the p38 MAPK and ERK1/2 pathways. Int J Mol Med. 32:917–925.
2013.PubMed/NCBI
|
|
43
|
Yan J, Young ME, Cui L, Lopaschuk GD, Liao
R and Tian R: Increased glucose uptake and oxidation in mouse
hearts prevent high fatty acid oxidation but cause cardiac
dysfunction in diet-induced obesity. Circulation. 119:2818–2828.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Liu AL, Wang XW, Liu AH, Su XW, Jiang WJ,
Qiu PX and Yan GM: JNK and p38 were involved in hypoxia and
reoxygenation-induced apoptosis of cultured rat cerebellar granule
neurons. Exp Toxicol Pathol. 61:137–143. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Lan A, Liao X, Mo L, et al: Hydrogen
sulfide protects against chemical hypoxia-induced injury by
inhibiting ROS-activated ERK1/2 and p38MAPK signaling pathways in
PC12 cells. PLoS One. 6:e259212011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Guo R, Lin J, Xu W, Shen N, Mo L, Zhang C
and Feng J: Hydrogen sulfide attenuates doxorubicin-induced
cardiotoxicity by inhibition of the p38 MAPK pathway in H9c2 cells.
Int J Mol Med. 31:644–650. 2013.PubMed/NCBI
|
|
47
|
Guo RM, Xu WM, Lin JC, et al: Activation
of the p38 MAPK/NF-κB pathway contributes to doxorubicin-induced
inflammation and cytotoxicity in H9c2 cardiac cells. Mol Med Rep.
8:603–608. 2013.
|
|
48
|
Chen L, Liu L, Yin J, Luo Y and Huang S:
Hydrogen peroxide-induced neuronal apoptosis is associated with
inhibition of protein phosphatase 2A and 5, leading to activation
of MAPK pathway. Int J Biochem Cell Biol. 41:1284–1295. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Tajmir P, Ceddia RB, Li RK, Coe IR and
Sweeney G: Leptin increases cardiomyocyte hyperplasia via
extracellular signal-regulated kinase- and phosphatidylinositol
3-kinase-dependent signaling pathways. Endocrinology.
145:1550–1555. 2004. View Article : Google Scholar
|