Introduction
Gastric carcinoma remains one of the most common and
lethal malignancies worldwide. With approximately one million cases
diagnosed annually, gastric cancer is one of the leading causes of
cancer-related mortality worldwide (1). Helicobacter pylori (H.
pylori) has been defined as a class I carcinogen by the World
Health Organization (2), and its
persistent colonization in the stomach significantly increases the
risk of gastric adenocarcinoma (3). During the course of H. pylori
infection, bacterial virulence factors, such as
cytotoxin-associated gene A (CagA), interact with numerous
signaling molecules and elicit a series of cellular events; one of
which is the H. pylori-induced activation of
mitogen-activated protein kinases (MAPK) which has been implicated
in the development of gastric adenocarcinoma (4,5).
MAPK consists of a family of ubiquitously expressed
proteins including extracellular signal-regulated kinases 1 and 2
(ERK1/2), p38 and c-Jun/SAPK N-terminal kinases (JNK) (6,7).
In addition to activating MAPK, H. pylori infection also
induces the expression of other inflammatory responsive genes
including nuclear factor-κB (NF-κB) (8), cyclooxygenase-2 (COX-2) (9) and aquaporin 3 (AQP3) (10). H. pylori has been reported
to stimulate the proliferation of gastric epithelial cells both
in vitro and in vivo (5,11,12) and to alter intercellular tight
junctions (13), thus
facilitating the malignant transformation of the gastric
mucosa.
Protein phosphatase 2A (PP2A) facilitates the
proteolytic degradation of oncoprotein Myc and prevents malignant
cell growth (14). Cancerous
inhibitor of protein phosphatase 2A (CIP2A) is a recently
identified human oncoprotein that stabilizes c-Myc protein by
inhibiting its degradation. CIP2A has been reported to promote the
proliferation of various types of cancer cells (15). Moreover, CIP2A is required for
malignant cell growth and the malignant transformation of human
cells (15). Overexpression of
CIP2A has been observed in many types of solid tumors such as head
and neck squamous cell carcinoma and gastric cancer (15,16). In human gastric cancer, a higher
level of CIP2A was noted in cancerous tissues when compared to the
level in non-cancerous mucosa, suggesting that CIP2A plays an
oncogenic role in the development of human gastric cancer (16,17). Previous research has shown that
JNK2 is a key regulator of CIP2A expression (18).
Persistent H. pylori infection is a
well-recognized condition that is closely linked to the development
of gastric cancer. It has been shown that H. pylori
infection upregulates the expression of CIP2A in gastric cancer
cells (13). CagA, a major
virulent factor of H. pylori, activates the JNK pathway and
is thus involved in promoting gastric cancer progression (4,19).
In addition, JNK2 was found to regulate CIP2A expression via
activation of transcription factor 2 (ATF2) (18). However, it is not clear whether
JNK2 is involved in the regulation of CIP2A expression following
H. pylori infection.
In the present study, we aimed to examine the effect
of H. pylori infection on JNK2 and CIP2A and the subsequent
biological impact on gastric cancer cells. We found that H.
pylori increased CIP2A expression and promoted cell
proliferation via the JNK2/ATF2 signaling pathway in MKN-45 cells.
Possible implications of CIP2A and its interaction with JNK
signaling in gastric cancer are discussed.
Materials and methods
Cell culture, RNA interference (RNAi) and
reagents
Human gastric cancer cell line MKN-45 (Chinese
Academy of Sciences, Shanghai, China) was cultured in RPMI-1640
(HyClone Laboratories, Inc., Logan, UT, USA) supplemented with 10%
fetal bovine serum (FBS) (HyClone Laboratories, Inc.) and 1%
penicillin and streptomycin (North China Pharmaceutical Co., Inc.,
Shijiazhuang, China) in a humidified atmosphere containing 5%
CO2 at 37°C. The specific siRNA against CIP2A and the
scrambled control siRNA were purchased from Invitrogen (Carlsbad,
CA, USA). The siRNA sequence for CIP2A was
5′-GACAACUGUCAAGUGUACCACU CUU-3′ (16). Lipofectamine™ 2000 was used to
transfect the siRNA sequences into MKN-45 cells according to the
manufacturer’s instructions (Invitrogen). SP600125, a JNK2
inhibitor, was purchased from InvivoGen (San Diego, CA, USA).
H. pylori culture and co-culture with
MKN-45 cells
The H. pylori strains used in this study were
CagA and VacA positive standard strain (NCTC11637) (Institute of
Pathogenic Biology, School of Basic Medical Science, Lanzhou
University, Lanzhou, China). H. pylori were cultured on
Columbia agar plates (Difco Laboratories, Detroit, MI, USA)
containing 5% sheep blood at 37°C for 72 h under microaerophilic
conditions using an anaerobic box (Mitsubishi Gas Chemical Co.,
Inc., Tokyo, Japan). The bacteria were harvested and resuspended in
antibiotic-free RPMI-1640 medium supplemented with 10% FBS. The
density of the bacteria was adjusted by measuring the optical
density (OD) at 600 nm, in which one unit of OD600
corresponds to 1×108 colony-forming units (CFU)/ml. For
the cell and bacterial co-culture, MKN-45 cells were cultured in
RPMI-1640 medium in the presence of H. pylori at a
cell-to-bacterium ratio of 1:100.
Quantitative real-time reverse
transcription-polymerase chain reaction (qRT-PCR) assay
Total RNA from the cells was prepared using RNAiso
Plus reagent (Takara Biotechnology Co., Dalian, China) under an
RNase-free condition, and cDNA was synthesized using the
Primescript™ Reverse Transcription (RT) Master Mix (Takara
Biotechnology Co.) according to the manufacturer’s protocols.
Reverse transcription reaction was performed at 37°C for 15 min
followed by 85°C for 5 sec. qRT-PCR amplifications were performed
with the Applied Biosystems 7500/7500 Fast Real-Time PCR Software
(Applied Biosystems) using the SYBR® Premix Ex Taq™ II
(Takara Biotechnology Co.). Reactions were carried out in a 20-μl
reaction volume following the manufacturer’s instructions. The
thermal cycle conditions were as follows: 95°C for 30 sec, followed
by 40 cycles of 95°C for 5 sec and 60°C for 34 sec. GAPDH was used
as an internal control. The primer sequences were: CIP2A sense,
5′-GGCACTTGGAGGTAATTTCT-3′ and antisense,
5′-CTGGTTTCAATGTCTACTGCTAG-3′ (GenBank accession no. NM020890);
GAPDH sense, 5′-AAG GCTGGGGCTCATTTG-3′ and antisense, 5′-AGGAGGCA
TTGCTGATGATC-3′ (GenBank accession no. NM002046). All primers were
synthesized by Takara Biotechnology Co. The mRNA expression level
was expressed as relative expression to the basal level without
H. pylori stimulation. Data were analyzed according to the
comparative Ct method.
Western blot assay
Treated cells were washed with ice-cold
phosphate-buffered saline (PBS) and then lysed using RIPA lysis
buffer (Beyotime Biotechnology, Haimen, China) supplemented with 1
mmol/l phenylmethanesulfonyl fluoride (PMSF). After centrifugation
at 12,000 rpm for 15 min at 4°C, the supernatant was collected and
the protein concentration was measured by the BCA protein assay
(Beyotime Biotechnology). An equal amount of protein (50 μg) from
each sample was separated on a 10% SDS polyacrylamide gel and
transferred onto polyvinylidene fluoride (PVDF) membranes. The
membranes were blocked with 5% nonfat dry milk in Tris-buffered
saline containing 0.1% Tween-20 (TBST) for 2 h at room temperature
and incubated with the respective primary antibodies overnight at
4°C. The following primary antibodies were used for the procedure
(all diluted at 1:1,000 in TBST): anti-CIP2A (2G10-3B5) (Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA), anti-phospho-ATF2 and
anti-ATF2 (Cell Signaling Technology, Inc., Danvers, MA, USA),
anti-phospho-JNK2 (G-7) and anti-JNK2 (G-7) (Santa Cruz
Biotechnology, Inc.) and anti-β-actin (Zhongshan Golden Bridge
Biotech, Beijing, China). β-actin was used as a loading control.
After overnight incubation, the membranes were washed three times
with TBST, and then incubated with the horseradish
peroxidase-conjugated secondary antibodies (Zhongshan Golden Bridge
Biotech) (1:10,000) at room temperature for 1 h. The signals were
detected using the SuperSignal West Pico Chemiluminescent Substrate
(Thermo Fisher Scientific, Inc., Rockford, IL, USA) and imaged
using a VersaDoc Imaging System (Bio-Rad Laboratories Co., Ltd.
Hercules, CA, USA). Data obtained from the western blotting
experiments were analyzed by Bio-Rad Quantity One Software v4.62
(Bio-Rad Laboratories Co., Ltd.).
MTT assay
Cell proliferation was measured using the
dimethylthiazolyl-2,5-diphenyltetrazolium bromide (MTT) assay.
Cells were plated at a density of 5,000 cells/well in 96-well
plates in 200 μl antibiotic-free culture medium and cultured for 12
h. After the appropriate treatment, MTT reagent was added (20
μl/well of 5 g/l solution in PBS) into each well. Subsequently,
incubation was carried out at 37°C for 4 h and then washing with
PBS. Next, 200 μl dimethyl sulfoxide (DMSO) was added to each well.
The absorbance was measured at 490 nm. All samples were assayed
repeatedly in 6 wells. The cell proliferation rate was quantified
as the fraction of cells surviving relative to the untreated
controls.
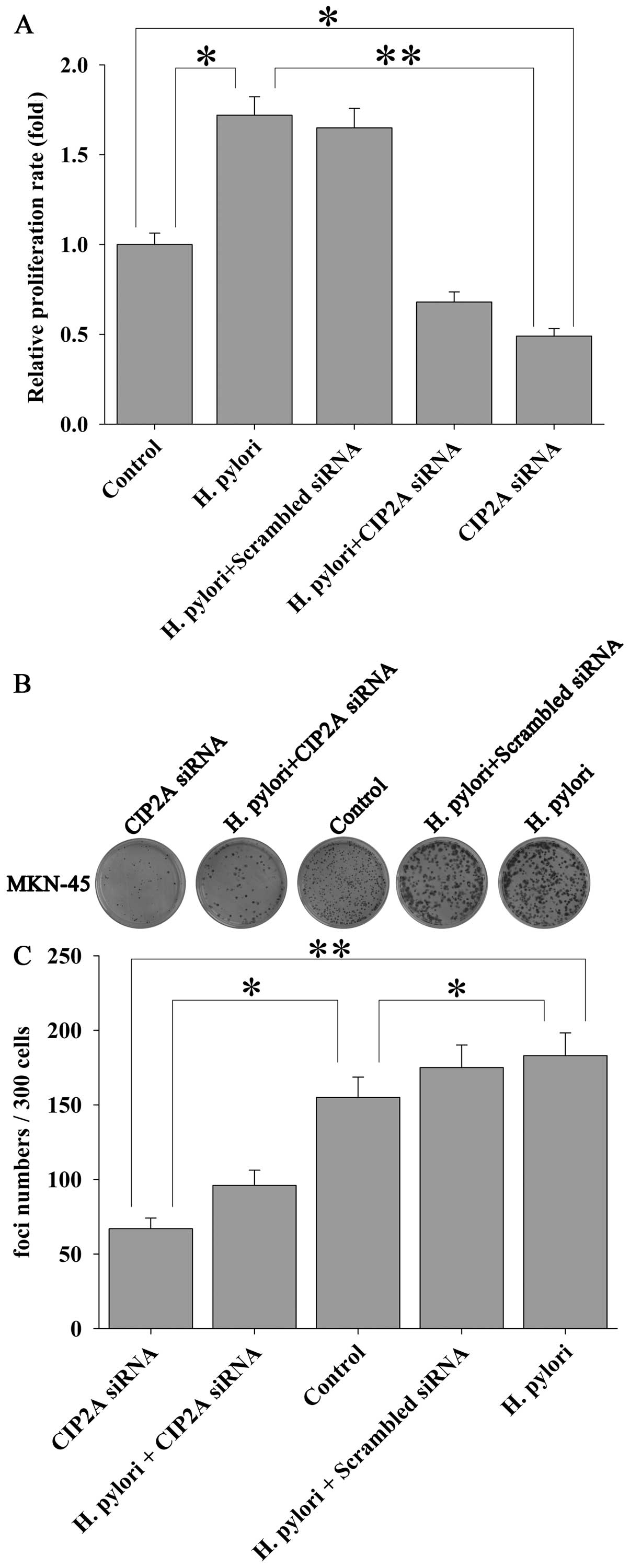
Colony formation assay
Anchorage-independent growth was measured by colony
formation assay. MKN-45 cells were transfected with siRNA against
CIP2A or scrambled siRNA for 72 h. The transfected cells were
infected with or without H. pylori for 3 h and planted into
6-well plates at low density (300 cells/well). After 14 days, cells
were fixed and stained with crystal violet. Foci and colonies
containing >50 cells were counted using a microscope.
Statistical analysis
All experiments were repeated three times, and data
are expressed as mean ± standard deviation (SD). Statistical
significance was determined with one-way ANOVA followed by the
Bonferroni correction. All statistical analyses were performed
using SPSS 19.0 (IBM, Armonk, NY, USA), and data values were
displayed using SigmaPlot 10.0 (Systat Software Inc., Chicago, IL,
USA). A P-value of <0.05 was considered to indicate a
statistically significant result.
Results
H. pylori upregulates CIP2A expression in
MKN-45 cells
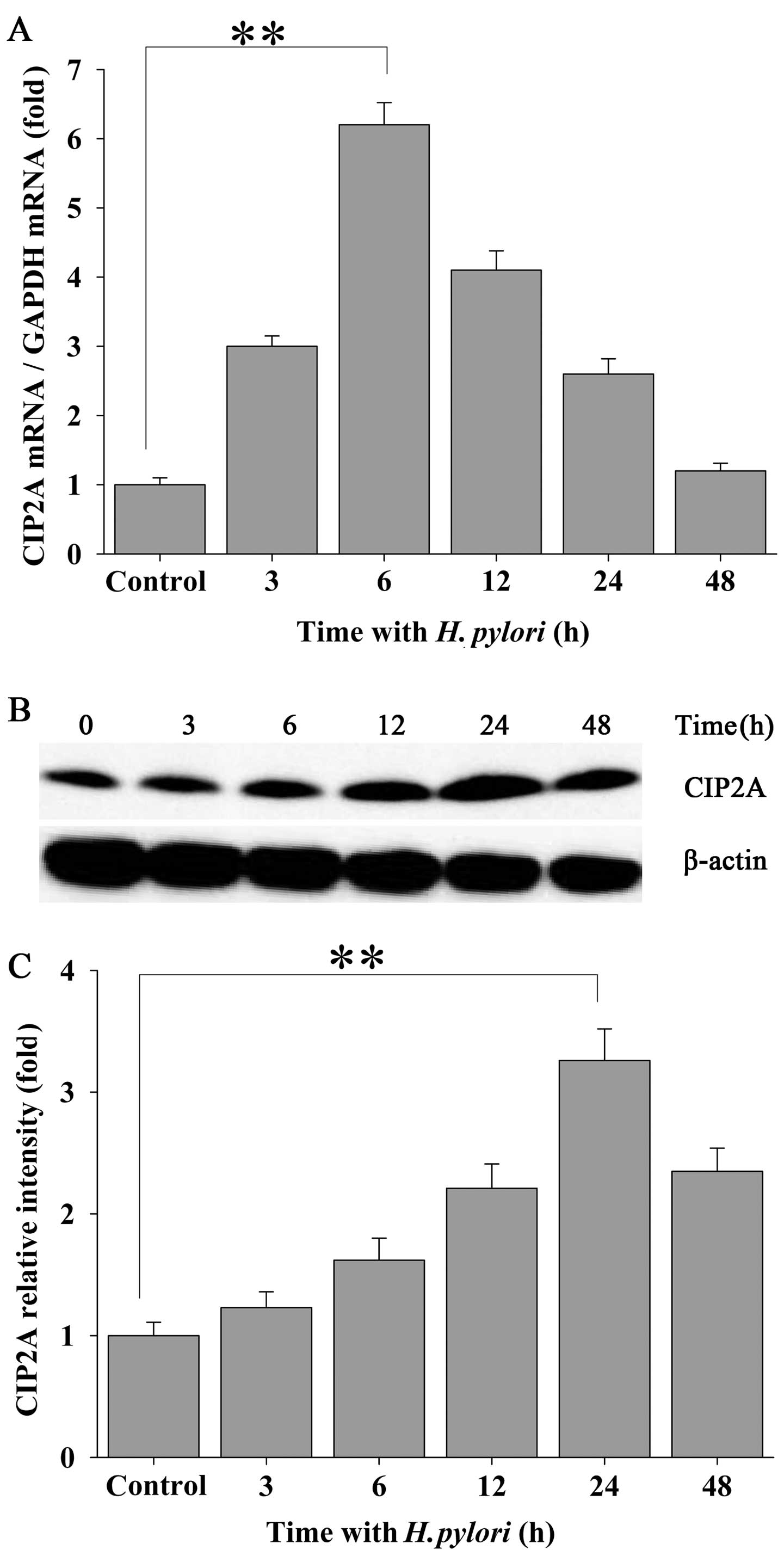
To investigate the effects of H. pylori
infection on CIP2A expression in human gastric carcinoma cells,
confluent MKN-45 cells were incubated with H. pylori at a
density of 100 bacteria per cell for 0, 3, 6, 12, 24 and 48 h.
qRT-PCR assays were performed to examine the mRNA transcript levels
of CIP2A in the MKN-45 cells. Treatment of the MKN-45 cells with
H. pylori led to a significantly elevated expression of
CIP2A at the mRNA level which peaked at 6 h of incubation and
declined thereafter while still maintaining an elevated level
(Fig. 1A). Increased expression
of CIP2A was also confirmed at the protein level by western blot
analysis (Fig. 1B and C), and
this level peaked at 24 h following bacterial infection.
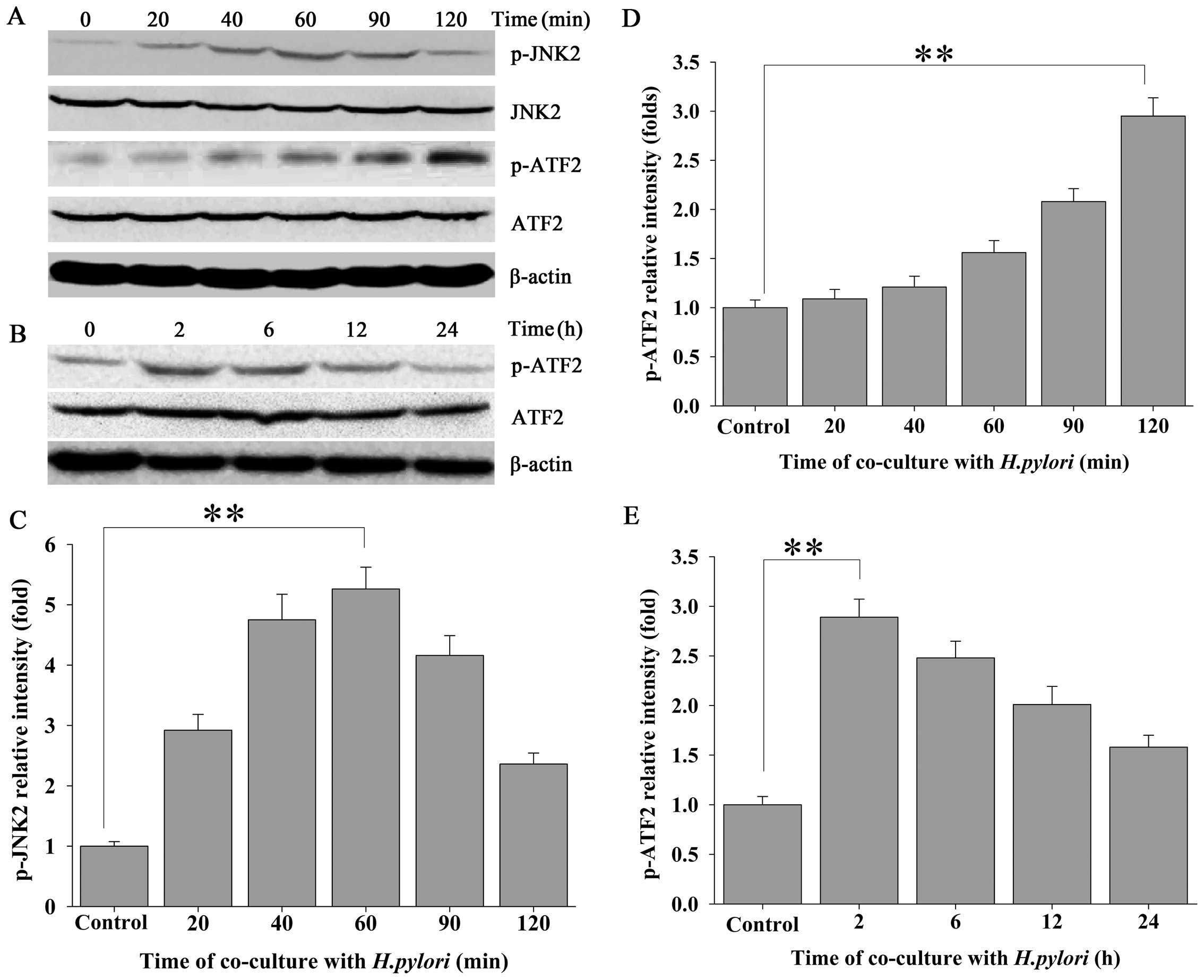
The JNK2 signaling pathway is involved in
H. pylori-regulated CIP2A expression in MKN-45 cells
To ascertain whether JNK2 signaling is involved in
the H. pylori-induced expression of CIP2A, the expression of
phosphorylated JNK2 (p-JNK2) in response to H. pylori
infection was evaluated by western blot analysis using a
phospho-specific antibody against JNK2. Approximately 20 min after
MKN-45 cells were challenged with H. pylori, a steady
increase in the expression of p-JNK2 was noted, which exhibited an
approximate time-dependent manner, whereas total JNK2 remained
unchanged (Fig. 2A and C). To
further confirm the H. pylori-induced activation of the JNK2
pathway in MKN-45 cells, we evaluated the expression of
phospho-ATF2 (p-ATF2), a substrate for p-JNK2. Activation of the
JNK2 pathway was followed by a transient increase in the expression
of p-ATF2 in the H. pylori-treated MKN-45 cells (Fig. 2A, B, D and E).
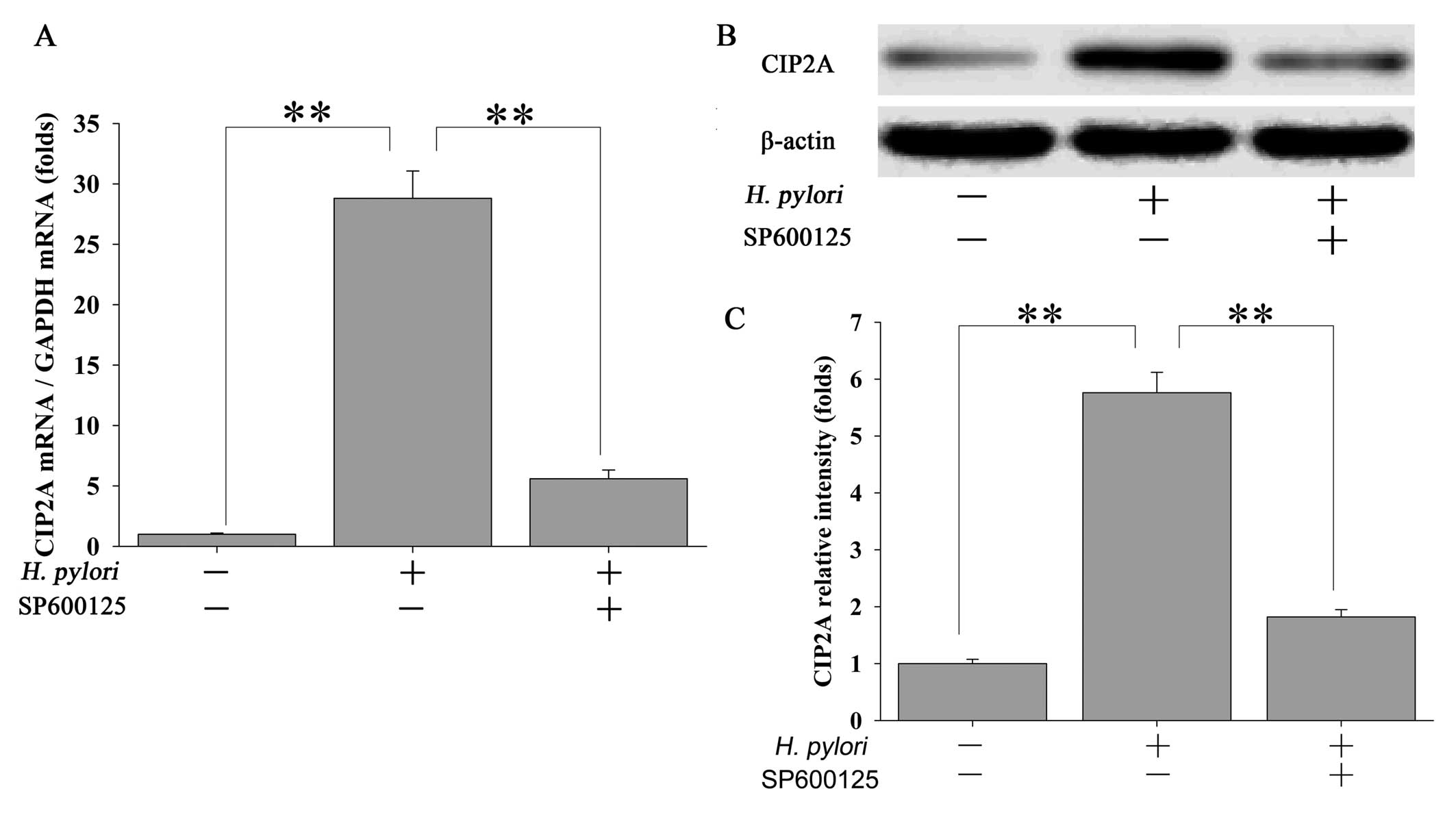
Inhibition of JNK2 attenuates H.
pylori-stimulated CIP2A expression in MKN-45 cells
To further ascertain whether the H.
pylori-induced upregulation of CIP2A is mediated through the
JNK2 pathway, MKN-45 cells were pretreated with 20 μM of the JNK2
inhibitor SP600125 for 2 h followed by treatment with H.
pylori. Inhibition of JNK2 signaling by SP600125 led to a
significant but transient reduction in CIP2A mRNA in response to
H. pylori infection (Fig.
3A). These changes were also reflected at the protein level
(Fig. 3B and C).
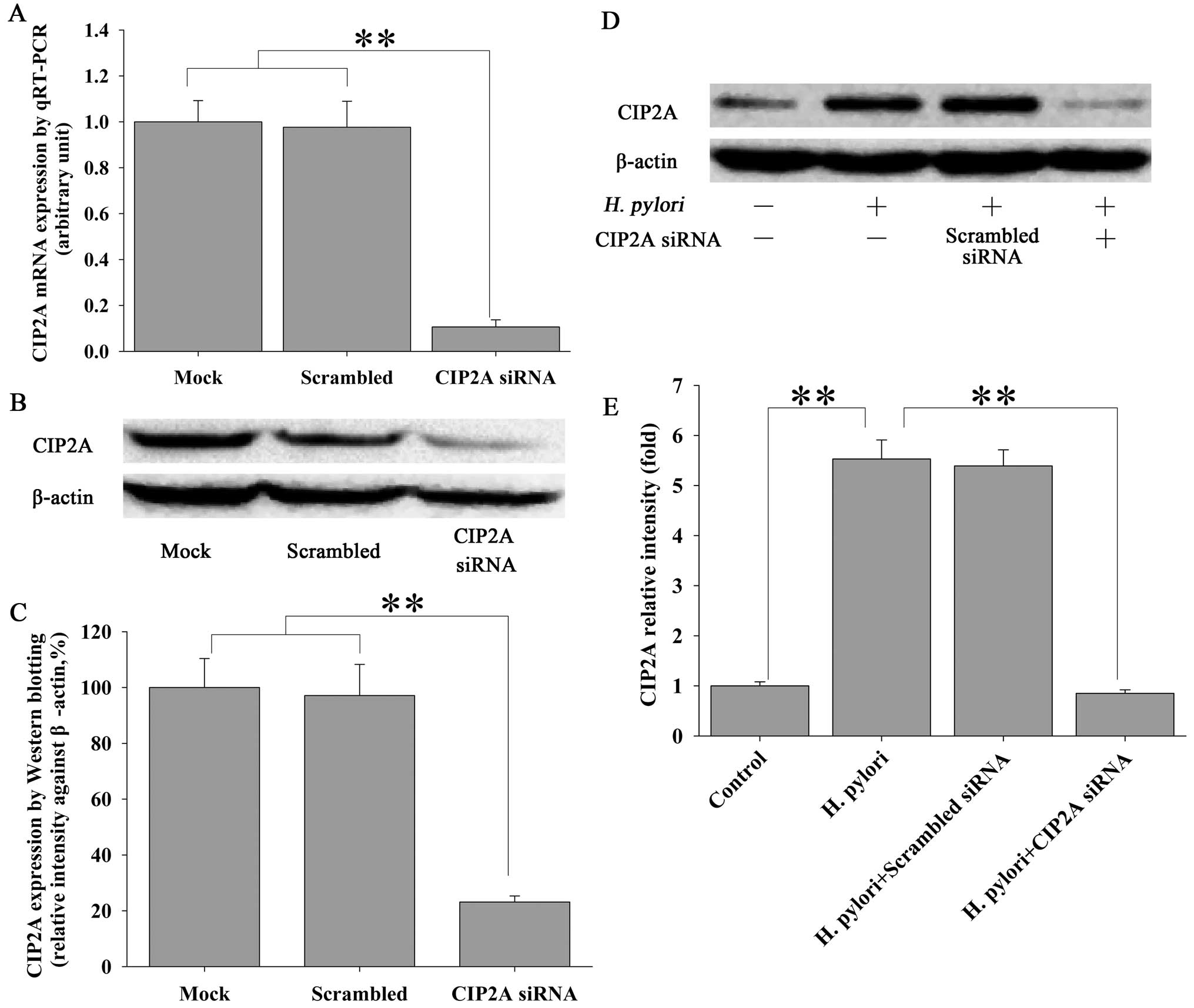
H. pylori induces proliferation of MKN-45
cells via CIP2A
In order to examine the functional impact of H.
pylori-induced upregulation of CIP2A in gastric cancer cells,
we utilized siRNA techniques to modulate the expression of CIP2A
and examined the effect on cell proliferation. MKN-45 cells were
transfected with siRNA against CIP2A or scrambled siRNA, and the
effect on the proliferation of MKN-45 cells was examined by MTT
assay. As expected, siRNA against CIP2A not only significantly
knocked down the basal level of CIP2A expression at the mRNA (by
90%) (Fig. 4A) and protein (by
82%) (Fig. 4B and C) levels, as
determined by qRT-PCR and western blot analysis, respectively, but
also the H. pylori-induced upregulation of CIP2A at the
protein level (Fig. 4D and E).
Parallel to these changes, H. pylori-induced proliferation
(Fig. 5A) and colony formation
(Fig. 5B and C) of the MKN-45
cells were significantly attenuated by CIP2A knockdown. These data
suggest that CIP2A is involved in H. pylori-induced
proliferation of MKN-45 cells.
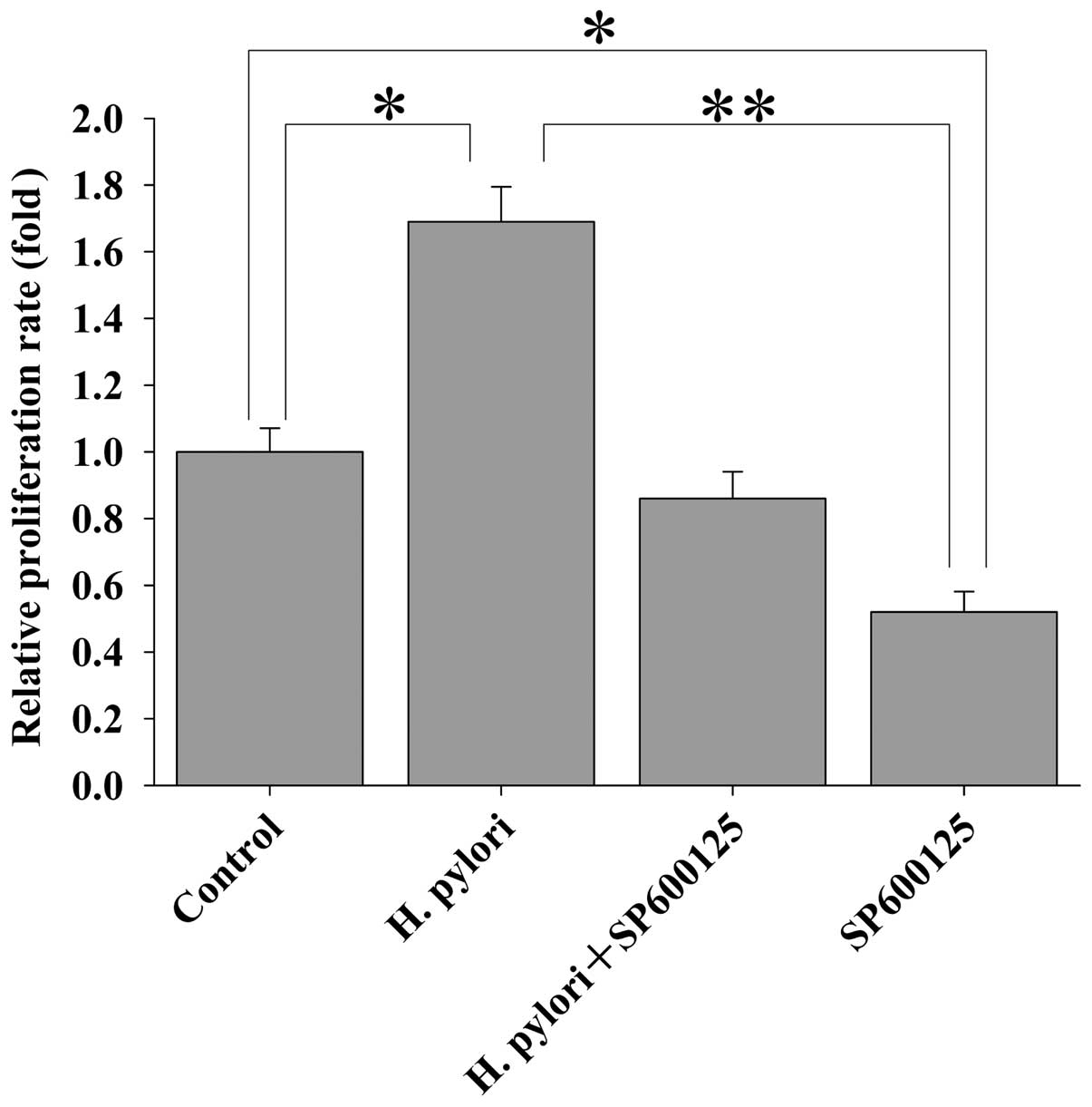
H. pylori induces the proliferation of
MKN-45 cells via JNK2 signaling
The results as reported above showed that H.
pylori-induced upregulation of CIP2A in MKN45 cells was
regulated via JNK2 signaling, and that H. pylori-induced
proliferation of MKN45 cells was mediated by CIP2A. Here, we
further investigated whether JNK2 signaling is involved in H.
pylori-induced proliferation of MKN-45 cells. MKN-45 cells were
treated with the JNK2 inhibitor SP600125 for 2 h prior to H.
pylori infection. Cell proliferation was examined by the MTT
assay. Treatment of MKN-45 cells with SP600125 attenuated the H.
pylori-stimulated cell proliferation (Fig. 6). These data indicate that H.
pylori-induced proliferation of MKN-45 cells is dependent upon
JNK2 signaling.
Discussion
H. pylori infection is the major risk factor
for gastric cancer, which is the second leading cause of
cancer-related mortality worldwide (20). The prevalence of H. pylori
infection varies according to geographical region, race, age and
socioeconomic status (21). A
high prevalence of H. pylori infection is present in several
regions of China such as Hexi Corridor where the incidence of
gastric cancer is high (22).
Clinical epidemiological data has clearly shown a strong
association between H. pylori infection and gastric
carcinogenesis (23–25), and eradication of H. pylori
was found to reduce the incidence of gastric cancer (26,27).
CIP2A was reported to promote cell proliferation and
tumor formation by stabilizing the oncogene c-Myc (15–17). Overexpression of CIP2A has been
shown in human gastric carcinoma tissues (16,17). A recent study revealed an
association between H. pylori infection and CIP2A expression
(13); yet, the biological impact
of H. pylori-induced CIP2A expression and its underlying
molecular mechanisms are not clearly defined.
It has been reported that H. pylori infection
activates the three major components of MAPK signaling in gastric
epithelial cells, namely ERK1/2, p38 and JNK (4,28,29). The MAPK pathway plays a crucial
role in mediating a number of events in host cell physiology,
including gene transcription, mitosis, motility, adhesion,
metabolism and apoptosis (6,7).
The JNK-mediated cascade is the basic signal transduction pathway
that regulates essential cellular processes (30,31). It has been established that H.
pylori induces gene expression in human gastric epithelial
cells by activating JNK signaling pathways (29,32–34). Among the JNK family, JNK2 is
essential for Ras signaling-induced cellular transformation of
human epithelial cells (35,36).
Previous studies have shown that JNK2 is essential
for the expression of CIP2A, and JNK2 regulates CIP2A transcription
via ATF2 (18). In the present
study, we further demonstrated that H. pylori infection
significantly increased the expression of CIP2A in MKN-45 cells,
both at the mRNA and protein levels. In addition, H.
pylori-induced CIP2A required functional JNK2, as inhibition of
JNK2 by its inhibitor SP600125 significantly decreased the
expression of CIP2A in response to H. pylori. Thus, JNK2
signaling plays an essential role in H. pylori-induced CIP2A
expression in gastric cancer cells.
Perhaps the most important implication of these
finding is the functional impact of CIP2A and JNK2 on gastric
epithelial cells during H. pylori infection. It is known
that both CIP2A and H. pylori infection enhance cell
proliferation (15,16,37–39). In the present study, blocking of
CIP2A by its specific siRNA significantly attenuated H.
pylori-induced cell proliferation, suggesting that H.
pylori induced the proliferation of gastric epithelial cells
through upregulation of CIP2A. This mechanism warrants further
investigation to ascertain whether CIP2A is suitable for use as a
therapeutic target for gastric cancer. As our study was conducted
using a single cell line, further studies using multiple cell
lines, particularly using normal gastric mucosa cells are needed,
and appropriate in vivo studies must be performed to further
verify the findings reported in the present study.
In conclusion, our results demonstrated that H.
pylori infection promotes MKN-45 gastric epithelial cell
proliferation through upregulation of CIP2A expression, and this
was likely mediated via the JNK2 signaling pathway. The data
presented here suggest that CIP2A plays a critical role in the
development of human gastric carcinoma, elucidating another
mechanism of H. pylori-induced carcinogenesis. Further
studies, particularly in vivo studies using appropriate
animal models, are needed to verify whether the interaction between
CIP2A and JNK2 signaling is indeed essential for the development of
H. pylori-induced gastric cancer formation, and if so, what
the potential is for possible gene therapy for this malignancy.
Acknowledgements
This study was supported by the Scientific Research
Funds of the Health Sector Projects in Gansu Province (grant no.
GWGL2010-17), the National Natural Science Foundation of China
(grant no. 31270532) and the Fundamental Research Funds of the
Central Universities, China (grant no. lzujbky-2013-m04).
Abbreviations:
|
CagA
|
cytotoxin-associated gene A
|
|
CIP2A
|
cancerous inhibitor of protein
phosphatase 2A
|
|
PP2A
|
protein phosphatase 2A
|
|
CFU
|
colony-forming units
|
|
ERK1/2
|
extracellular signal-regulated kinases
1 and 2
|
|
FBS
|
fetal bovine serum
|
|
H. pylori
|
Helicobacter pylori
|
|
JNK
|
c-Jun/SAPK N-terminal kinase
|
|
ATF2
|
activating transcription factor 2
|
|
MAPK
|
mitogen-activated protein kinases
|
|
RNAi
|
RNA interference
|
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar
|
|
2
|
Konturek PC, Konturek SJ and Brzozowski T:
Helicobacter pylori infection in gastric cancerogenesis. J
Physiol Pharmacol. 60:3–21. 2009.
|
|
3
|
Polk DB and Peek RM Jr: Helicobacter
pylori: gastric cancer and beyond. Nat Rev Cancer. 10:403–414.
2010. View
Article : Google Scholar
|
|
4
|
Ding SZ, Smith MF Jr and Goldberg JB:
Helicobacter pylori and mitogen-activated protein kinases
regulate the cell cycle, proliferation and apoptosis in gastric
epithelial cells. J Gastroenterol Hepatol. 23:e67–e78. 2008.
View Article : Google Scholar
|
|
5
|
Chen YC, Wang Y, Li JY, Xu WR and Zhang
YL: H. pylori stimulates proliferation of gastric cancer
cells through activating mitogen-activated protein kinase cascade.
World J Gastroenterol. 12:5972–5977. 2006.
|
|
6
|
Johnson GL and Lapadat R:
Mitogen-activated protein kinase pathways mediated by ERK, JNK, and
p38 protein kinases. Science. 298:1911–1912. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chang L and Karin M: Mammalian MAP kinase
signalling cascades. Nature. 410:37–40. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Brandt S, Kwok T, Hartig R, König W and
Backert S: NF-kappaB activation and potentiation of proinflammatory
responses by the Helicobacter pylori CagA protein. Proc Natl
Acad Sci USA. 102:9300–9305. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li Q, Liu N, Shen B, et al:
Helicobacter pylori enhances cyclooxygenase 2 expression via
p38MAPK/ATF-2 signaling pathway in MKN45 cells. Cancer Lett.
278:97–103. 2009. View Article : Google Scholar
|
|
10
|
Wang G, Gao F, Zhang W, et al: Involvement
of Aquaporin 3 in Helicobacter pylori-related gastric
diseases. PLoS One. 7:e491042012.
|
|
11
|
de Paulis A, Prevete N, Rossi FW, et al:
Helicobacter pylori Hp(2–20) promotes migration and
proliferation of gastric epithelial cells by interacting with
formyl peptide receptors in vitro and accelerates gastric mucosal
healing in vivo. J Immunol. 183:3761–3769. 2009.
|
|
12
|
Nagy TA, Wroblewski LE, Wang D, et al:
β-Catenin and p120 mediate PPARδ-dependent proliferation induced by
Helicobacter pylori in human and rodent epithelia.
Gastroenterology. 141:553–564. 2011.
|
|
13
|
Zhao D, Liu Z, Ding J, et al:
Helicobacter pylori CagA upregulation of CIP2A is dependent
on the Src and MEK/ERK pathways. J Med Microbiol. 59:259–265. 2009.
View Article : Google Scholar
|
|
14
|
Seshacharyulu P, Pandey P, Datta K and
Batra SK: Phosphatase: PP2A structural importance, regulation and
its aberrant expression in cancer. Cancer Lett. 335:9–18. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Junttila MR, Puustinen P, Niemelä M, et
al: CIP2A inhibits PP2A in human malignancies. Cell. 130:51–62.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li W, Ge Z, Liu C, et al: CIP2A is
overexpressed in gastric cancer and its depletion leads to impaired
clonogenicity, senescence, or differentiation of tumor cells. Clin
Cancer Res. 14:3722–3728. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Khanna A, Böckelman C, Hemmes A, et al:
MYC-dependent regulation and prognostic role of CIP2A in gastric
cancer. J Natl Cancer Inst. 101:793–805. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mathiasen DP, Egebjerg C, Andersen SH, et
al: Identification of a c-Jun N-terminal kinase-2-dependent signal
amplification cascade that regulates c-Myc levels in ras
transformation. Oncogene. 31:390–401. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wandler AM and Guillemin K: Transgenic
expression of the Helicobacter pylori virulence factor CagA
promotes apoptosis or tumorigenesis through JNK activation in
Drosophila. PLoS Pathog. 8:e10029392012.PubMed/NCBI
|
|
20
|
Thun MJ, DeLancey JO, Center MM, Jemal A
and Ward EM: The global burden of cancer: priorities for
prevention. Carcinogenesis. 31:100–110. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Fock KM and Ang TL: Epidemiology of
Helicobacter pylori infection and gastric cancer in Asia. J
Gastroenterol Hepatol. 25:479–486. 2010.
|
|
22
|
Zhang DH, Zhou LY, Lin SR, et al: Recent
changes in the prevalence of Helicobacter pylori infection
among children and adults in high- or low-incidence regions of
gastric cancer in China. Chin Med J (Engl). 122:1759–1763.
2009.PubMed/NCBI
|
|
23
|
Wang XQ, Yan H, Terry PD, et al:
Interaction between dietary factors and Helicobacter pylori
infection in noncardia gastric cancer: a population-based
case-control study in China. J Am Coll Nutr. 31:375–384. 2012.
|
|
24
|
Zhang Y, Sun LP, Xing CZ, et al:
Interaction between GSTP1 Val allele and H. pylori
infection, smoking and alcohol consumption and risk of gastric
cancer among the Chinese population. PLoS One.
7:e471782012.PubMed/NCBI
|
|
25
|
Epplein M, Zheng W, Xiang YB, et al:
Prospective study of Helicobacter pylori biomarkers for
gastric cancer risk among Chinese men. Cancer Epidemiol Biomarkers
Prev. 21:2185–2192. 2012.
|
|
26
|
Fukase K, Kato M, Kikuchi S, et al: Effect
of eradication of Helicobacter pylori on incidence of
metachronous gastric carcinoma after endoscopic resection of early
gastric cancer: an open-label, randomised controlled trial. Lancet.
372:392–397. 2008.PubMed/NCBI
|
|
27
|
Fuccio L, Zagari RM, Eusebi LH, et al:
Meta-analysis: can Helicobacter pylori eradication treatment
reduce the risk for gastric cancer? Ann Intern Med. 151:121–128.
2009.
|
|
28
|
Jang SH, Lim JW and Kim H: Beta-carotene
inhibits Helicobacter pylori-induced expression of inducible
nitric oxide synthase and cyclooxygenase-2 in human gastric
epithelial AGS cells. J Physiol Pharmacol. 60(Suppl 7): S131–S137.
2009.
|
|
29
|
Lim JW and Kim H: Role of
protease-activated receptor-2 on cell death and DNA fragmentation
in Helicobacter pylori-infected gastric epithelial cells. J
Transl Med. 8:852010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhao C, Ma H, Bu X, Wang W and Zhang N:
SFRP5 inhibits gastric epithelial cell migration induced by
macrophage-derived Wnt5a. Carcinogenesis. 34:146–152. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhong J, Zhao M, Luo Q, et al: CCDC134 is
down-regulated in gastric cancer and its silencing promotes cell
migration and invasion of GES-1 and AGS cells via the MAPK pathway.
Mol Cell Biochem. 372:1–8. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Choi IJ, Fujimoto S, Yamauchi K, Graham DY
and Yamaoka Y: Helicobacter pylori environmental
interactions: effect of acidic conditions on H.
pylori-induced gastric mucosal interleukin-8 production. Cell
Microbiol. 9:2457–2469. 2007. View Article : Google Scholar
|
|
33
|
Ding S-Z, Olekhnovich IN, Cover TL, Peek
RM Jr, Smith MF Jr and Goldberg JB: Helicobacter pylori and
mitogen-activated protein kinases mediate activator protein-1
(AP-1) subcomponent protein expression and DNA-binding activity in
gastric epithelial cells. FEMS Immunol Med Microbiol. 53:385–394.
2008. View Article : Google Scholar
|
|
34
|
Matsumoto A, Isomoto H, Nakayama M, et al:
Helicobacter pylori VacA reduces the cellular expression of
STAT3 and pro-survival Bcl-2 family proteins, Bcl-2 and Bcl-XL,
leading to apoptosis in gastric epithelial cells. Dig Dis Sci.
56:999–1006. 2011. View Article : Google Scholar
|
|
35
|
Ke H, Harris R, Coloff JL, et al: The
c-Jun NH2-terminal kinase 2 plays a dominant role in human
epidermal neoplasia. Cancer Res. 70:3080–3088. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Cellurale C, Sabio G, Kennedy NJ, et al:
Requirement of c-Jun NH(2)-terminal kinase for Ras-initiated tumor
formation. Mol Cell Biol. 31:1565–1576. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Li N, Han L, Chen J, Lin X, Chen H and She
F: Proliferative and apoptotic effects of gastric epithelial cells
induced by coccoidHelicobacter pylori. J Basic Microbiol.
53:147–155. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Feng Y, Wang L, Zeng J, et al: FoxM1 is
overexpressed in Helicobacter pylori-induced gastric
carcinogenesis and is negatively regulated by miR-370. Mol Cancer
Res. 11:834–844. 2013.
|
|
39
|
Himaya SWA, Dewapriya P and Kim SK: EGFR
tyrosine kinase inhibitory peptide attenuates Helicobacter
pylori-mediated hyper-proliferation in AGS enteric epithelial
cells. Toxicol Appl Pharmacol. 269:205–214. 2013. View Article : Google Scholar : PubMed/NCBI
|