Introduction
Coronary artery disease [(CAD), also known as
atherosclerotic heart disease or ischemic heart disease] due to
atherosclerosis of the coronary arteries is a major cause of
mortality worldwide. The process of atherosclerosis involves
chronic inflammation of the vessels (1). In response to endothelial injury,
vascular smooth muscle cells and fibroblasts migrate and
proliferate into the intima layer of the arterial wall (1). The progression of the disease
involves the recruitment of immune cells from the circulation, as
well as the deposition of blood lipids (1). In the vessel wall, the process may
further involve the increased production of extracellular matrix
(2). The latter is of particular
importance in the process of plaque (lesion) destabilization, which
may ultimately lead to the leakage of plaque debris into the
circulation, the formation of thrombi and myocardial infarction
(3). Post-infarction remodelling
can then lead to heart failure (4). Heart failure represents a growing
clinical challenge, possibly due to a steadily aging population and
the improved treatment of patients with CAD, thus increasing their
survival rate. The process of remodelling of the heart involves
cardiomyocyte growth, cardiomyocyte death through apoptosis and
autophagy, the deposition of extracellular matrix and the
activation of fetal gene programs (5). Vascular and cardiac remodelling are
distinct processes, but have a number of similarities. When
evaluating how a new treatment regimen for CAD may influence cells
(cellular remodelling), one could in parallel assess its effects on
the vessel wall (vascular remodelling).
Retinoic acid (RA) metabolites are the active
derivatives of vitamin A (retinol), exhibiting multiple effects on
cell growth, differentiation and proliferation (6,7).
RA metabolites have been shown to delay the development of
atherosclerosis in ApoE-deficient mice, and prevent reocclusion
following percutaneous coronary intervention (8). They have also been shown to modulate
proliferative ability and promote reendothelialization following
arterial injury (9,10). The improved understanding of the
effects of RA on cells that are involved in different aspects of
cardiovascular remodelling may lead to the identification of novel
agents for the prevention and treatment of cardiac disorders.
All-trans retinoic acid (ATRA) is used for the treatment of
certain proliferative diseases, such as leukemia (10). It is appealing to clinically
determine whether ATRA also shows antiproliferative properties in
diseases of the cardiovascular system.
Vitamin A is stored in the liver and is transported
to target cells bound to the plasma retinol binding protein (RBP).
It enters the cells via the membrane receptor for plasma RBP, known
as stimulated by retinoic acid gene 6 (STRA6). Intracellularly,
retinol is converted to 9-cis retinoic acid (9cisRA),
13-cis retinoic acid (13cisRA), and ATRA in a two-step
oxidative process. First, retinol is converted into retinal by
retinol dehydrogenases (RDHs), and then retinal is converted into
RA by retinal dehydrogenases (RALDHs or ALDHs). Synthesized RA
binds to nuclear retinoic acid receptors (RARs) and retinoic X
receptors (RXRs). Intracellular concentrations of RA metabolites
are strictly regulated by the cytochrome P450 superfamily of
enzymes (CYP26A1, CYP26B1 and CYP26C1), which convert ATRA into its
hydroxylated inactive forms (11). Cellular retinol and RA binding
proteins (CRBP and CRABP, respectively) deliver RA to the RARs
(12,13).
While a great deal of progress has been made in
identifying the stimuli that promote cell proliferation, the
mechanisms that inhibit or suppress proliferative ability are less
well characterized. Thus, the aim of this study was to investigate
whether the RA metabolic pathway is activated in patients with CAD
showing different levels of heart functionality, as well as in
human atherosclerotic plaques. Furthermore, we examined the effects
of ATRA on the expression of RA target genes and on the
proliferation of endothelial cells, smooth muscle cells,
cardiofibroblasts and cardiomyocytes.
Materials and methods
Ethics
All experiments were performed according to the
Declaration of Helsinki, and the experiments were approved by the
local ethics committee for animal and human research. Written
informed consent was obtained from all patients or their family
members prior to enrollment. The latter was the case when biopsies
were obtained from explanted hearts of individuals who died from a
cause not related to cardiac disease; the valves of these
individuals were used for homograft preparations.
Human heart biopsies
Different sources of biopsy material were used for
the measurement of RA metabolites and the expression of genes
involved in RA metabolism and transport (RA target genes): i) A
study performed at the National Hospital, Oslo, Norway, where
biopsies of the left ventricular (LV) free wall were collected from
still-beating explanted human hearts from patients with end-stage
heart failure undergoing cardiac transplantation due to CAD (CAD
group, n=9) or non-ischemic dilated cardiomyopathy (CMP group,
n=15). The patients were classified as class IV based on the New
York Heart Association (NYHA) functional classification system,
with an LV ejection fraction of <35%. Donor hearts were obtained
from gender- and age-matched healthy individuals with no history of
heart disease (donor group, n=7). ii) A published study conducted
at the Tampere University Hospital in Finland (14), where biopsies from the LV free
wall were collected from patients undergoing elective coronary
artery bypass grafting (CABG group, n=11). Tru-cut needle biopsies
were performed following cannulation for extracorporeal
circulation, but prior to cardioplegic arrest. The elective CABG
patients had CAD associated with mild cardiac dysfunction and were
classified as NYHA classes II–III, with an LV ejection fraction of
>60%. From this study, material was sufficient for mRNA
extraction, but not for the measurement of RA metabolites. Tissues
were snap-frozen in liquid nitrogen, and stored at −80°C until
use.
Human artery biopsies
In a third study performed at the Karolinska
Institutet, Stockholm and at Örebro University, Örebro, Sweden,
atherosclerotic plaques were collected from 9 patients during
endarterectomy due to >80% carotid artery stenosis (15). Additional biopsies of
non-sclerotic renal arteries were collected via nephrectomy on 5
patients who had no reported history of cardiovascular disease;
this material was described in a previous study (15). Surgically-removed carotid and
renal arteries were immediately rinsed in ice-cold sterile
RNase-free phosphate-buffered saline (PBS) and transversely cut
across the most prominent region. Three quarters of the specimen
were instantly frozen on dry ice, to be later used for total RNA
isolation. The remaining quarter was embedded and frozen in optimal
cutting temperature (OCT) compound to be used for morphological
confirmation of atherosclerosis using immunofluorescence (data not
shown), and then frozen for RNA extraction, as previously described
(16).
Measurement of RA metabolites
Human heart LV samples of CMP, CAD and healthy
donors were homogenized in ice-cold PBS with a motorized
homogenizer (Pro Scientific, Inc., Oxford, CT, USA). Retinoids were
extracted from the homogenates by the addition of 2 volumes of
ice-cold acetonitrile (10 ng/ml) containing ATRA (Sigma-Aldrich,
St. Louis, MO, USA) labeled with 13C as an internal
standard (IS). After vortexing for 10 min and centrifugation at
3,200 × g for 10 min, an aliquot of 75 ml was injected into a 4000
QTRAP liquid chromatography-mass spectrometry (LC-MS/MS) instrument
with APCI ionization (Applied Biosystems, Foster City, CA, USA).
The LC-MS/MS conditions were as described previously (17), except that the separating column
was an ABZ Plus (Supelco/Sigma-Aldrich, Bellefonte, PA, USA) with a
75 by 3 mm inner diameter and 3-μm particles. The entire procedure
was performed under red light. Calibration graphs were constructed
by linear least-squares regression analysis, plotting peak area
ratios of the analyte concentration and the IS against the
corresponding concentrations. Quantification was carried out by
interpolation and linear least-squares regression, as previously
described (17).
Effects of RA on gene expression and
proliferation of vascular cells
Human aortic smooth muscle cells (AOSMCs) were
purchased from Cambrex Bio Science Walkersville, Inc.
(Walkersville, MD, USA), while primary human umbilical vein
endothelial cells (HUVECs) were purchased from Invitrogen
(Stockholm, Sweden). AOSMCs from passages 4–8 and HUVECs from
passages 3–7 were seeded in 12-well plates at a density of
2×104 cells/well. AOSMCs were cultured in supplemented
human smooth muscle cell medium (M-231 with M-231–500; Cascade
Biologics). HUVECs were cultured in VascuLife VEGF Medium (LL-0003;
Lifeline Cell Technology). In order to investigate proliferation,
cells were stimulated with 1 μM ATRA dissolved in DMSO or 1 μM DMSO
alone for 24 and 72 h, trypsinized and counted by a haemocytometer
(Burker). Counting was performed in triplicates of 5 independent
experiments. Additionally, AOSMCs and HUVECs were seeded in 6-well
plates at a density of 2×105 and stimulated with ATRA 1
μM or the vehicle alone (DMSO). Cells were harvested serially for
total RNA extraction and the evaluation of expression of RA target
genes by quantitative PCR after 24, 48, 72 and 96 h.
Isolation and culture of adult mouse
cardiomyocytes and cardiofibroblasts
Adult mouse cardiomyocytes and cardiofibroblasts
were isolated from hearts of C57BL6 mice using the method described
by O’Connell et al (18).
Hearts were explanted, cannulated through the aorta, purged of
blood with perfusion buffer, and then perfused with digestion
buffer containing collagenase II (Worthington Biochemical Corp.,
Lakewood, NJ, USA). Digested hearts were mechanically disrupted and
suspended in perfusion buffer supplemented with 12.5 μM
CaCl2 and 5% fetal calf serum (FCS). Cardiomyocytes were
separated from cardiofibroblasts by serial centrifugations as
previously described (18).
Cardiofibroblasts were preserved in the supernatant from the first
centrifugation and transferred to a separate tube for resuspension
in Minimum Essential Medium (MEM) with Hank’s balanced salt
solution (Gibco-BRL) and additives as previously described
(19). Non-coated 6-well plates
were plated equally with cell suspension. Cardiomyocytes were
resuspended in MEM with Hank’s balanced salt solution with
additives at 37°C in an atmosphere supplied with 2% CO2
for 24 h.
RA effects on gene expression and
proliferation of cardiac cells
Cardiomyocytes and cardiofibroblasts were isolated
from the hearts of 5 C57BL6 mice for the evaluation of RA target
gene expression. Cardiofibroblasts were incubated up to 96 h in
medium supplemented with 1 μM ATRA dissolved in ethanol. Control
experiments were performed using medium supplemented with 1 μM
ethanol only. Cells were harvested serially for total RNA
extraction and the evaluation of the expression of RA target genes
by quantitative PCR after 24, 48, 72 and 96 h. Cardiomyocytes were
stimulated with 0,1 or 1 μM ATRA dissolved in ethanol or ethanol
alone as the control. Cells were observed only for 24 h for gene
expression evaluation, as these cells do not survive long once
isolated and are not proliferative cells. Cells were harvested in
600 μl RLT buffer (Qiagen, Hilden, Germany), using a cell scraper,
snap-frozen in liquid nitrogen, and stored at −80°C. Additional
cardiofibroblasts were isolated from 4 C57BL6 mouse hearts. Cells
were incubated up to 72 h in medium supplemented with 1 μM ATRA
dissolved in ethanol (Sigma-Aldrich) and 10 μM
5-ethynyl-2′-deoxyuridine (EdU; nucleoside analogue to thymidine
which is incorporated into DNA during active DNA synthesis). ATRA
and EdU supplemented medium was changed every 24 h. Control
experiments were performed using ethanol only. Cells were harvested
for proliferation evaluation by EdU incorporation. Cardiofibroblast
proliferation was determined using the Click-iT™ cell proliferation
kit for flow cytometry following the manufactorers instructions
(C-10418; Invitrogen). The events of 1,000 were analyzed using a
LSR II flow cytometer (BD Biosciences). Pacific Blue™ was detected
with logarithmic amplification using 406 nm excitation with violet
(450/50 nm) emission filter. Side- and forward-scatter was
monitored and used to exclude debris and doublets. Experiments were
performed in duplicates from 4 independent isolations.
RNA isolation and cDNA synthesis
RNA was extracted using an RNeasy mini kit (Qiagen)
with an additional phenol-chloroform extraction step and in-column
DNase treatment (Qiagen). Random hexamers for priming (3 min at
70°C) were used for reverse transcription of 1 mg of RNA from whole
heart extracts, carotid arteries lesion, normal kidney arteries,
human aortic smooth muscle cells and human umbilical vein
endothelial cells and 200 ng of RNA from isolated cardiomyocytes
and cardiac fibroblasts. Reverse transcription was followed by a
modified First Strand cDNA Synthesis Protocol with Superscript III
(Invitrogen) and RNasin (Promega Corp., Madison, WI, USA) enzymes
and cDNAs were amplified using quantitative PCR.
Quantitative polymerase chain
reaction
RA target genes retinol binding protein 1 (RBP1),
RBP2, RBP4, STRA6, aldehyde dehydrogenase 1 family, member A1
(ALDH1A1), ALDH1A2, cellular retinoic acid binding protein 1
(CRABP1), CRABP2, CYP26B1, RARα, RARβ and RARγ were amplified from
human hearts and arterial biopsies, human aortic smooth muscle
cells, human umbilical vein endothelial cells, and murine
cardiomyocytes and cardiofibroblasts using real-time PCR. For
amplification of human genes, predesigned primers and probes from
Applied Biosystems were used (Table
I). β-actin was used as an endogenous control for HUVECs,
AOSMCs, and amplification of arterial RNA: β-actin forward, 5-CTG
GCT GCT GAC CGA GG-3, reverse, 5-GAA GGT CTC AAA CAT GAT CTG GGT-3
and probe, 5-CCT GAA CCC CAA GGC CAA CCG-3. (TaqMan Gene Expression
Assays; Applied Biosystems). For human heart material, hypoxanthine
phosphoribosyl transferase (HPRT) was used as endogenous control.
HPRT was purchased with ready-made primers and probe (assay ID
4310890E; Applied Biosystems). Mouse primers were designed with
Primer Express 3.0 software and custom made by Eurofins on the
basis of published cDNA sequences (primer sequences are available
upon request) (Table II).
SYBR-Green (Applied Biosystems) was used for detection. 18S rRNA
predesigned Real-Time PCR primer and probe (Applied Biosystems;
assay ID 4310893E) was used as endogenous control for mouse
materials (20–22), while wells without cDNA were used
as negative controls. PCR-amplification mixtures contained sample 2
μl cDNA, 10 μl of Power SYBR-Green PCR Master Mix (Applied
Biosystems), and primers at a final concentration of 900 nM diluted
with Milli-Q water in a total volume of 20 μl. The PCR reaction had
the standard amplification scheme: one cycle of 2 min at 50°C
(AmpErase UNG activation), one cycle of 10 min at 95°C (Gold
AmpliTaq activation, AmpErase UNG inactivation), followed by 40
cycles of denaturation for 15 sec at 95°C and annealing/extension
for 1 min at 60°C in a ABI 7900 HT Sequence Detection System
(Applied Biosystems). Ct cycle values were correlated to a standard
curve. The resulting mRNA levels relative to the 18S rRNA, HPRT, or
β-actin were calculated according to the standard formula
2−ΔΔCt, where ΔΔCt =
(CtTarget_sample−Ctendogenous control_sample)
− (CtTarget_calibrator−Ctendogenous
control_calibrator) (Applied Biosystems).
 | Table ITaqMan Gene Expression Assay IDs used
for the amplification of human genes. |
Table I
TaqMan Gene Expression Assay IDs used
for the amplification of human genes.
| Gene name | Abbreviation | Assay ID |
|---|
| Retinol binding
protein 1 | RBP1 | Hs00161252_m1 |
| Retinol binding
protein 4 | RBP4 | Hs00198830_m1 |
| Aldehyde
dehydrogenase 1 family, member A1 | ALDH1A1 | Hs00167445_m1 |
| Aldehyde
dehydrogenase 1 family, member A2 | ALDH1A2 | Hs00180254_m1 |
| Stimulated by
retinoic acid gene 6 | STRA6 | Hs00223621_m1 |
| Cellular retinoic
acid binding protein 2 | CRABP2 | Hs00275636_m1 |
| Cytochrome P450,
family 26, subfamily B, polypeptide 1 | CYP26B1 | Hs00175627_m1 |
| Retinoic acid
receptor γ | RARγ | Hs00171273_m1 |
| Hypoxanthine
phosphoribosyl transferase | HPRT | 4310893E |
| Table IIPrimer sequences of the investigated
mouse genes. |
Table II
Primer sequences of the investigated
mouse genes.
| Mouse gene | Forward primer | Reverse primer | NCBI Accession
no. |
|---|
| RBP1 |
GCGCTCGACGTCAACGT |
GCCATCCTTGCACGATCTCTT | NM_011254.5 |
| CRABP1 |
TCACTGCACACAGACACTTCTTGA |
GGCGCCAAATGTCAGGATTA | NM_001162475.1 |
| CRABP2 |
GCGAGCAGAGGCTTCTGAAG |
CATTGTCAGGATCAGCTCTCCAT | NM_007759.2 |
| ALDH1A2 |
GCTCACCAGGGTGTGTTCTTC |
CCTCATAGATGGATTCCACAA | NM_009022.4 |
| CYP26B1 |
TGCCACCCGCGACAA |
GGAACCCTGTAGCAACCAGTGA | NM_175475.2 |
| RARα |
CTGGACTGCTCAGTGCCATCT |
TGCAGCATGTCCACCTTGTC | NM_001177302.1 |
| RARβ |
GGGTAAATACACCACGAATTCCA |
TGTCCCAGAGGCCCAAGTC | NM_011243.1 |
| RARγ |
CCAGGAGACTTTTCCCTCACTCT |
GCTGCACCCGGTGATCTG | NM_011244.3 |
Statistical analysis
One-way analysis of variance (ANOVA) with Dunnett’s
adjustment for multiple comparisons was used to evaluate RA
metabolite measurements and gene expression in human hearts. The
Student’s t-test was used to compare gene expression and cell
proliferation data. Data are presented as the means ± SD.
Differences were considered significant at P<0.05.
Results
Altered RA metabolism in human hearts
with end-stage failure
Left ventricular biopsies of explanted human hearts
collected during heart transplantation were homogenized for
measurement of RA metabolites with LC-MS/MS. The ATRA concentration
was significantly higher in the patients with end-stage heart
failure due to CAD (CAD group) compared to the healthy donor
hearts. 13cisRA expression was decreased in the hearts with
non-ischemic dilated CMP (CMP group) compared to the healthy donor
hearts (Fig. 1). Retinol and
retinal levels were unaltered (data not shown). The samples from
patients undergoing elective CABG (CABG group) were not sufficient
for metabolite determination.
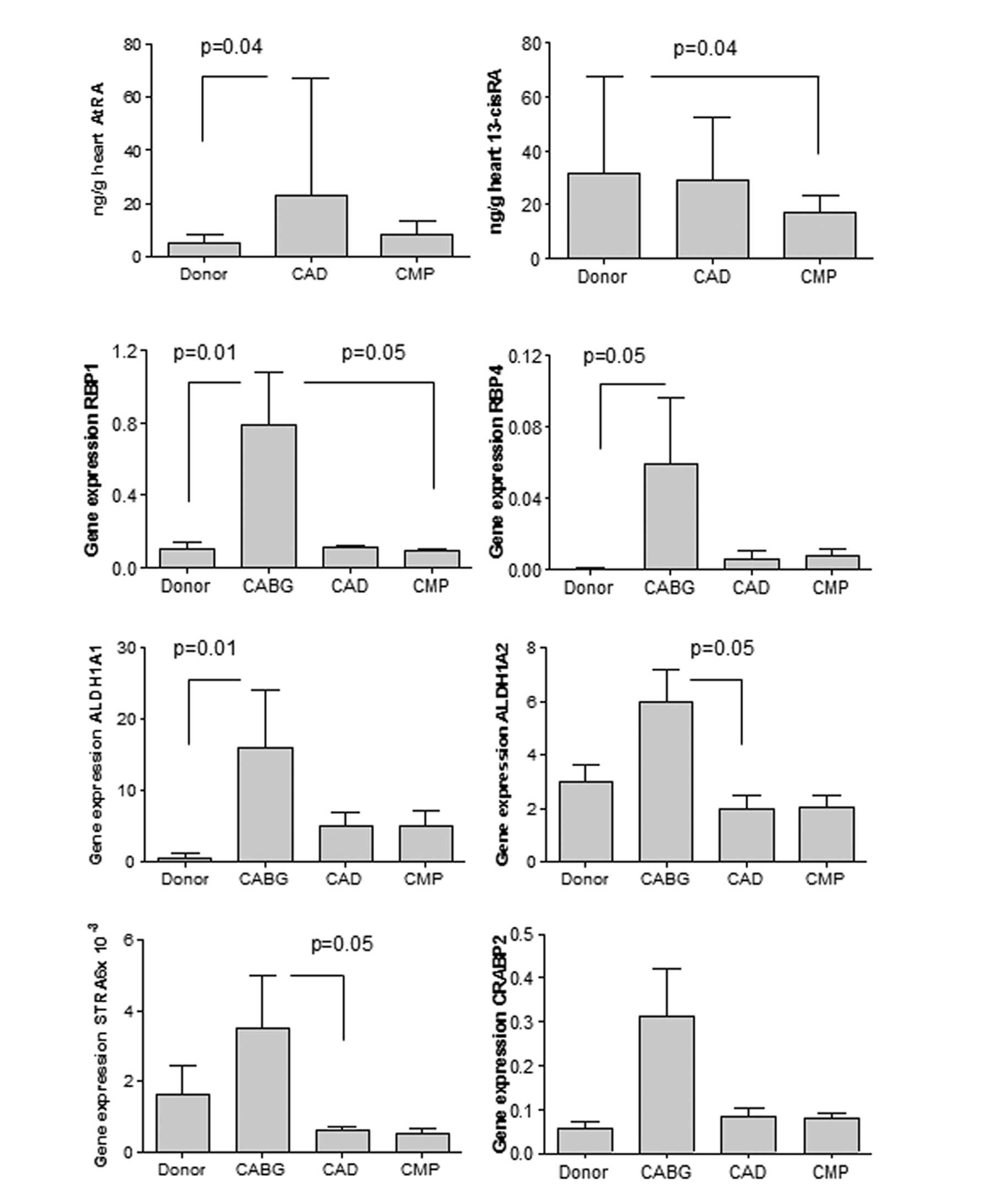 | Figure 1Left ventricular biopsies were
collected from still-beating explanted human hearts from patients
with end-stage heart failure undergoing cardiac transplantation due
to dilated cardiomyopathy (CMP, n=15) or coronary artery disease
(CAD, n=9). Explanted healthy donor hearts (donor, n=7) were used
as the controls. The endogenous retinoids, all-trans
retinoic acid; (ATRA) and 13-cis retinoic acid (13cisRA)
were quantified by triple-stage liquid chromatography/tandem mass
spectrometry. The expression of retinoic acid target genes was
investigated on biopsies from patients undergoing coronary artery
bypass grafting (CABG, n=11), as well as from CAD and CMP patients,
using qRT-PCR. RBP1, retinol binding protein 1; RBP4, retinol
binding protein 4; STRA6, stimulated by retinoic acid gene 6;
ALDH1A1, aldehyde dehydrogenase 1 family, member A1; ALDH1A2,
aldehyde dehydrogenase 1 family, member A2; CRABP2, cellular
retinoic acid binding protein 2. Data are presented as the means ±
SD. P-values indicate comparisons between DMSO-stimulated cells and
the corresponding ATRA stimulation. |
Increased cardiac expression of RA target
genes in CAD
The expression of RA target genes was investigated
in the LV biopsies of patients undergoing transplantation due to
end-stage heart failure from CAD (CAD group) or due to CMP, as well
as in biopsies obtained during open heart surgery with CABG (CABG
group). Note that both groups of suffered from CAD, but patients in
the CABG group had a good heart function with an ejection fraction
>60%, as opposed to <35% in the CAD group. Hearts of the CABG
group generally showed an elevated expression of genes coding for
RA target enzymes, compared to the hearts of the CAD or CMP groups
that came from patients classified in higher NYHA classes, or to
healthy donor hearts. The expression of genes involved in the
delivery of retinol into the cell (RBP1, RBP4 and STRA6), and that
of genes involved in the transformation of intracellular retinol
into the active forms of RA (ALDH1A1 and ALDH1A2) was significantly
increased in patients undergoing CABG (p<0.05) compared to the
other groups. The apparent increase of CRABP2 was not
significant (Fig. 1).
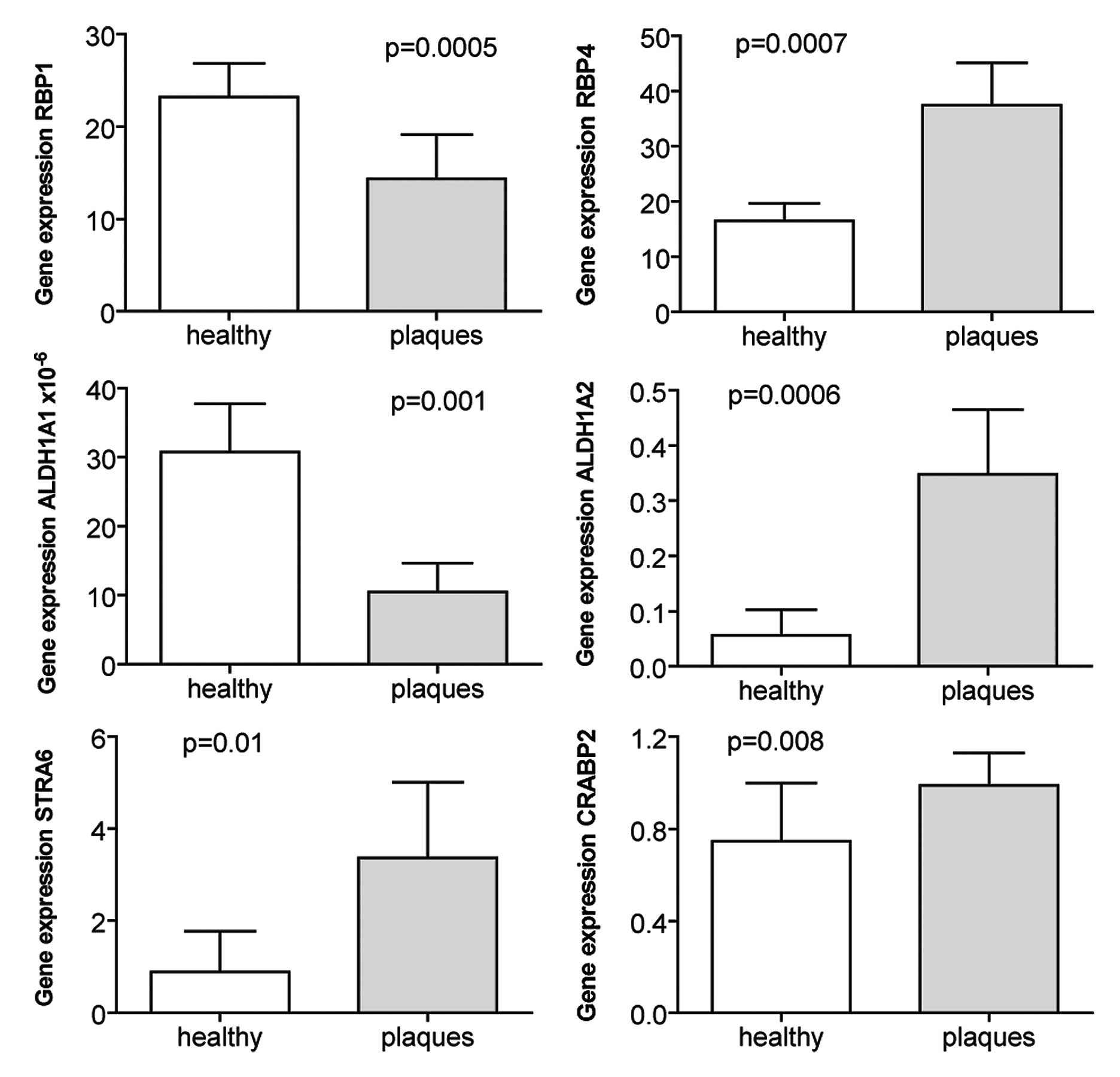
Increased expression of RA target genes
in atherosclerotic plaques
When the expression of the same genes was
investigated in atherosclerotic plaques harvested from carotid
arteries and compared to renal arteries without atherosclerosis,
the expression of RBP4, STRA6, CRABP2 and ALDH1A2 was increased
(Fig. 2). By contrast, the
expression of RBP1 and ALDH1A1 was higher in the healthy renal
arteries compared to the atherosclerotic plaques (Fig. 2).
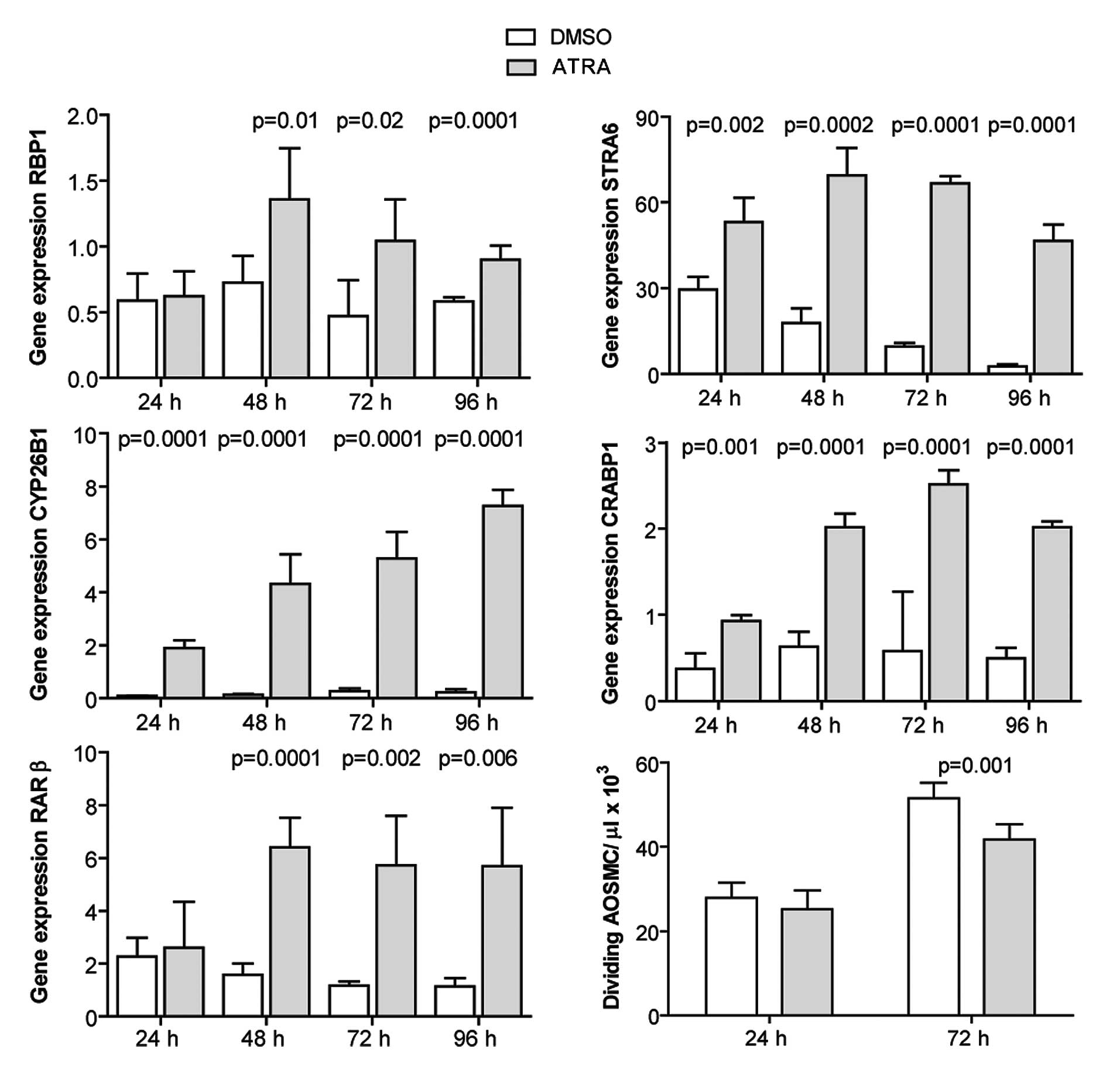
ATRA induces the expression of RA target
genes and inhibits the proliferation of human AOSMCs
AOSMCs were cultured and incubated with 1 μM ATRA
for 24–96 h. The Expression of RA target genes was quantified with
by qRT-PCR. ATRA induced the expression of the investigated genes
at all time points, with a slightly different profile of induction
observed for each gene (Fig. 3).
Cell proliferation was evaluated at 24 and 72 h post-ATRA treatment
by cell counting. ATRA inhibited the proliferation of AOSMCs at 72
h (Fig. 3).
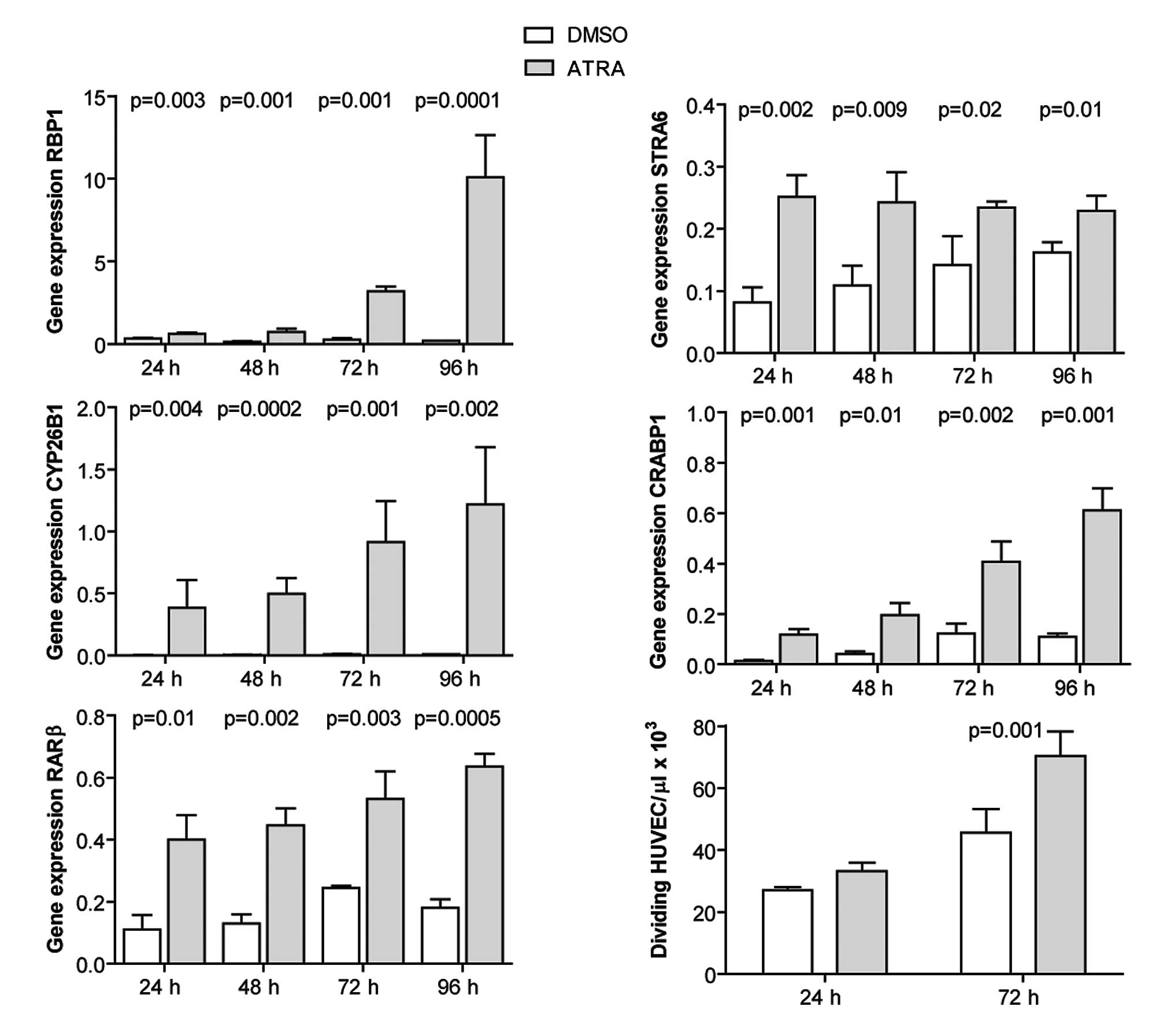
ATRA induces the expression of RA target
genes and promotes the proliferation of HUVECs
HUVECs were cultured and incubated with 1 μM ATRA
for 24–96 h, and expression of RA target genes was quantified by
qRT-PCR. ATRA induced the expression of the investigated genes at
all time points (Fig. 4). Cell
proliferation was evaluated at 24 and 72 h post-ATRA treatment by
cell counting. ATRA promoted the proliferation of HUVECs at 72 h
post-treatment (Fig. 4).
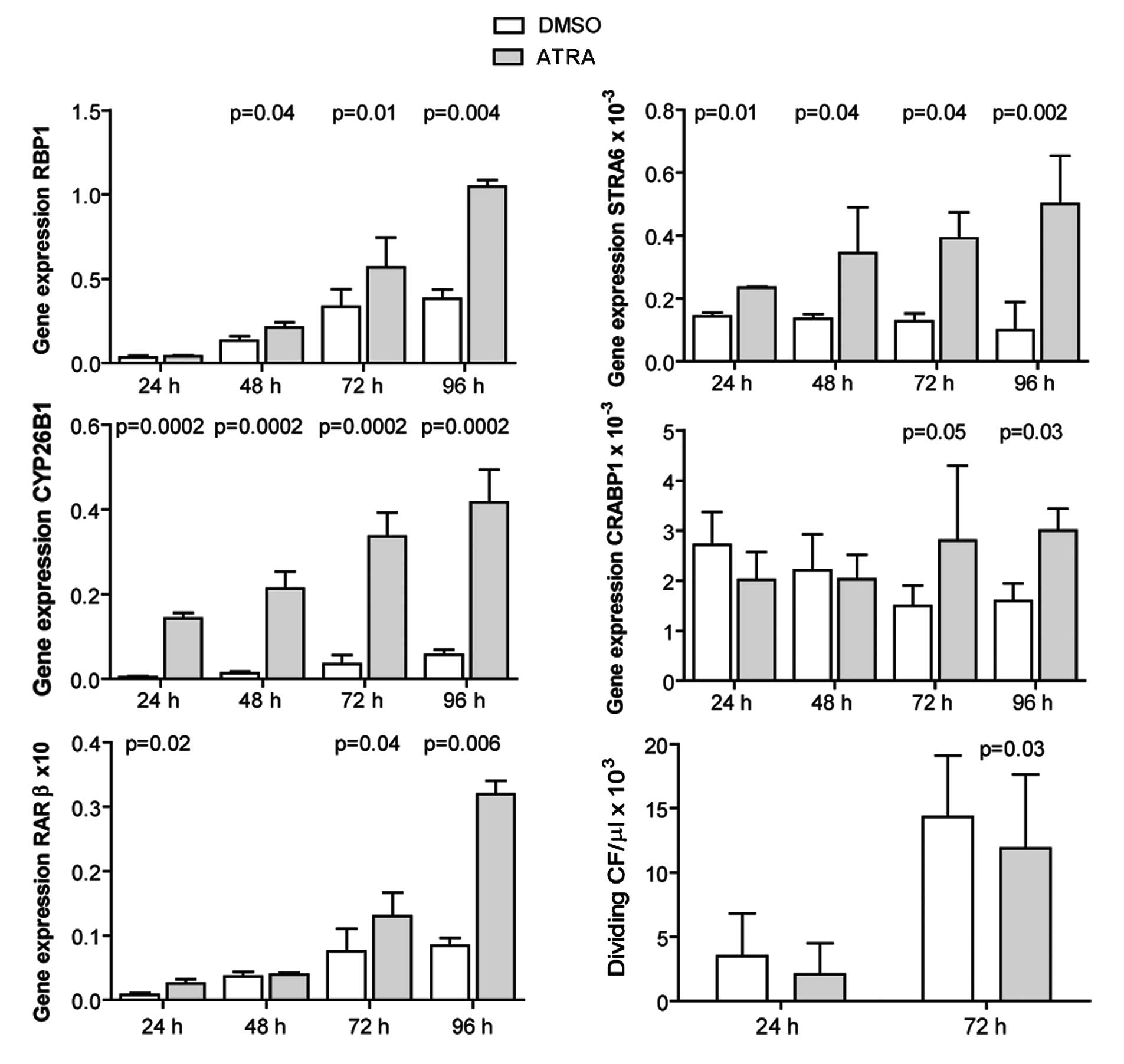
ATRA induces the expression of RA target
genes and inhibits the proliferation of adult murine
cardiofibroblasts
Cardiofibroblasts were isolated from adult mouse
hearts, and incubated with 1 μM ATRA for 24–96 h. The expression of
RA target genes was evaluated by qRT-PCR. ATRA induced the
expression of the investigated genes, apart from CRABP1 at 24 and
48 h post-treatment (Fig. 5).
Cell proliferation was evaluated at 24 and 72 h by measuring EdU
incorporation. ATRA inhibited the proliferation of
cardiofibroblasts (Fig. 5).
ATRA induces the expression of RA target
genes in adult murine cardiomyocytes
Cardiomyocytes were isolated from adult mouse
hearts, and were treated either with dissolvent alone (ethanol), or
with 0.1 or 1 μM ATRA for 24 h prior to harvest. qRT-PCR of RA
target genes revealed that ATRA induced the expression of RBP1,
STRA6 and CYP26B1 in a dose-dependent manner. ALDH1A2 and CRBP1
expression was not affected by ATRA stimulation, while RARβ showed
increased expression levels only after treatment with the lowest
ATRA dose (Fig. 6). Cell
proliferation was not examined.
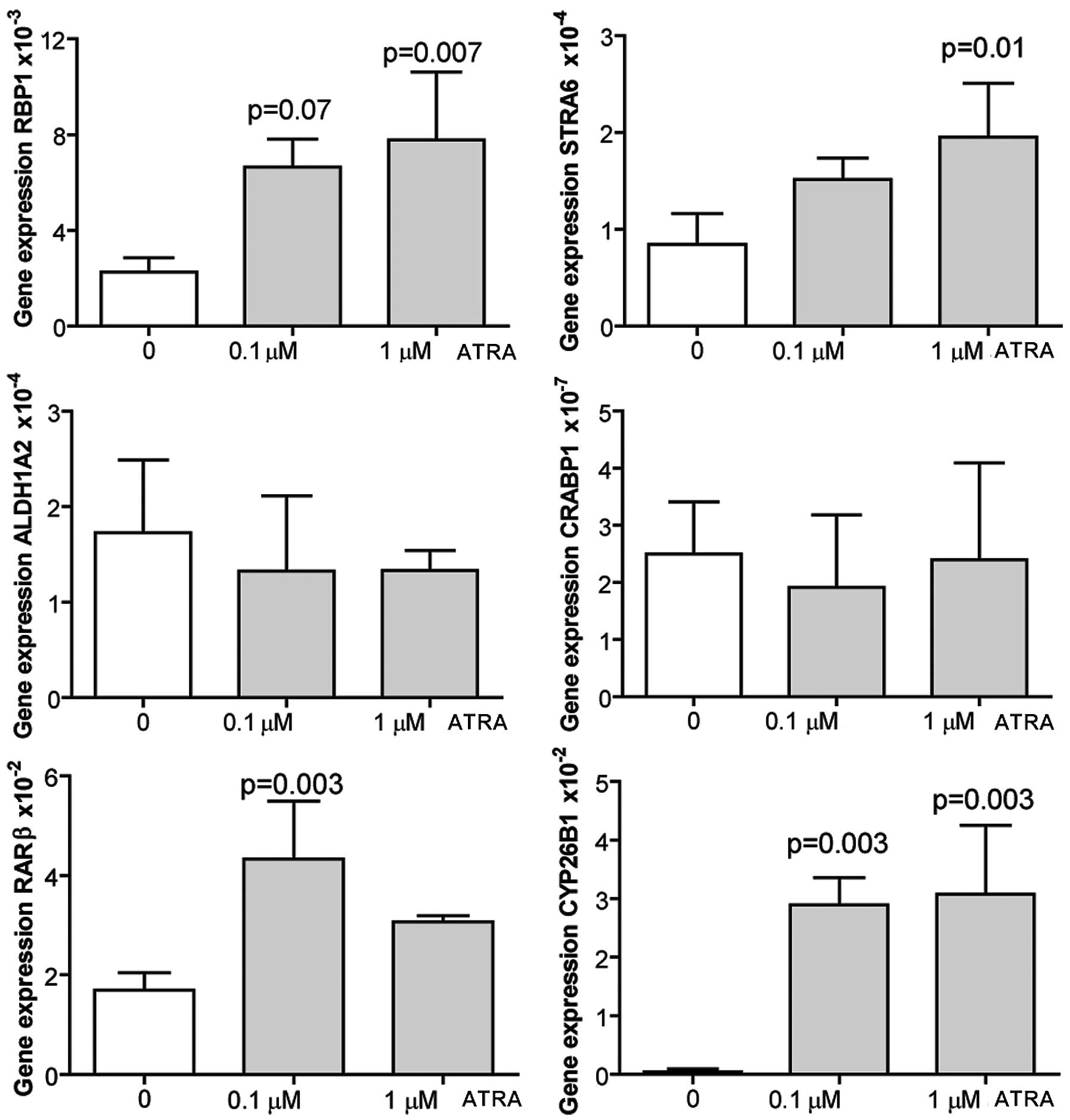 | Figure 6Adult murine cardiomyocytes were
isolated from C57BL6 mouse hearts and cultured in medium
supplemented with 0,1 μM all-trans retinoic acid (ATRA) or 1
μM ATRA dissolved in ethanol, or ethanol alone. Cells were
harvested 24 h later for evaluation of expression of retinoic acid
target genes by qRT-PCR. Data are presented as the means ± SD of 4
independent experiments performed in duplicate. RBP1, retinol
binding protein 1; STRA6, stimulated by retinoic acid gene 6;
CYP26B1, cytochrome P450, family 26, subfamily B, polypeptide 1;
CRABP1, cellular retinoic acid binding protein 1; RARβ, retinoic
acid receptor β; ALDH1A2, aldehyde dehydrogenase 1 family, member
A2. P-values indicate comparisons between DMSO-stimulated cells and
the corresponding ATRA stimulation. |
Discussion
To our knowledge, this is the first study on the
expression of endogenous metabolites and RA target genes in
patients with CAD. Changes were observed in cardiac RA metabolites
in patients with end-stage heart failure. The expression of RA
target genes was clearly higher in patients with CAD undergoing
CABG (CABG group) compared to patients with end-stage heart failure
due to CAD (CAD group) or non-ischemic dilated CMP (CMP group), as
well as in atherosclerotic plaques compared to healthy arteries.
However, we could not obtain atherosclerotic plaques from the
coronary arteries, and we did not have access to healthy carotid
arteries for comparisons of gene expression in the same vascular
bed. We hypothesized that the increased expression of RA target
genes observed in the hearts of CABG patients may reflect tissue
repair occurring in these patients where heart function is still
good, compared to hearts of CAD or CMP patients, where the repair
potential of the hearts is virtually non-existent.
The gene expression patterns observed in this study
indicate increased RA signaling in CAD, with activation of genes
involved in RA transport, uptake, conversion and delivery to
nuclear receptors. The net effect of these changes may be the
accumulation of RA derivatives. The hearts of patients of the CAD
group showed an accumulation of ATRA, in contrast to those of the
CMP group. The latter showed reduced levels of 13cisRA, a finding
which is difficult to interpret. The cardiac cells in the hearts of
CMP patients may be transformed from dilation to a degree that
renders the comparison with other types of hearts of limited value.
We were not able to measure RA metabolites in the hearts of CABG
patients, since the biopsies obtained with a tru-cut needle were of
insufficient size. Gene expression was examined instead. The
upregulation of RBP1, RBP4 and STRA6 genes strongly indicates the
increased activity of the RBP-STRA6 system, involved in
physiological vitamin A uptake (23). The STRA6 protein, which is induced
by RA metabolites, has previously been suggested to be a good
predictor of the retinol cellular status (24). The increased expression of ALDH1A2
suggests the increased synthesis of biologically active forms of RA
metabolites, while the increased expression of CRABP2 promotes the
delivery of ATRA to the nucleus (25). In a model of permanent left
coronary artery ligation in mice, we demonstrated that endogenous
RA signaling is activated, concomitant with the accumulation of
retinol in the heart during myocardial ischemia and remodelling
(26). In this model, the
intensity of gene expression and activity of RA response elements
was most evident shortly after myocardial infarction, gradually
decreasing over time. This pattern may be analogous to the
increased expression of RA target genes in the CABG patients
observed in the present study. CABG patients can be considered as a
pathophysiological intermediate between normality and heart
failure, and we speculate that their hearts undergo remodelling. In
accordance with this hypothesis, another study demonstrated an
accumulation of exogenously administrated RA in post-infarcted
hearts of rats (27). The authors
speculated that it would be beneficial to increase the antioxidant
content in the ischemic hearts.
One of the beneficial roles of RA in myocardial
ischemia may be mediated by the reduction in cardiofibroblast
fibrosis, since treatment with ATRA reduced fibroblast
proliferation in our study. Additional evidence for the role of RA
in heart remodelling has been provided by other studies: rats with
tissue insufficiency of vitamin A showed increased cardiac
remodelling and increased ventricular dysfunction (27,28). Adverse LV remodelling was
intensified when myocardial infarction was induced in rats with
tissue vitamin A deficiency (28). In addition, the administration of
RA to rats in a model of tobacco smoke-induced LV remodelling
prevented adverse remodelling (29). RA administration also prevented
remodelling induced by left coronary artery ligation (30). In our study, in the
atherosclerotic plaques, we observed an increase in the expression
of STRA6 and ALDH1A2 genes, suggesting an increased uptake of
retinol and the synthesis of ATRA in the plaque. This could
potentially delay plaque development, since we demonstrated that
ATRA inhibited smooth muscle cell proliferation. In agreement with
this result, we previously demonstrated that the increased
catabolism of ATRA caused by a single nucleotide polymorphism (SNP)
in the enzyme CYP26B1 is associated with larger atherosclerotic
plaques (31).
Human hearts mainly contain cardiomyocytes,
cardiofibroblasts, smooth muscle cells and endothelial cells. Thus,
we investigated the effects of ATRA on these cell populations. The
treatment of human smooth muscle cells with ATRA induced the
expression of RBP1, STRA6, CYP26B1, CRABP1 and RARβ and inhibited
cell proliferation at 72 h after treatment compared to time-matched
control cells. These findings are in agreement with those of other
studies, which reported that RA limits the proliferation of human
coronary smooth muscle cells (6)
and smooth muscle cells from the rat aorta (9). A number of studies have reported the
beneficial effects of ATRA on intimal hyperplasia in models of
arterial injury (6,9). ATRA signaling is tightly regulated
by CYP26B1. siRNA-mediated silencing of the CYP26B1 gene and
reduction of the enzymatic activity of CYP26B1 using the inhibitor,
R115866, has been shown to increase ATRA-mediated signaling and to
result in decreased smooth muscle cell proliferation (32).
In our study, the treatment of HUVECs with ATRA had
similar effects on gene expression, but different biological
effects. ATRA induced the proliferation of HUVECs, which is in
accordance with observations made by others (33). In a previous study, in
vivo, treatment with ATRA after balloon injury of the rat aorta
increased reendothelialization, and enhanced endothelial-dependent
relaxation of the injured aorta (34). Thus, RA metabolites may be
important for endothelial recovery and the formation of new
vessels.
When cardiofibroblasts and cardiomyocytes were
treated with ATRA, the increase in gene expression was similar to
that observed in endothelial and smooth muscle cells. The
cardiomyocytes could only be studied in a short timeframe after
treatment, since they do not survive long ex vivo. In the
present study, the proliferative ability of cardiofibroblasts was
inhibited by ATRA. We were unable to find similar studies performed
by other research groups to assess the validity of this result.
Notably, it was previously reported that ATRA inhibits angiotensin
II-induced increase in cell growth and collagen secretion in
neonatal cardiac fibroblasts (7).
The effect of ATRA on fibroblast proliferation is assumed to be
beneficial, since it may reduce the in vivo scar formation
in post-infarcted tissue. The current therapy of CAD is based on
the reduction of risk factors (by lowering triglycerides,
increasing HDL, decreasing LDL, decreasing blood pressure or
controlling hyperglycaemia), but not on the inhibition of the
proliferative ability of cells involved in cardiovascular
remodelling. The antiproliferative properties of ATRA are already
used in clinical practice against acute promyelocytic leukemia and
some forms of acute myeloid leukemia. The data from the present
study suggest that due to its antiproliferative properties, ATRA
may be used to delay remodelling, reduce restenosis, and preserve
the functions of the cardiovascular system.
Acknowledgements
Torun Flatebø and Stian Weiseth are gratefully
acknowledged for their excellent work. This study was supported by
grants from the Novo Nordisk Foundation, the Norwegian Health
Association, The Center for Heart Failure Research and the
University of Oslo.
References
|
1
|
Hansson GK and Hermansson A: The immune
system in atherosclerosis. Nat Immunol. 12:204–212. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tyagi SC, Meyer L, Schmaltz RA, Reddy HK
and Voelker DJ: Proteinases and restenosis in the human coronary
artery: extracellular matrix production exceeds the expression of
proteolytic activity. Atherosclerosis. 116:43–57. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fishbein MC: The vulnerable and unstable
atherosclerotic plaque. Cardiovasc Pathol. 19:6–11. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
McKay RG, Pfeffer MA, Pasternak RC, Markis
JE, Come PC, Nakao S, et al: Left ventricular remodeling after
myocardial infarction: a corollary to infarct expansion.
Circulation. 74:693–702. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Valen G: Innate immunity and remodelling.
Heart Fail Rev. 16:71–78. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wakino S, Kintscher U, Kim S, Jackson S,
Yin F, Nagpal S, Chandraratna RA, Hsueh WA and Law RE: Retinoids
inhibit proliferation of human coronary smooth muscle cells by
modulating cell cycle regulators. Arterioscler Thromb Vasc Biol.
21:746–751. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
He Y, Huang Y, Zhou L, Lu LM, Zhu YC and
Yao T: All-trans retinoic acid inhibited angiotensin
II-induced increase in cell growth and collagen secretion of
neonatal cardiac fibroblasts. Acta Pharmacol Sin. 27:423–429.
2006.
|
|
8
|
Gidlof AC, Ocaya P, Krivospitskaya O and
Sirsjo A: Vitamin A: a drug for prevention of
restenosis/reocclusion after percutaneous coronary intervention?
Clin Sci (Lond). 114:19–25. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Miano JM, Kelly LA, Artacho CA, Nuckolls
TA, Piantedosi R and Blaner WS: All-trans-retinoic acid
reduces neointimal formation and promotes favorable geometric
remodeling of the rat carotid artery after balloon withdrawal
injury. Circulation. 98:1219–1227. 1998.
|
|
10
|
Lee CW, Park SJ, Park SW, Kim JJ, Hong MK
and Song JK: All-trans-retinoic acid attenuates neointima
formation with acceleration of reendothelialization in
balloon-injured rat aorta. J Korean Med Sci. 15:31–36. 2000.
|
|
11
|
Fujii H, Sato T, Kaneko S, Gotoh O,
Fujii-Kuriyama Y, Osawa K, Kato S and Hamada H: Metabolic
inactivation of retinoic acid by a novel P450 differentially
expressed in developing mouse embryos. EMBO J. 16:4163–4173. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dong D, Ruuska SE, Levinthal DJ and Noy N:
Distinct roles for cellular retinoic acid-binding proteins I and II
in regulating signaling by retinoic acid. J Biol Chem.
274:23695–23698. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Blomhoff R, Green MH, Berg T and Norum KR:
Transport and storage of vitamin A. Science. 250:399–404. 1990.
View Article : Google Scholar
|
|
14
|
Czibik G, Wu Z, Berne GP, Tarkka M, Vaage
J, Laurikka J, Järvinen O and Valen G: Human adaptation to ischemia
by preconditioning or unstable angina: involvement of nuclear
factor kappa B, but not hypoxia-inducible factor 1 alpha in the
heart. Eur J Cardiothorac Surg. 34:976–984. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Olofsson PS, Sheikine Y, Jatta K, Ghaderi
M, Samnegard A, Eriksson P and Sirsjö A: A functional interleukin-1
receptor antagonist polymorphism influences atherosclerosis
development. The interleukin-1beta:interleukin-1 receptor
antagonist balance in atherosclerosis. Circ J. 73:1531–1536. 2009.
View Article : Google Scholar
|
|
16
|
Olofsson PS, Söderström LA, Jern C, Sirsjö
A, Ria M, Sundler E, de Faire U, Wiklund PG, Ohrvik J, Hedin U,
Paulsson-Berne G, Hamsten A, Eriksson P and Hansson GK: Genetic
variants of TNFSF4 and risk for carotid artery disease and stroke.
J Mol Med (Berl). 87:337–346. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gundersen TE, Bastani NE and Blomhoff R:
Quantitative high-throughput determination of endogenous retinoids
in human plasma using triple-stage liquid chromatography/tandem
mass spectrometry. Rapid Commun Mass Spectrom. 21:1176–1186. 2007.
View Article : Google Scholar
|
|
18
|
O’Connell TD, Rodrigo MC and Simpson PC:
Isolation and culture of adult mouse cardiac myocytes. Methods Mol
Biol. 357:271–296. 2007.
|
|
19
|
Le Tallec LP, Korwin-Zmijowska C and
Adolphe M: Limitations of alginate gels as a culture model for the
study of the effects of UVA radiation on human dermal fibroblasts.
Cell Biol Toxicol. 13:95–102. 1997.PubMed/NCBI
|
|
20
|
Vinet L, Rouet-Benzineb P, Marniquet X,
Pellegrin N, Mangin L, Louedec L, Samuel JL and Mercadier JJ:
Chronic doxycycline exposure accelerates left ventricular
hypertrophy and progression to heart failure in mice after thoracic
aorta constriction. Am J Physiol Heart Circ Physiol. 295:H352–H360.
2008. View Article : Google Scholar
|
|
21
|
Brattelid T, Winer LH, Levy FO, Liestol K,
Sejersted OM and Andersson KB: Reference gene alternatives to Gapdh
in rodent and human heart failure gene expression studies. BMC Mol
Biol. 11:222010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Seeland U, Selejan S, Engelhardt S, Muller
P, Lohse MJ and Böhm M: Interstitial remodeling in beta1-adrenergic
receptor transgenic mice. Basic Res Cardiol. 102:183–193. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sun H and Kawaguchi R: The membrane
receptor for plasma retinol-binding protein, a new type of
cell-surface receptor. Int Rev Cell Mol Biol. 288:1–41. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wolf G: Identification of a membrane
receptor for retinol-binding protein functioning in the cellular
uptake of retinol. Nutr Rev. 65:385–388. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Blomhoff R and Blomhoff HK: Overview of
retinoid metabolism and function. J Neurobiol. 66:606–630. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bilbija D, Haugen F, Sagave J, Baysa A,
Bastani N, Levy FO, Sirsjö A, Blomhoff R and Valen G: Retinoic acid
signalling is activated in the postischemic heart and may influence
remodelling. PloS One. 7:e447402012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Palace VP, Hill MF, Khaper N and Singal
PK: Metabolism of vitamin A in the heart increases after a
myocardial infarction. Free Radic Biol Med. 26:1501–1507. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Azevedo PS, Minicucci MF, Chiuso-Minicucci
F, Justulin LA Jr, Matsubara LS, Matsubara BB, Novelli E, Seiva F,
Ebaid G, Campana AO, Zornoff LA and Paiva SA: Ventricular
remodeling induced by tissue vitamin A deficiency in rats. Cell
Physiol Biochem. 26:395–402. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Oliveira LC, Azevedo PS, Minicucci ME,
Rafacho BP, Duarte DR, Matsubara LS, Matsubara BB, Paiva SA and
Zornoff LA: Retinoic acid prevents ventricular remodelling induced
by tobacco smoke exposure in rats. Acta Cardiol. 66:3–7.
2011.PubMed/NCBI
|
|
30
|
Paiva SA, Matsubara LS, Matsubara BB,
Minicucci MF, Azevedo PS, Campana AO and Zornoff LA: Retinoic acid
supplementation attenuates ventricular remodeling after myocardial
infarction in rats. J Nutr. 135:2326–2328. 2005.PubMed/NCBI
|
|
31
|
Krivospitskaya O, Elmabsout AA, Sundman E,
Söderström LA, Ovchinnikova O, Gidlöf AC, Scherbak N, Norata GD,
Samnegård A, Törmä H, Abdel-Halim SM, Jansson JH, Eriksson P,
Sirsjö A and Olofsson PS: A CYP26B1 polymorphism enhances retinoic
acid catabolism which may aggravate atherosclerosis. Mol Med.
18:712–718. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ocaya PA, Elmabsout AA, Olofsson PS, Torma
H, Gidlof AC and Sirsjo A: CYP26B1 plays a major role in the
regulation of all-trans-retinoic acid metabolism and
signaling in human aortic smooth muscle cells. J Vasc Res.
48:23–30. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Saito A, Sugawara A, Uruno A, Kudo M,
Kagechika H, Sato Y, Owada Y, Kondo H, Sato M, Kurabayashi M,
Imaizumi M, Tsuchiya S and Ito S: all-trans retinoic acid
induces in vitro angiogenesis via retinoic acid receptor: possible
involvement of paracrine effects of endogenous vascular endothelial
growth factor signaling. Endocrinology. 148:1412–1423. 2007.
|
|
34
|
Lee CW, Park SJ, Park SW, Kim JJ, Hong MK
and Song JK: All-trans-retinoic acid attenuates neointima
formation with acceleration of reendothelialization in
balloon-injured rat aorta. J Korean Med Sci. 15:31–36. 2000.
|