Introduction
Alzheimer’s disease (AD) is a devastating
neurodegenerative disorder that usually affects individuals of an
older age (1). Human life
expectancy has increased tremendously over the years due to
improved public health programs and advances in clinical medicine.
At the same time, the incidence of AD is rapidly increasing. It has
been reported that >15 million individuals are suffering from AD
and this number is expected to more than double in the next
generation (2). Studies on the
pathogenesis of AD require the use of animal models that develop
some degree of amyloid pathologies in the brain (3). The utilization of animal models is
crucial for any biomedical research on human disease processes at
the cellular and molecular levels, and for developing novel
therapies. A transgenic animal model may also be used to examine
the pathogenic mechanisms of AD and related disorders, such as
frontotemporal dementia. Furthermore, these models may aid in the
development of vaccines or improve current treatment
strategies.
AD is considered a multifactorial and polygenic
disease in which environmental and genetic factors play a major
role. Although several candidate genes have been surveyed, three
are believed to be responsible for the rare early-onset familial
form of the disease. These include the amyloid precursor protein
(APP), as well as the presenilin 1 (PSEN1) and
presenilin 2 (PSEN2) genes (1). Genetically modified mice, flies,
fish and worms have been developed that reproduce different AD
histopathologies, such as plaques containing β-amyloid (Aβ) and
tau-containing neurofibrillary tangles (NFTs) (4). Previusly, the production of
transgenic mini-pigs carrying the APP695sw transgene using a
handmade cloning procedure was reported (4). However, to our knowledge, AD-like
symptoms in a transgenic canine model have not been published to
date.
One main purpose of an animal model of AD is to
replicate the symptoms, lesions, or causes of the disease. Numerous
transgenic murine lines have successfully been used to partially
reproduce AD lesions, such as extracellular deposits of the Aβ
peptide and the intracellular accumulation of the tau protein
(5). Mutated human APP
(mhAPP) transgenes result in the deposition of the Aβ
peptide, similar but not identical to the plaques observed in
senile humans (6). Canines are
extensively used in various biomedical studies as model animals.
The development of successful somatic cell nuclear transfer (SCNT)
procedures in canines has made considerable progress. Consequently,
small, medium and large breed dogs have been cloned from cultured
cells as the genetic donor or long-term cryopreserved somatic cells
(5,7–9).
Therefore, it has become possible to use donor cells to create
knockout and knockin canines or animals overexpressing the gene of
interest. In the present study, we established a somatic cell line
overexpressing mhAPP to generate a canine model of AD by
SCNT. The transgenic canines were confirmed to have an increased
mhAPP expression in the brain, resulting in the accumulation
of Aβ, enlarged ventricles, an atrophied hippocampus and abnormal
behavior.
Materials and methods
Animal care and welfare
Standard procedures established by the SooAm Biotech
Research Foundation (Seoul, Korea) for the Accreditation of
Laboratory Animal Care were followed strictly when caring for the
dogs used in our study. All experiments and surgical procedures
were conducted in accordance with the Guidelines for the Care and
Use of Laboratory Animals published by the SooAm Biotech Research
Foundation. Canines at the pro-estrus stage (mixed breed, 1–7 years
old, 20–25 kg body weight) were obtained from a breeder and reared
indoors in separated cages up until oocyte recovery. Following
oocyte recovery, the canines were sent back to the breeder. None of
the dogs were used repeatedly. The pregnant recipient canines were
kept in the research facility while the others were returned to the
breeder.
Chemicals
All chemicals were purchased from Sigma-Aldrich (St.
Louis, MO, USA), unless otherwise stated.
Cell culture and establishment of canine
fibroblasts
Human IMR-32 neuroblastoma cells (Korean Cell Line
Bank, Seoul, Korea) were maintained in Dulbecco’s modified Eagle’s
medium (DMEM) containing penicillin and streptomycin, and 10% fetal
bovine serum (FBS) (all from Invitrogen, Carlsbad, CA, USA).
Primary culture of canine adult and fetal fibroblasts was performed
as described in a previous study (8). Briefly, fetal fibroblasts were
obtained from artificially inseminated embryos on the 30th day of
pregnance by trypsinization. Fibroblasts were cultured in DMEM
containing 10% FBS.
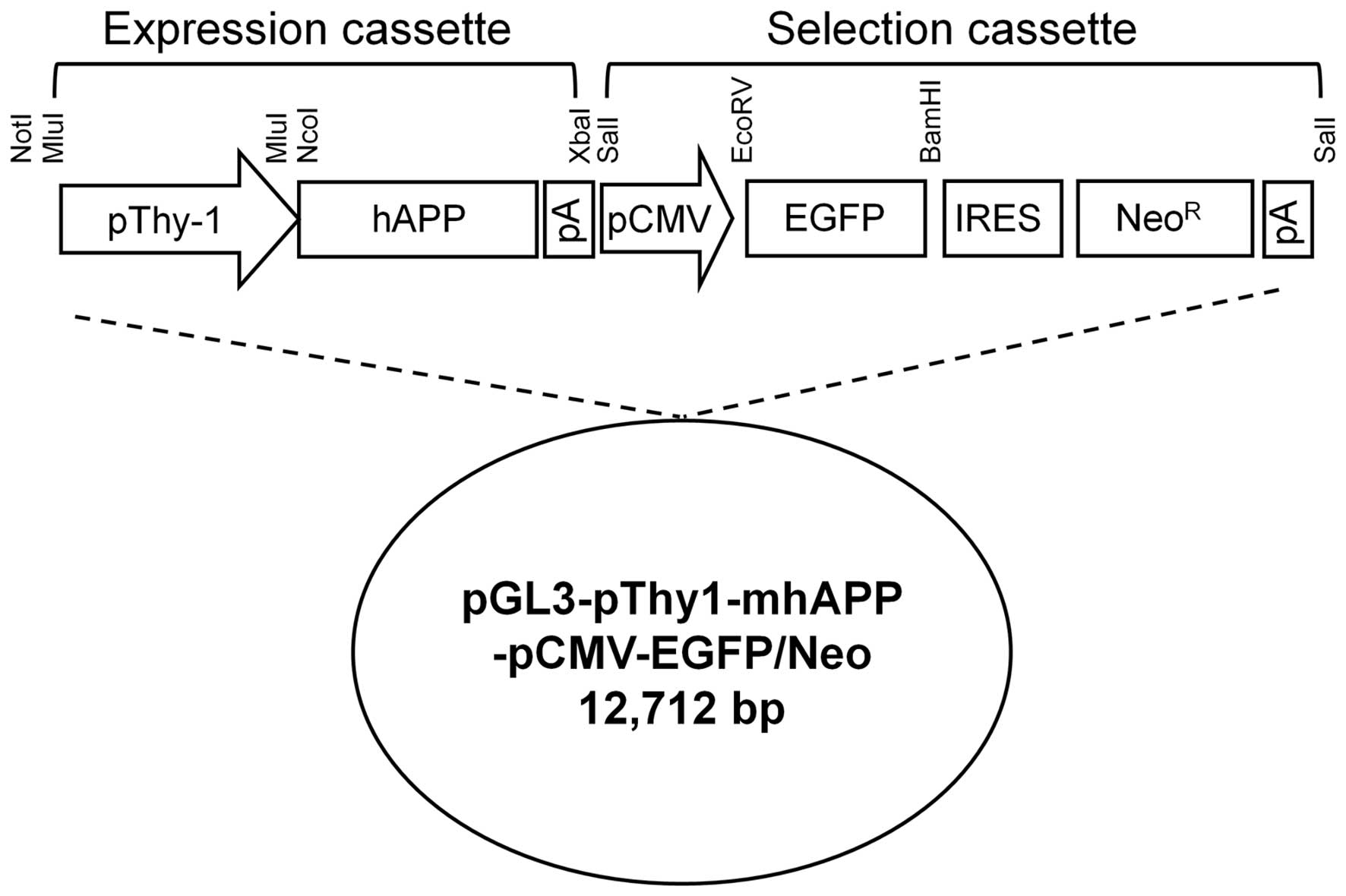
Construction of transgenic vector
Various regions of the Thy-1 promoter [from
nucleotides −2.0 to +2.3 kilonucleotides (knts), +1 = the
transcriptional start site] were isolated by a long-range
polymerase chain reaction (PCR) (LA Taq; Takara Bio, Inc., Shiga,
Japan) using genomic DNA from beagle fibroblasts as a template.
Amplified fragments were ligated into the MluI site of the
promoterless pGL3-Basic vector (Promega, Madison, WI, USA). Human
APP cDNA was prepared by PCR using the full-length clone
from the Mammalian Gene Collection (cat. no. 6152423; Invitrogen)
as a template. The amplified APP gene was mutated by two
primer sets containing Swedish mutation (KM to NL) or Indiana
mutation (V to F) indicated in Table
I using a Site-Directed Mutagenesis kit (Stratagene, Inc., La
Jolla, CA, USA). The mutated APP gene were further ligated into the
NcoI and XbaI sites of the recombinant pGL3-Basic
vector containing the Thy-1 promoter. An enhanced green
fluorescent protein (EGFP) gene from the internal ribosomal entry
site (pIRES)2_EGFP (Clontech Laboratories, Inc., Madison, WI, USA)
was ligated into the EcoRV and BamHI sites of the
pIRES_Neo vector (Clontech Laboratories, Inc.). This selection
cassette was inserted into the SalI site of the backbone
plasmid containing the dog Thy-1 promoter region and mutant
human APP gene. The final construct
(pGL3-pThy1-mhAPP-pCMV-EGFP/Neo) was linearized by NotI
digestion (Fig. 1). All amplified
products obtained at each step of the construct production were
analyzed by sequencing analysis (Genotech Co. Ltd., Daejeon,
Korea).
 | Table IPrimer sequences. |
Table I
Primer sequences.
| Name | Direction | Sequences (5′ to
3′) |
|---|
| Human APP
cDNA | Forward | CGC GCA GGG TCG CGA
TGC TGC CCG |
| Reverse | CTG TGG CGG GGG TCT
AGT TCT GCA |
| Thy-1
promoter region | Forward | GAC TGG ACC ATC CTT
GCA GCT CAT |
| Reverse | GCT CAG TCC TTG ATC
TGG GGG TGG |
| KM to NL (Swedish
mutation) | Forward | CGA GAT TCT GAA GTG
AAC CTG GAT GCA GAA TTC CGA CAT G |
| Reverse | CAT GTC GGA ATT CTG
CAT CCA GGT TCA CTT CAG AAT CTC G |
| V to F (Indiana
mutation) | Forward | GGA CTC ATG GTG GGC
GGT TTT GTC ATA GCG ACA GTG ATC |
| Reverse | GAT CAC TGT CGC TAT
GAC AAA ACC GCC CAC CAT GAG TCC |
| EGFP | Forward | CAC AAC CAT GGT GAG
CAA GGG CGA |
| Reverse | TTA CTT GTA CAG CTC
GTC CAT GCC |
| Confirming primer
A | Forward | CCT TGT GCT GTC TCC
CCC TC |
| Confirming primer
B | Reverse | TCA CAA AGT GGG GAT
GGG TC |
| Confirming primer
C | Forward | CAT GAA GCA GCA CGA
CTT CT |
| Confirming primer
D | Reverse | CCT AGG AAT GCT CGT
CAA GA |
| Confirming primer
for human APP | Forward | GAG GTG GAA GAA GAA
GAA GCC |
| Reverse | GTC AAC GGC ATC AGG
GGT ACT |
| Confirming primer
for canine APP | Forward | GAT GTT GAG GAA GAG
GAA GCT GAG G |
| Reverse | GGG AAG AGG TTC CTG
GGT TG |
Transient transfection and reporter gene
assay
To control for different transfection efficiencies
of the various luciferase constructs, a Rous sarcoma virus
(RSV)-lacZ plasmid was used to co-transfect the IMR-32 cells along
with the Thy-1 promoter-luciferase construct. Briefly,
3×105 IMR-32 cells were seeded in six-well tissue
culture plates one day prior to transfection. Thy-1
promoter-luciferase constructs (3.5 μg) and 0.5 μg of the RSV-lacZ
plasmid were used to transiently transfect the cells using
Lipofectamine™ 2000 (Invitrogen) followed by incubation for an
additional 48 h. Cellular lysates were prepared using 150 μl of
reporter lysis buffer and assayed for luciferase activity using the
Luciferase Assay System (both from Promega). Luminescence was
measured using a GloMax 20/20 Luminometer (Promega) and
β-galactosidase activity was measured using a β-galactosidase
Enzyme Assay System (Promega). Promoter activity was expressed as a
percentage of relative luciferase activity (RLA %;
luciferase/β-galactosidase activity).
Laparotomy and oocyte collection
Mature canine oocytes were collected in vivo
by laparotomy at 72–84 h after ovulation using standard procedures
(8,9). The time of ovulation was determined
by the serum progesterone concentration and vaginal cytology
(8). All the operative and
post-operative procedures and care were performed by licensed
veterinarians. The reproductive tract was exposed during
mid-ventral laparotomy under a general anesthesia. The fimbriated
end of the oviduct was canalized using a six-gauge bulbed needle
held in place by a surgical ligature. The oviductal lumen at the
base of the utero-oviductal junction was cannulated using a
24-gauge hypodermic needle (Angiocath TM Plus; Becton-Dickinson,
Franklin Lakes, NJ, USA). Approximately 10 ml of TCM 199 collection
medium supplemented with HEPES (Sigma-Aldrich) was used to flush
the hypodermic needle. The medium passed through the oviductal
lumen and bulbed needle before being deposited into a sterile
plastic Petri dish. After flushing both oviducts, each ovarian
bursa was incised to pull the ovary out and the corpora lutea were
counted. Finally, the abdominal incision was closed using a
two-layer method followed by the application of a surgical adhesive
along the skin incision.
SCNT and embryo transfer
SCNT was performed according to a previously
described procedure (5,9) with some modifications. Briefly,
enucleation of the stripped oocytes was performed under an inverted
microscope with epifluorescence (TE2000-E; Nikon Corp., Tokyo,
Japan). Using an injection pipette, a trypsinized fibroblast with a
smooth surface was transferred into the perivitelline space of an
enucleated oocyte. The couplets were fused with two DC pulses of
1.75 kV/cm for 15 μsec from a BTX Electro-Cell Manipulator 2001
(BTX Inc., San Diego, CA, USA) and transferred to naturally
synchronized recipients The embryos were loaded into a Sovereign
Tom Cat catheter (Sherwood Medical, St. Louis, MO, USA) with a
minimal volume of medium and gently transferred into the 2/3 to
deep distal portion of the oviduct without insufflating air.
Diagnosis of pregnancy
Approximately 30 days after embryo transfer, the
recipients were examined using a portable real-time ultrasonography
machine with a 3.5-MHz curved transducer (SSD-900; Aloka Co. Ltd.,
Tokyo, Japan). To detect embryonic or fetal death, sizes and shapes
of the chorionic cavities along with embryonic or fetal heartbeats
were examined. After confirming pregnancy, this examination was
performed every seven days to monitor fetal development until
birth.
Genomic DNA extraction and PCR
Canine genomic DNA was isolated using a G-DEX™ II
Genomic DNA Extraction kit (Intron Biotechnology, Inc., Suwon,
Korea). Genomic DNA (100 ng) or plasmid DNA (10 ng) was amplified
in a 50-μl PCR reaction containing 1 U of LA™ or Ex™-Taq polymerase
(Takara Bio, Inc.) and 10 pmol of specific primers. PCR was carried
out for 35 cycles of denaturation for 30 sec at 94°C, and annealing
and extension for 1–3 min at 68°C followed by a final extension for
30 min at 72°C. All primers are presented in Table I. PCR products were separated on
an agarose gel, stained with ethidium bromide and photographed
under UV illumination. The images were scanned using a Gel Doc EQ
system (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
RNA preparation and quantitative
RT-PCR
Total RNA was isolated from the heart, pancreas,
liver, lungs, kidneys, spleen and cerebrum of two puppies (AM144
and AM145) using TRIzol reagent (Invitrogen). DNA contamination was
further eliminated by treatment with DNAase (Invitrogen). Total RNA
(1 μg) was reverse transcribed into first-strand cDNA using M-MLV
reverse transcriptase (Invitrogen) and random primers (9-mer;
Takara Bio, Inc.). The GAPDH gene was amplified to evaluate
RNA degradation and was used to control for variation in mRNA
concentration in the RT-PCR reactions. GAPDH and human
APP genes were quantified using 28 and 30 cycles,
respectively. cDNA was amplified in a 20-μl PCR reaction containing
1 U TaqDNA polymerase (Intron Biotechnology, Inc.) and 10 pmol of
the specific primers (Table I).
The PCR procedure included denaturation at 95°C for 30 sec,
annealing at 58°C for 30 sec and extension at 72°C for 30 sec. The
PCR products (8 μl) were separated on a 2% agarose gel, stained
with ethidium bromide and photographed under UV illumination. The
images were scanned using a Gel Doc EQ system (Bio-Rad
Laboratories, Inc.).
Western blot analysis
Proteins (40 μg) were separated by electrophoresis
on SDS-PAGE and then transferred onto a PVDF membrane. The membrane
was incubated with antibodies against the following proteins: APP
and Aβ (1:1,000; cat. nos. 2450 and 2454; Cell Signaling
Technology, Beverly, MA, USA) or β-actin (1:2,000; Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA). Immunoreactive proteins
were visualized by exposure to X-ray film. Band intensities were
quantified by image scanning using a Gel Doc EQ system (Bio-Rad
Laboratories, Inc.), corrected by background subtraction, and
normalized using β-actin as an internal control.
Immunohistochemical staining
Brain tissues were embedded in paraffin. Sections
(5-μm thick) were deparaffinized and hydrated in descending grades
of ethanol. The sections were subsequently incubated at room
temperature for 2 h with an anti-Aβ antibody (diluted 1:100, #2454;
Cell Signaling Technology) and rabbit anti-ionized calcium-binding
adapter molecule 1 (Iba1, diluted 1:500, #019–19741; Wako Pure
Chemical Industries Ltd., Osaka, Japan) in 10% normal goat serum.
The sections were reacted with biotinylated goat anti-rabbit IgG
(Vector ABC Elite kit; Vector Laboratories, Inc., Burlingame, CA,
USA) for 45 min at room temperature. Immunoreactivity was assessed
using the avidin-biotin peroxidase complex (Vector ABC Elite kit)
prepared according to the manufacturer’s instructions. The
peroxidase reaction was developed using a diaminobenzidine
substrate kit (SK-4100; Vector Laboratories, Inc.). The primary
antibodies were omitted for a few test sections in each experiment
as a control. The sections were counterstained with Harris’s
hematoxylin before mounting.
Results
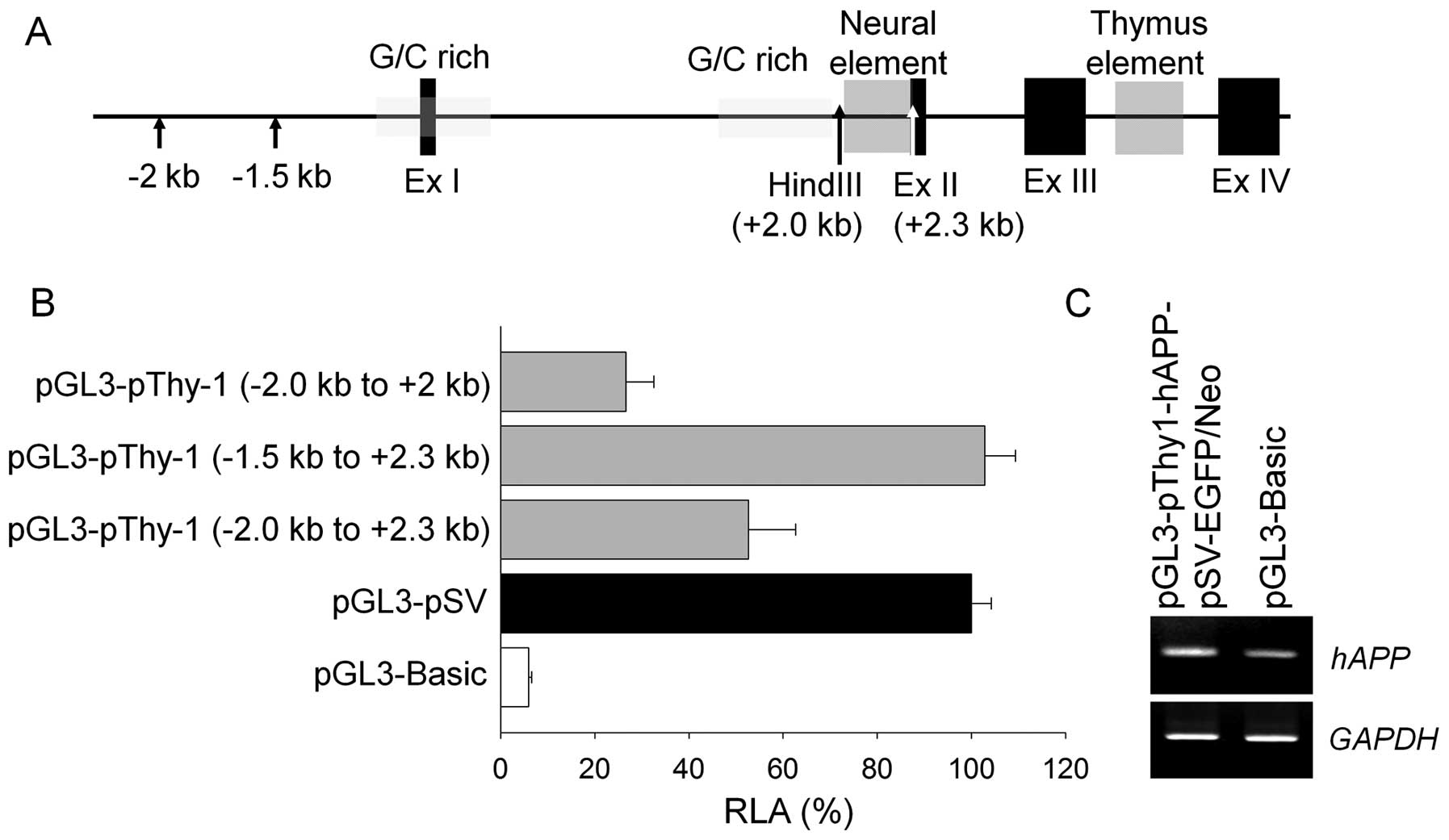
Construction and functional activity of
the transgenic vector
We generated a human APP gene expressing a
cassette containing two well-characterized mutations, Swedish
(K670N and M671L) and Indiana FAD (V717F), which successfully
induces AD-like pathological symptoms in mouse models (10–12). The mhAPP gene was
controlled by a canine Thy-1 promoter for selective
expression in neural tissues. Specific regions of the Thy-1
promoter were selected by a luciferase activity assay in human
IMR-32 neuroblastoma cells (Fig. 2A
and B). We deleted the upstream portion (−2.0 to −1.5 knts, +1
= transcriptional start) of the Thy-1 promoter as it
possessed unknown inhibitory elements, and the neural element
(between −2.0 to −2.3 knts) in intron A was added as it was
essential for promoter activity in the IMR-32 cells. Finally, the
Thy-1 promoter (−1.5 to +2.3 knts) was found to have maximum
activities similar to the SV promoter in neural cells. The
transgenic vector was transiently expressed in IMR-32 cells and
induced expression levels of APP transcripts two-fold higher
compared to the empty pGL3-Basic Vector (Fig. 2C). The transgenic vector also
contained a selection cassette including EGFP and neomycin
resistance genes linked by IRES, and were controlled by the CMV
promoter to facilitate selection for further processes.
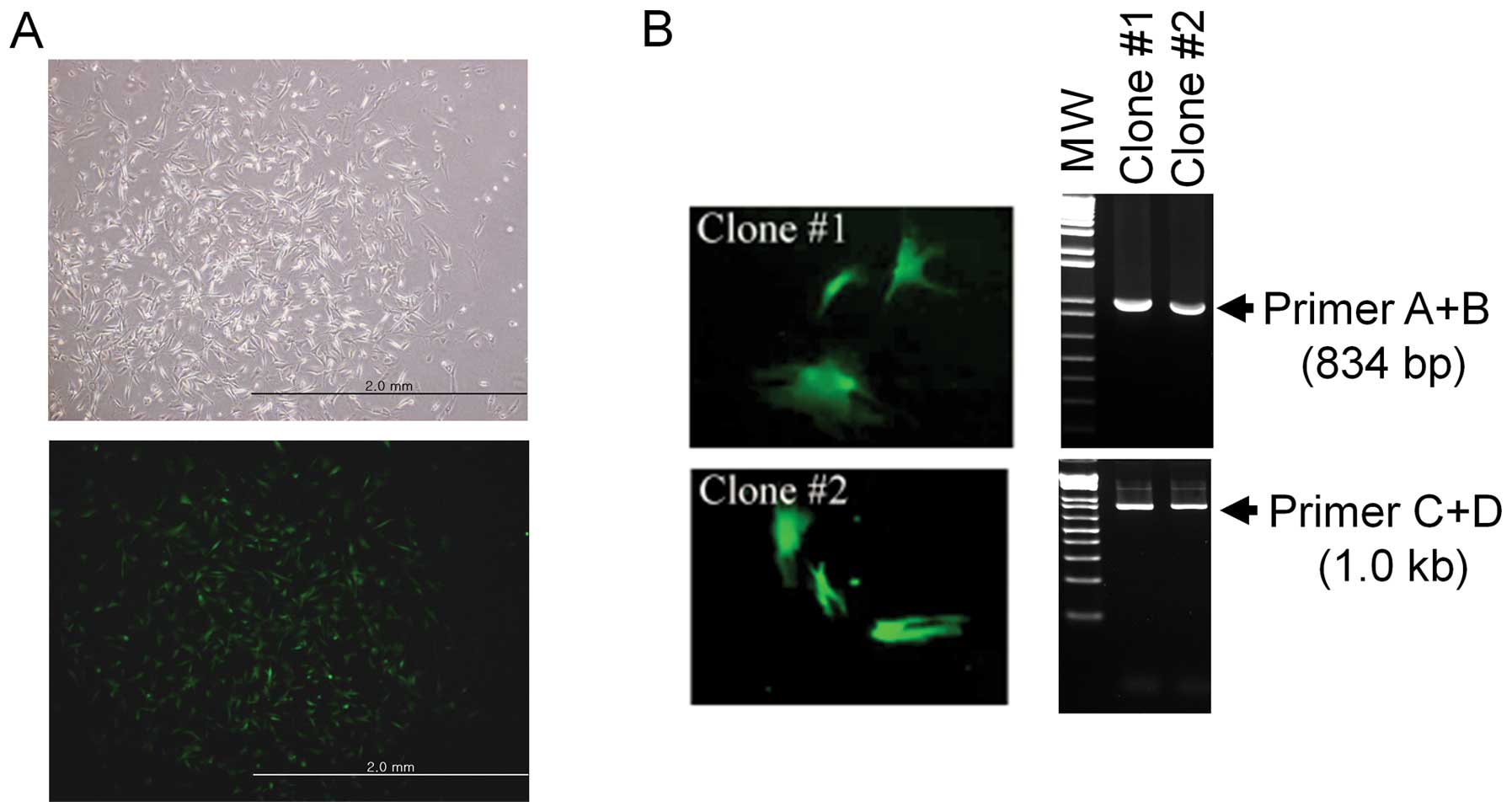
Establishment and confirmation of the
somatic cell line as a nuclear donor
Canine fibroblasts were transfected with the
linearized transgenic vector and cultured in medium containing 350
μg/ml of G418 to select a transgenic clone. The cells resistant to
G418 for four weeks were further confirmed to express EGFP by
fluorescence microscopy (Fig.
3A). The insertion of the transgenic vector in the transgenic
cells was confirmed by PCR (Fig.
3B). The positive cells were isolated and frozen until
SCNT.
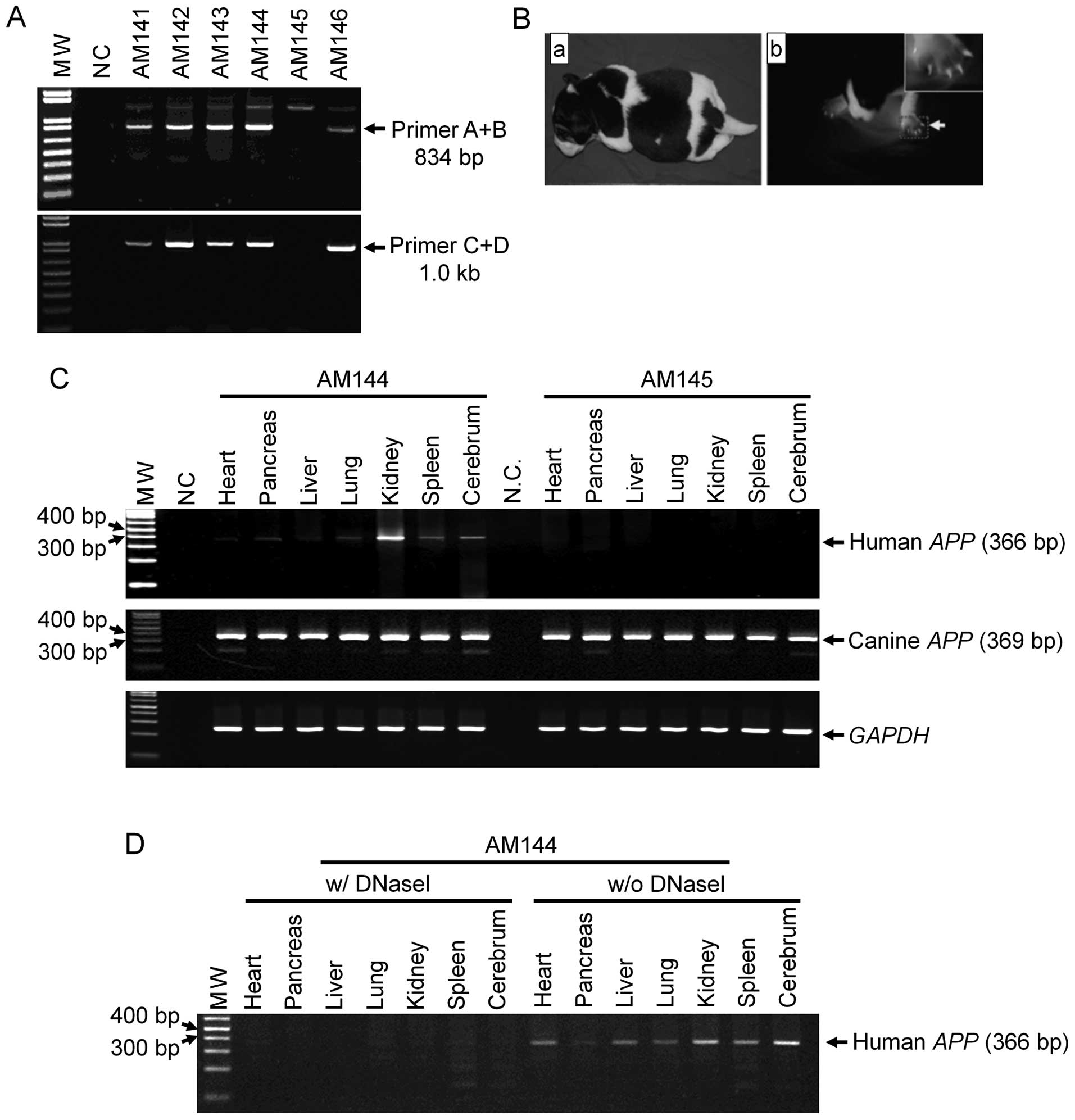
Production and characterization of
transgenic puppies
SCNT was performed using the transgenic cell line
and 332 oocytes matured in vivo from 29 donors. Six puppies
(AM141 to 146) were born from four out of 17 surrogates. Five
puppies (AM141, 142, 143, 144 and 146) were confirmed to be
transgenic animals by PCR (Fig.
4A), and observed to express EGFP in their nails, toes and fur
(Fig. 4B).
An EGFP-positive puppy (AM144) and EGFP-negative
littermate (AM145) were sacrificed to measure mhAPP
expression inserted by SCNT. To eliminate genomic DNA
contamination, we treated DNase I on total RNAs before the
reverse-transcriptase reaction. In addition, we designed specific
primers to distinguish between human and canine APP
transcripts on their mRNA sequences. The expression of the
mhAPP gene was only observed in the organs of the
EGFP-positive puppy (Fig. 4C).
The amplified mhAPP fragments were confirmed not to be due
to genomic DNA contamination (Fig.
4D). Endogenous canine APP mRNA was detected in the
organs of both animals. Amplified mhAPP transcription was
further confirmed by sequence analysis. Thus, the transgenic
puppies we created were found to successfully express the exogenous
mhAPP gene.
AD-associated pathology in transgenic
canines
One of the mhAPP transgenic canines (AM146)
was mated with a wild-type female dog (AF165) to produce more
puppies for observing the development of AD-like symptoms and
stabilizing of the mhAPP gene. This mating produced four
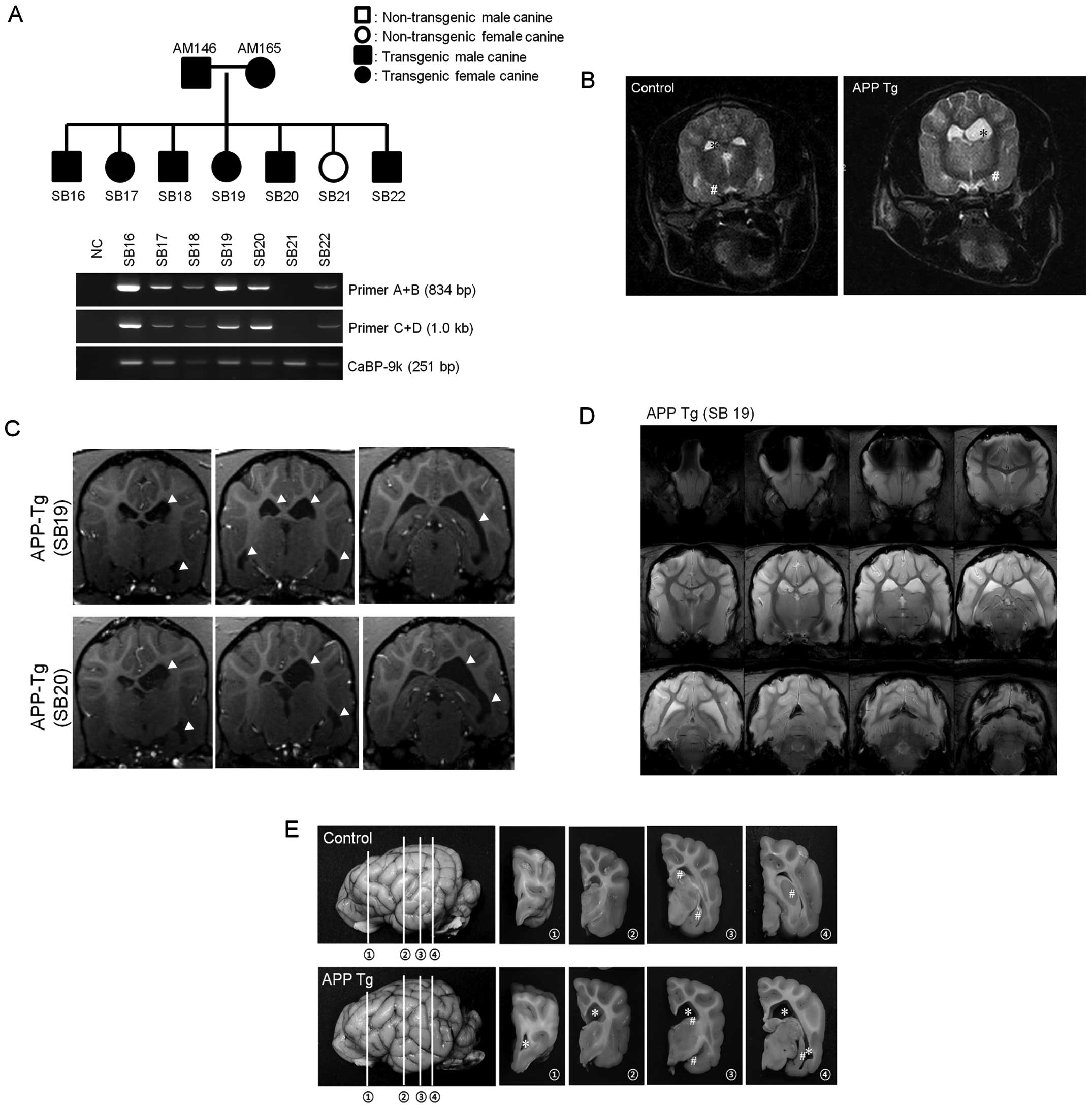
male and three female puppies (SB16 to 22). All canines had the
transgenic vector in their genomic DNA apart from SB21 (Fig. 5A). When the puppies were six
months old, one transgenic puppy (SB17) presented recurrent
behavioral disorders, such as tetanic convulsions. Although
seizures are not a common phenotype of AD, recent reports have
indicated that seizures may be one of the phenotypes of AD
(13–17). The brain of SB17 was further
examined by magnetic resonance imaging (MRI, 7.0 T). AD-like
symptoms, including enlarged ventricles and an atrophied
hippocampus, were observed (Fig.
5B). The symptoms are not common in canines; however, emerging
evidence has indicated that ventricle enlargement and hippocampal
atrophy are phenotypes of human AD (18–21). Although the other littermates did
not develop the behavioral disorders observed in SB17, an MRI image
of SB19 and SB20 showed results similar to those of SB17,
indicating that these puppies also had enlarged ventricles and an
atrophied hippocampus (Fig. 5C and
D). Our results indicated that the transgenic puppies had
developed human AD-like symptoms.
SB17 was sacrificed and pathological AD phenotypes
in the entire brain were analyzed. The cerebral ventricles were
greatly enlarged (Fig. 5E). The
hippocampus had also atrophied and became degenerated in the
transgenic puppy compared to the non-transgenic animal, as
previously documented by an MRI. To further characterize this
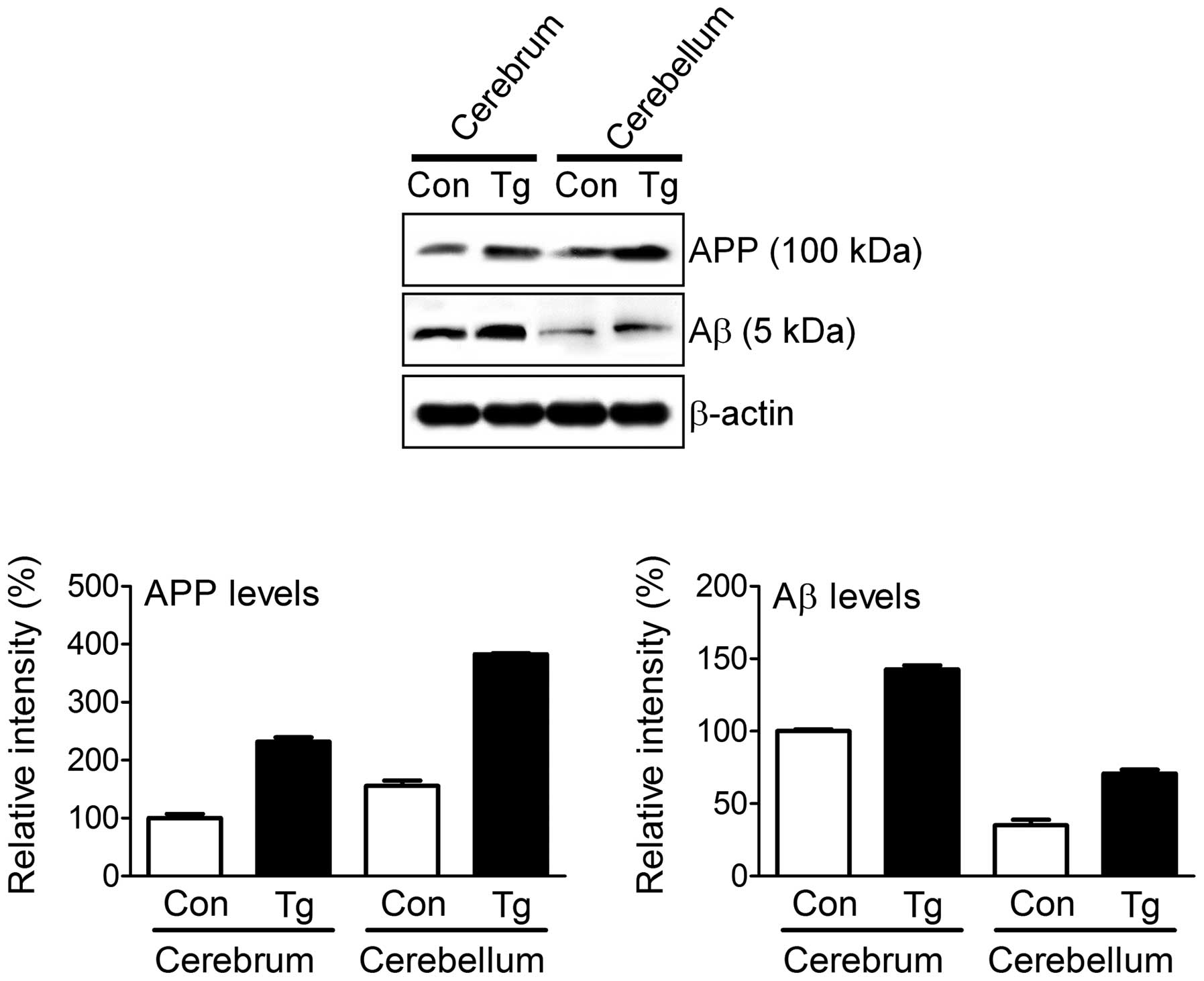
animal, we examined the molecular mechanisms underlying the AD-like
symptoms. APP was highly expressed in the cerebrum and
cerebellum of the transgenic puppy compared to the control animal
(Fig. 6). However, we could not
distinguish the expression of mhAPP from that of endogenous
canine APP with the currently available anti-APP sera.
Increased levels of Aβ, hallmarks of AD, were also observed in the
transgenic puppy. This finding suggests that the overexpression of
mhAPP in canines induces an accumulation of the hallmarks of
AD (Aβ accumulation in the brain, resulting in AD-like symptoms
such as enlarged ventricles, an atrophied hippocampus and abnormal
behavior).
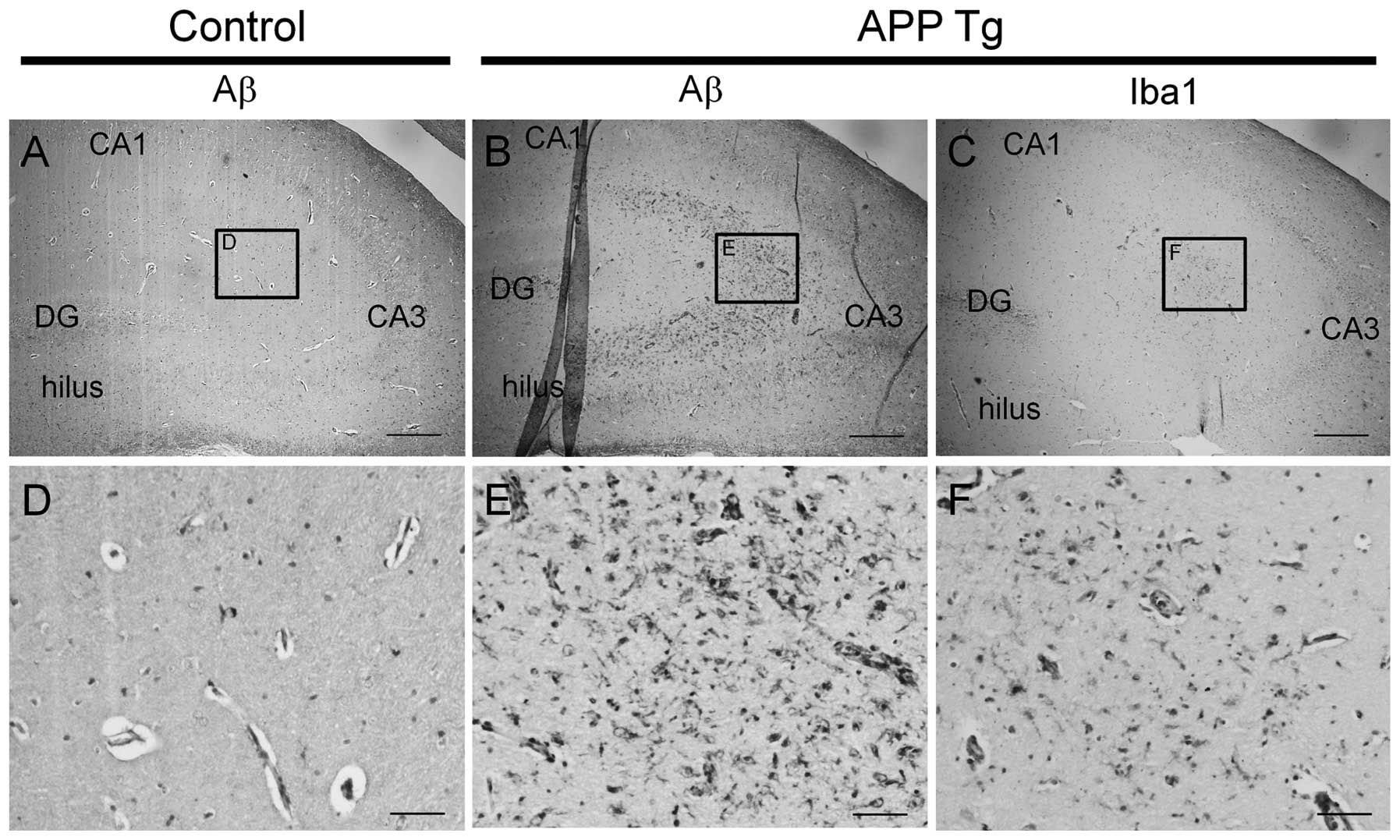
For additional histopathological characterization,
Aβ accumulation in the hippocampal regions was further measured
with an immunohistochemical assay. In the hippocampus, Aβ-positive
immunoreactivity was rarely detected in non-transgenic canines
(Fig. 7A and D), whereas the
immunoreactivity was observed to a great extent in the transgenic
canines (Fig. 7B and E).
Additionally, microglial activation was observed in the hippocampus
of the transgenic canines where Aβ accumulation was present
(Fig. 7C and F). These findings
suggest that Aβ accumulation in cells induces an immune response,
as previously described (22).
Discussion
In this study, we generated transgenic canines
overexpressing the human APP gene containing two well-known
AD-associated mutations, Swedish (K670N and M671L) and Indiana FAD
(V717F), which inhibited proteolytic processing by α-secretase. The
animals were produced with a non-viral gene delivery system to
avoid unknown side-effects of viral protein contamination in the
chromosome. mhAPP was successfully overexpressed in the
brain of the transgenic animals and the protein end-product, Aβ,
accumulated in the brain. The transgenic canines developed abnormal
neurological symptoms, such as tetanic convulsions along with
enlarged ventricles and an atrophied hippocampus, similar to humans
with AD. In addition, Aβ plaque-like structures, a
histopathological hallmark of AD, were detected in the frontal
cortex regions, and significant Aβ accumulation was further
observed in the cortex and hippocampus. Thus, the transgenic
canines we produced are good candidates for replacing the present
rodent model for studying the pathophysiological characteristics of
AD and performing pharmaceutical research for the treatment of this
disease.
AD is the most common dementia disorder in humans.
The major pathological hallmarks of this disease are an abnormal
accumulation of extracellular amyloid plaques and intracellular
NFTs in vulnerable brain regions. The tangles are primarily
composed of paired helical filaments of hyper-phosphorylated forms
of the microtubule-associated protein, tau. The main constituent of
the plaques is Aβ, a peptide with 40–42 amino acids derived from
APP by sequential proteolytic cleavage by β- and γ-secretase
(23–25). Numerous mouse lines that develop
Aβ deposits have been produced (6). MhAPP transgenes in mice cause
Aβ deposition, although these lesions are not identical to the Aβ
protein in human AD plaques. Aside from Aβ deposition, axon
dystrophy and dendrite alterations have been observed (6). All APP mutations in
transgenic mice increase Aβ42 levels, although the overexpression
of wild-type APP alone does not induce Aβ deposition in mice
(6). Double transgenic mice
(expressing both APP and PS1) develop lesions
earlier. The development of APP transgenic mice has raised
new questions concerning the mechanisms of neuronal loss, the
accumulation of Aβ protein in the body of neurons, inflammation and
gliosis, and dendritic alterations. These data from transgenic mice
have provided some insight into the kinetics of the pathogenesis of
AD. The connections between AD-associated symptoms, lesions and
increased levels of Aβ oligomers have been difficult to unravel.
Thus far, the best animal model for studying AD has been transgenic
mice. However, the transgenic canines presenting AD-like symptoms
we generated may prove to be useful for studying AD in humans.
Successful studies of human diseases require an
appropriate animal model. Common animal models for AD research are
transgenic mice that overexpress a mutant form of the human Aβ
precursor protein and/or enzymes implicated in its metabolic
processing (26). However, rodent
models are not sufficient to fully elucidate the pathogenesis of AD
due to genetic, physiological and anatomic differences between mice
and humans. Canines are more suitable for examining human disorders
than mice, as canines have evolved physiologically and genetically
in close proximity to humans (27,28). In addition, canine models
naturally develop an age-related cognitive dysfunction that
reproduces several aspects of AD (27,28). Numerous studies using canine
cohorts examining several behavioral paradigms have revealed
subsets of aged canines with learning and memory impairments
(29–31). Thus, canine models have been
identified as a unique model for studying a human disorders, such
as AD.
Although a canine model is ideal for human disease
studies, to our knowledge, no such model has been reported to date,
due to the lack of canine embryonic stem cells, which are generally
used for gene targeting, and proper protocols for producing mature
oocytes in vitro. From the molecular point of view, the
canine is a suitable AD model animal as APP and most of the
enzymatic machinery for processing this factor are highly
homologous in canines and humans. This genetic similarity allows us
to expect concordance in gene expression and development of the
disease phenotype in the transgenic puppies. This may facilitate
fundamental studies of the disease process and phenotype
development as a function of time.
In a previous study, Hong et al (7) reported the production of a
transgenic canine expressing red fluorescent protein using
retroviral gene delivery methods. Although the efficient genetic
modification of donor cells is a key prerequisite to produce
transgenic animals, viral delivery systems are associated a minor
risk of neoplastic transformation (32). To prevent these unwanted effects,
in this study, we generated five genetically engineered puppies
expressing mhAPP using a liposomal transfection method. The
stable transfection of canine fibroblasts with a liposomal reagent
is a very effective method that can replace viral gene delivery
techniques. In this study, the expression and selection cassettes
were successfully integrated into genomic DNA and effectively
expressed in tissues following SCNT. This non-viral, SCNT-mediated
gene delivery system is a safer protocol for creating animal
disease models than viral gene delivery.
Canines naturally develop AD and senile plagues
similar to humans, and the plaques are initially observed between
the ages of eight and nine years (33). The pathological features in aged
canines having AD-like symptoms are quite similar to human
phenotypes, such as neuronal loss and cognitive deficits. These
progressive studies suggest that canines are the best model for
studying human AD. Although the aged canine model has several
advantages, the fact that over eight years are required to develop
AD symptoms is still a major obsticle for general usage in the
research field. Hence, we suggest the use of transgenic canines
carrying mhAPP to induce the early onset of AD phenotypes,
as performed in the present study.
In conclusion, we generated a canine model of AD to
replace the current rodent models. This is important as canines
possess a higher intelligence than mice and are large enough to
undergo surgical procedures in pre-clinical studies. This canine
model expressing the mhAPP gene produced by SCNT may be used
for understanding the pathological and developmental
characteristics of AD, and may aid in the development of novel
therapeutic modalities for AD.
Acknowledgements
We thank Zang-Hee Cho and Young-Bo Kim (Neuroscience
Research Institute, Gachon University of Medicine and Science,
Incheon, Republic of Korea) for helping with the analyses of the
brain MRI scans of the transgenic canines. This study was supported
by grants from the Next-Generation BioGreen 21 Program [nos.
PJ008323 and PJ009489], Rural Development Administration (RDA),
Republic of Korea.
References
|
1
|
Rocchi A, Pellegrini S, Siciliano G and
Murri L: Causative and susceptibility genes for Alzheimer’s
disease: a review. Brain Res Bull. 61:1–24. 2003.
|
|
2
|
Götz J, Streffer JR, David D, et al:
Transgenic animal models of Alzheimer’s disease and related
disorders: histopathology, behavior and therapy. Mol Psychiatry.
9:664–683. 2004.
|
|
3
|
Head E, Pop V, Sarsoza F, et al:
Amyloid-beta peptide and oligomers in the brain and cerebrospinal
fluid of aged canines. J Alzheimers Dis. 20:637–646.
2010.PubMed/NCBI
|
|
4
|
Kragh PM, Nielsen AL, Li J, et al:
Hemizygous minipigs produced by random gene insertion and handmade
cloning express the Alzheimer’s disease-causing dominant mutation
APPsw. Transgenic Res. 18:545–558. 2009.PubMed/NCBI
|
|
5
|
Jang G, Kim MK, Oh HJ, et al: Birth of
viable female dogs produced by somatic cell nuclear transfer.
Theriogenology. 67:941–947. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Duyckaerts C, Potier MC and Delatour B:
Alzheimer disease models and human neuropathology: similarities and
differences. Acta Neuropathol. 115:5–38. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hong SG, Kim MK, Jang G, et al: Generation
of red fluorescent protein transgenic dogs. Genesis. 47:314–322.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hossein MS, Kim MK, Jang G, et al: Effects
of thiol compounds on in vitro maturation of canine oocytes
collected from different reproductive stages. Mol Reprod Dev.
74:1213–1220. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lee BC, Kim MK, Jang G, et al: Dogs cloned
from adult somatic cells. Nature. 436:6412005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Spanopoulou E, Giguere V and Grosveld F:
The functional domains of the murine Thy-1 gene promoter. Mol Cell
Biol. 11:2216–2228. 1991.PubMed/NCBI
|
|
11
|
Dudal S, Krzywkowski P, Paquette J, et al:
Inflammation occurs early during the Abeta deposition process in
TgCRND8 mice. Neurobiol Aging. 25:861–871. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Spires TL and Hyman BT: Transgenic models
of Alzheimer’s disease: learning from animals. NeuroRx. 2:423–437.
2005.
|
|
13
|
Friedman D, Honig LS and Scarmeas N:
Seizures and epilepsy in Alzheimer’s disease. CNS Neurosci Ther.
18:285–294. 2012.
|
|
14
|
Irizarry MC, Jin S, He F, et al: Incidence
of new-onset seizures in mild to moderate Alzheimer disease. Arch
Neurol. 69:368–372. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Larner AJ and Marson AG: Epileptic
seizures in Alzheimer’s disease: another fine MESS? J Alzheimers
Dis. 25:417–419. 2011.
|
|
16
|
Pandis D and Scarmeas N: Seizures in
Alzheimer disease: clinical and epidemiological data. Epilepsy
Curr. 12:184–187. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Picco A, Archetti S, Ferrara M, et al:
Seizures can precede cognitive symptoms in late-onset Alzheimer’s
disease. J Alzheimers Dis. 27:737–742. 2011.PubMed/NCBI
|
|
18
|
Apostolova LG, Green AE, Babakchanian S,
et al: Hippocampal atrophy and ventricular enlargement in normal
aging, mild cognitive impairment (MCI), and Alzheimer Disease.
Alzheimer Dis Assoc Disord. 26:17–27. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chou YY, Leporé N, Avedissian C, et al:
Mapping correlations between ventricular expansion and CSF amyloid
and tau biomarkers in 240 subjects with Alzheimer’s disease, mild
cognitive impairment and elderly controls. Neuroimage. 46:394–410.
2009.PubMed/NCBI
|
|
20
|
Ferrarini L, Palm WM, Olofsen H, et al:
MMSE scores correlate with local ventricular enlargement in the
spectrum from cognitively normal to Alzheimer disease. Neuroimage.
39:1832–1838. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Nestor SM, Rupsingh R, Borrie M, et al:
Ventricular enlargement as a possible measure of Alzheimer’s
disease progression validated using the Alzheimer’s disease
neuroimaging initiative database. Brain. 131:2443–2454.
2008.PubMed/NCBI
|
|
22
|
Glass CK, Saijo K, Winner B, Marchetto MC
and Gage FH: Mechanisms underlying inflammation in
neurodegeneration. Cell. 140:918–934. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dyrks T, Weidemann A, Multhaup G, et al:
Identification, transmembrane orientation and biogenesis of the
amyloid A4 precursor of Alzheimer’s disease. EMBO J. 7:949–957.
1988.PubMed/NCBI
|
|
24
|
Selkoe DJ: Alzheimer’s disease: genes,
proteins, and therapy. Physiol Rev. 81:741–766. 2001.
|
|
25
|
Sisodia SS: Beta-amyloid precursor protein
cleavage by a membrane-bound protease. Proc Natl Acad Sci USA.
89:6075–6079. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sarasa M and Pesini P: Natural
non-trasgenic animal models for research in Alzheimer’s disease.
Curr Alzheimer Res. 6:171–178. 2009.PubMed/NCBI
|
|
27
|
Head E: Combining an antioxidant-fortified
diet with behavioral enrichment leads to cognitive improvement and
reduced brain pathology in aging canines: strategies for healthy
aging. Ann NY Acad Sci. 1114:398–406. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Siwak-Tapp CT, Head E, Muggenburg BA,
Milgram NW and Cotman CW: Region specific neuron loss in the aged
canine hippocampus is reduced by enrichment. Neurobiol Aging.
29:39–50. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Head E, Mehta R, Hartley J, et al: Spatial
learning and memory as a function of age in the dog. Behav
Neurosci. 109:851–858. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Adams B, Chan A, Callahan H, et al: Use of
a delayed non-matching to position task to model age-dependent
cognitive decline in the dog. Behav Brain Res. 108:47–56. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Smith DR, Hoyt EC, Gallagher M, Schwabe RF
and Lund PK: Effect of age and cognitive status on basal level AP-1
activity in rat hippocampus. Neurobiol Aging. 22:773–786. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sokol DL and Gewirtz AM: Gene therapy:
basic concepts and recent advances. Crit Rev Eukaryot Gene Expr.
6:29–57. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Martin SB, Dowling AL and Head E:
Therapeutic interventions targeting Beta amyloid pathogenesis in an
aging dog model. Curr Neuropharmacol. 9:651–661. 2011. View Article : Google Scholar : PubMed/NCBI
|