Introduction
Pancreatic carcinoma (PC) is one of the most lethal
and aggressive of all malignancies. It is the sixth leading cause
of mortality from malignant disease in China and the fourth leading
cause of cancer-related mortality in the United States (1,2).
The median overall survival is >6 months and the 5-year survival
rate is dismal, ranging between 2 and 6% (3). Despite advances in chemotherapy and
radiotherapy over the past few decades, the overall prognosis of PC
has remained essentially unchanged (4). Recently, the classical categories of
oncogenes and tumor suppressor genes have been expanded to include
a novel class of small non-coding RNAs, termed microRNAs (miRNAs or
miRs), which can regulate a number of protein-coding genes,
including tumor-related genes (5).
miRNAs regulate genes expression by base pairing
with partially complementary messenger RNAs (mRNAs), resulting in
the degradation of target mRNAs or the inhibition of their
translation. The expression patterns of miRNAs appear to be
tissue-specific and more accurate than mRNA-based profiling in
tumor diagnostics. To date, a number of deregulated miRNAs have
been found in human malignancies, including breast cancer, lung
cancer, hepatocellular carcinoma (HCC) and colon cancer (6–8).
Among these miRNAs, miR-375 was originally reported to be an islet
cell-specific miRNA and to regulate insulin secretion (9). Recent studies have demonstrated the
downregulation of miR-375 in gastric cancer (10), HCC (11) and colorectal cancer (12). However, the function and
mechanisms of action of miR-375 in PC have not yet been fully
elucidated.
To determine the function of miR-375 as a tumor
suppressor gene, we searched for physiological targets using
TargetScan (http://www.targetscan.org/) and found that
3-phosphoinositide-dependent protein kinase 1 (PDK1) is a putative
target of miR-375. As a pleckstrin homology (PH) domain-containing
protein, PDK1 has emerged as an important oncogene in multiple
types of cancer (13). It has
been reported that PDK1 is overexpressed in human PC and promotes
cancer cell proliferation, growth and invasion (14). However, knowledge of the
mechanisms that regulate PDK1 expression during tumor progression
is limited.
In this study, we investigated the biological roles
and the potential mechanisms of action of miR-375 in PC.
Furthermore, we identified PDK1 as a putative target of miR-375 by
computational prediction and luciferase reporter assays. Our
findings demonstrated that miR-375 may have great potential for use
as molecular targets in the diagnosis and treatment of PC.
Materials and methods
Clinical samples
Tissue samples from 50 patients with PC were
collected during surgical resections performed at the First
Affiliated Hospital of Soochow University, Suzhou, China from
January 2010 to December 2012. Tumor tissues and adjacent non-tumor
tissues were frozen immediately after surgical removal in liquid
nitrogen and stored at −80°C. The patients had not received any
pre-operative chemotherapy, radiotherapy or immunotherapy. All
samples were obtained after receiving patient consent from and
approval from the Ethics Committee of Soochow University.
Cell culture and transfection
The human PC cell lines, PANC-1, SW1990, Capan-1,
Patu8988, AsPC-1 and BxPC-3 obtained from the Shanghai Institute of
Cell Biology (Shanghai, China) were maintained in DMEM or RPMI-1640
supplemented with 10% fetal bovine serum (all from Gibco, Grand
Island, NY, USA) and 100 μg/ml each of penicillin and streptomycin
(Invirtrogen, Carlsbad, CA, USA) in 5% CO2 at 37°C.
Cells (1×104) per well were cultured in
6-well plates overnight until they reached 60–70% confluence. Due
to their different expression levels of miR-375 (PANC-1 cells
express low levels of miR-375 and BxPC-3 cells express high levels)
the PANC-1 and BxPC-3 cells were selected and transfected with
miR-375 mimics (mimics group), miR-375 inhibitor (inhibitor group)
or a negative control (NC group) (GeneChem, Shanghai, China) using
Lipofectamine 2000 (Invitrogen, Carslbad, CA, USA) as the
transfection reagent. Non-transfected cells were used as the blank
control (mock group).
Quantitative reverse transcription
polymerase chain reaction (qRT-PCR)
Total RNA was extracted the from tissues and cells
using TRIzol reagent (Invitrogen). The expression levels of miR-375
were measured using a TaqMan microRNA assay (Applied Biosystems,
Foster City, CA, USA) specific for hsa-miR-375 in the PCR reactions
of 40 cycles (95°C, 15 sec; 60°C, 60 sec; 70°C, 30 sec), with U6
RNA as an internal control. PDK1 expression was detected with the
following primers: forward, 5′-GTGTAGATTAGAGGGATG-3′ and reverse,
5′-AAGGAATAGTGGGTTAGG-3′ and β-actin was used as an internal
control with the following primers: forward,
5′-AGCGAGCATCCCCCAAAGTT-3′ and reverse, 5′-GGGCACGAAGGCTCATCATT-3′.
The PCR reactions consisted of 40 cycles (95°C, 15 sec; 62°C, 45
sec; 72°C, 30 sec). The amplified segments were analyzed on 2.5%
agarose gels.
Western blot analysis
The cells were collected and lysed in lysis buffer
on ice. Total proteins were separated by 10% SDS-PAGE and blotted
onto PVDF membranes. The membranes were then blocked with 10%
non-fat milk powder at room temperature for 2 h and incubated with
the following primary antibodies: anti-PDK1 antibody (1:100),
anti-GAPDH antibody (1:300) (both from Santa Cruz Biotechnology,
Inc., Santa Cruz, CA, USA), anti-extracellular signal-regulated
kinase (ERK)1/2 antibody (1:100), anti-Akt antibody (1:100),
anti-p38 antibody (1:100) (all from CST Technologies, Inc.,
Chicago, IL, USA) at 4°C overnight. After 3 washes, the membranes
were incubated with a horseradish peroxidase-conjugated goat
anti-mouse IgG (1:2,000; Santa Cruz Biotechnology, Inc.) for 2 h at
room temperature. Reactive bands were detected using ECL Western
Blotting Detection Reagent (GE Healthcare, Pittsburgh, PA,
USA).
Luciferase reporter assay
To verify that PDK1 is a direct target of miR-375,
the wild-type 3′UTR (WT 3′UTR) of PDK1 containing the miR-375
binding site was amplified using the following primers: forward,
5′-ACCCAACCACACAAAGAACAAAA-3′ and reverse,
5′-TTTTGTTCTTTGTGTGGTTGGGT-3′. As a negative control, the mutated
binding site of the 3′UTR (MUT 3′UTR) was amplified using the
following primers: forward, 5′-ACCCAACCACACAAAGACACAAA-3′ and
reverse, 5′-TTTACAAGATTGTGTGGTTGGGT-3′. Products were cloned into
the luciferase reporter vector, pMIR-Report (Ambion, Austin, TX,
USA). PANC-1 cells were seeded in 24-well plates and co-transfected
with the luciferase reporter vector and miR-375 mimics or
miR-negative control (NC) as described above. The cells were
harvested 48 h after transfection and luciferase activity was
measured using the Dual-Luciferase assay system (Promega, Madison,
WI, USA).
Cell proliferation assay
The CCK8 assay was performed to determine cell
growth. Following transfection, the cells were seeded in 96-well
plates at 5×103 cells per well, followed by the addition
of 20 μl CCK8 (Dojindo Laboratories, Kumamoto, Japan) and
incubation at 37°C for an additional 2 h. An ultraviolet
spectrophotometer was used to measure the absorbance of each well
at 450 nm, and each experiment was performed in triplicate and
repeated 3 times.
Analysis of cell apoptosis
After 72 h of transfection, the cells were seeded
and a volume of 100 μl of cell suspension (1×106
cells/ml) was labeled with 10 μl of propidium iodided (PI) and 5 μl
of Annexin V/FITC (both from BD Biosciences, Franklin Lakes, NJ,
USA). The cells were incubated in the dark for 15 min at room
temperature and the number of early apoptotic cells was determined
using a FACSCalibur flow cytometer (BD Biosciences).
Terminal dUTP nick end-labeling (TUNEL)
assays
The cells were plated on glass coverslips in 24-well
plates and fixed in 4% paraformaldehyde for 60 min. After washing
with phosphate-buffered saline, 50 μl/well Cy3-labeling TUNEL
(Beyotime, Shanghai, China) reaction mixture were added to the
cells followed by incubation for 60 min at 37°C in the dark. The
TUNEL-positive cells emitted red fluorescence under a fluorescence
microscope. The number of apoptotic cells was calculated by
counting the number of positive cells in at least 5 randomly
selected microscopic fields (magnification, ×200).
Statistical analysis
SPSS software version 17.0 was used for statistical
analysis. Data re expressed as the means ± standard deviation (SD).
One-way analysis of variance (one-way Anova) or the t-test was
performed for inter-group comparisons. Pearson’s correlation
analysis was used to compare miR-375 expression with PDK1
expression. A value of P<0.05 was considered to indicate a
statistically significant difference.
Results
miR-375 is downregulated and inversely
correlates with PDK1 in PC
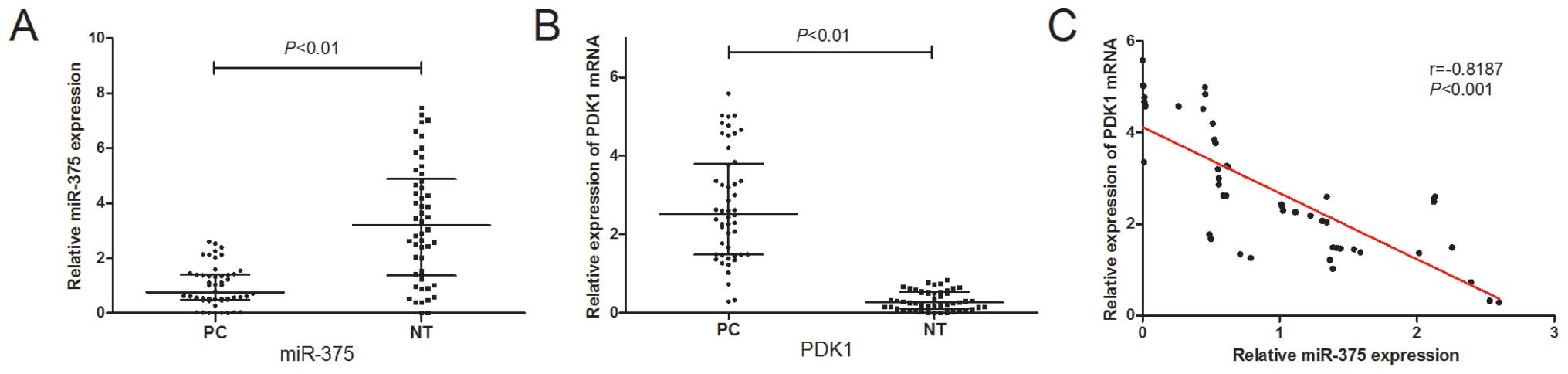
To determine the expression levels of miR-375 in PC,
we examined miR-375 expression in 50 pairs of PC tissues and
matched adjacent non-tumor tissues by qRT-PCR. As shown in Fig. 1A, the miR-375 expression levels
were significantly suppressed in the PC tissues (P<0.01). Of
note, the expression of miR-375 inversely correlated with that of
PDK1, a potential target of miR-375 (Fig. 1B and C).
miR-375 is a post-transcriptional
regulator of PDK1
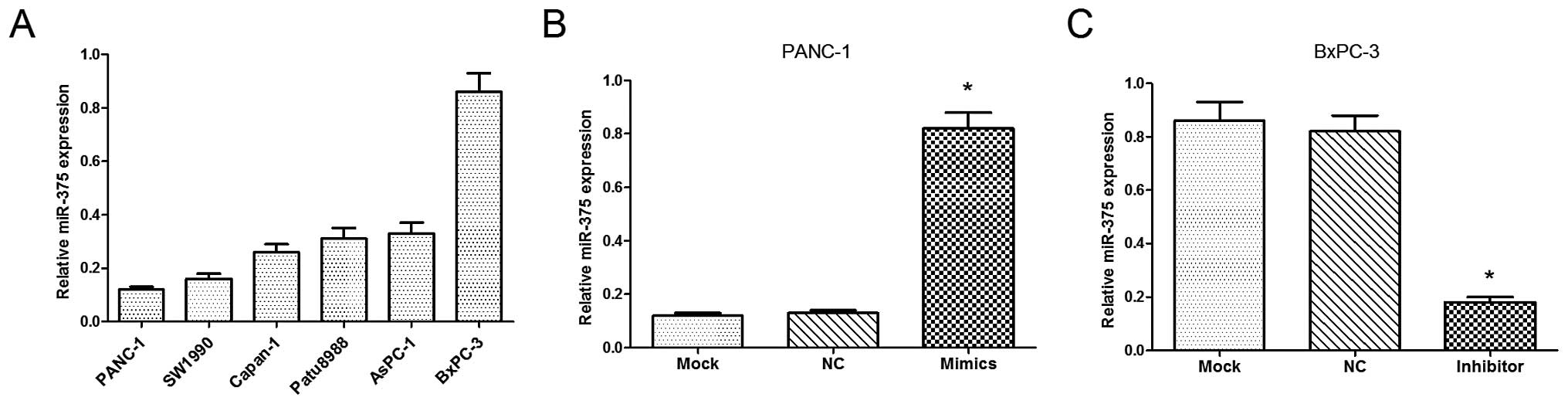
We examined the relative expression of miR-375 in 6
PC cell lines (PANC-1, SW1990, Capan-1, Patu8988, AsPC-1 and
BxPC-3), from which the PANC-1 and BxPC-3 cells were selected, as
they were found to have a low and high expression of miR-375,
respectively (Fig. 2A). As shown
in Fig. 2B and C, the levels of
miR-375 were significantly upregulated in the PANC-1 cells
following transfection with miR-375 mimics (P<0.05), while the
levels of miR-375 were signficantly downregulated in the BxPC-3
cells following transfection with the miR-375 inhibitor
(P<0.05).
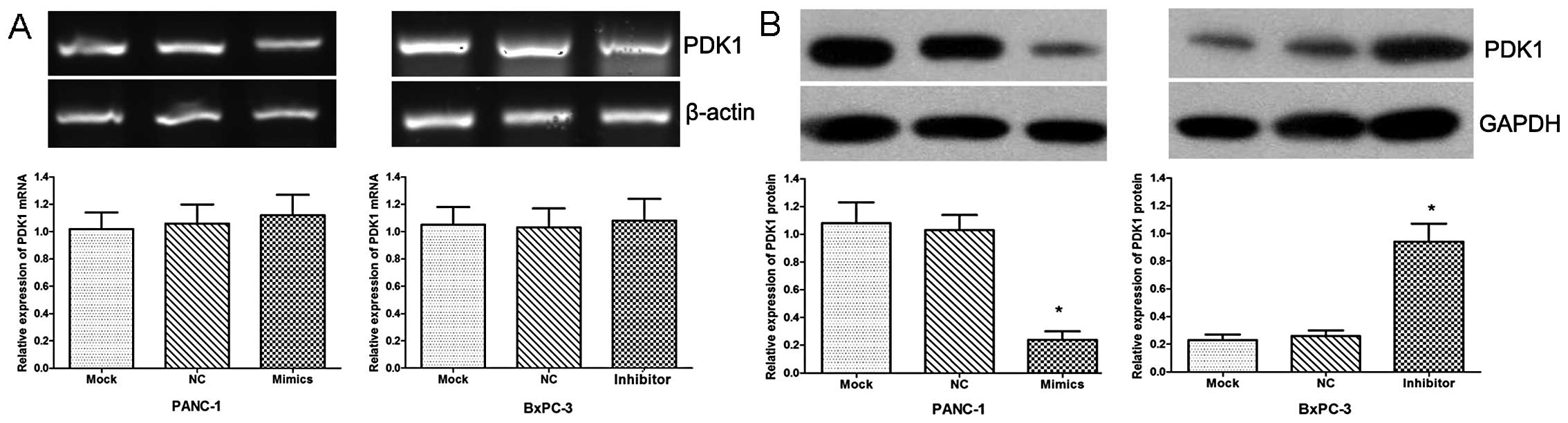
Furthermore, the expression of PDK1 was examined by
qRT-PCR and western blot analysis in these 2 cell lines. Our data
revealed no apparent change in PDK1 expression at the transcript
level (Fig. 3A), while the PDK1
protein level was significantly downregulated in the cells
transfected with miR-375 mimics and upregulated in the cells
transfected with the miR-375 inhibitor compared with the
NC-transfected cells (Fig. 3B;
both P<0.05).
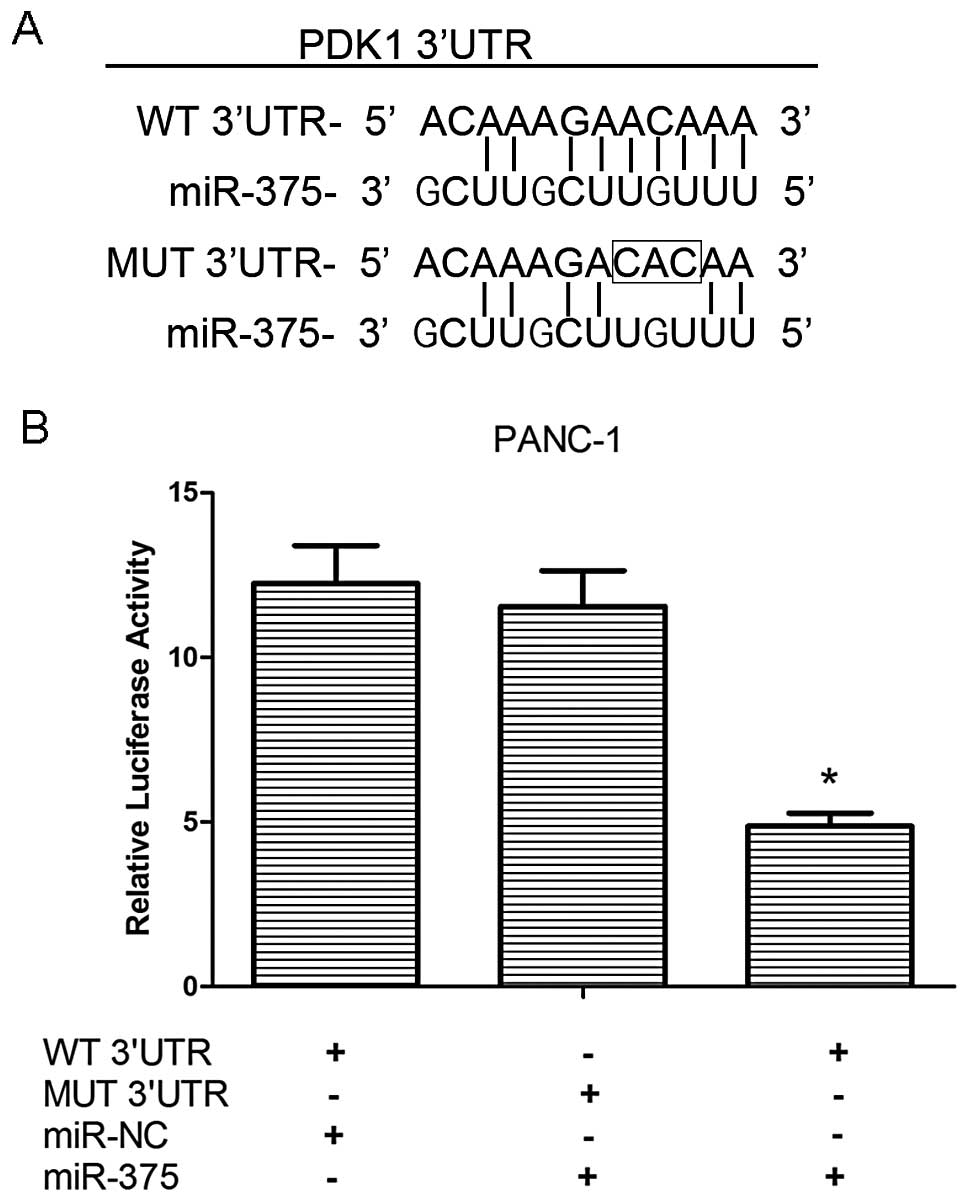
PDK1 is a direct target of miR-375
To confirm the direct targeting of PDK1 by miR-375,
we integrated a firefly luciferase reporter plasmid containing a
region of PDK1 3′UTR harboring the miR-375 target site, or a
fragment whose target site was mutated (Fig. 4A), and used these to co-transfect
the PANC-1 cells with miR-375 mimics or miR-NC. As shown in
Fig. 4B, our results demonstrated
that relative luciferase activity was decreased in the WT
3′UTR-transfected PANC-1 cells that were co-transfected with
miR-375 mimics as compared with those cells co-transfected with
miR-NC (P<0.05), while the mutation of the miR-375 binding site
blocked this suppressive effect. These results strongly suggest
that miR-375 negatively regulates the expression of PDK1 by
directly targeting the 3′UTR of the PDK1 transcript.
Effect of miR-375 on proliferation and
apoptosis of PC cells
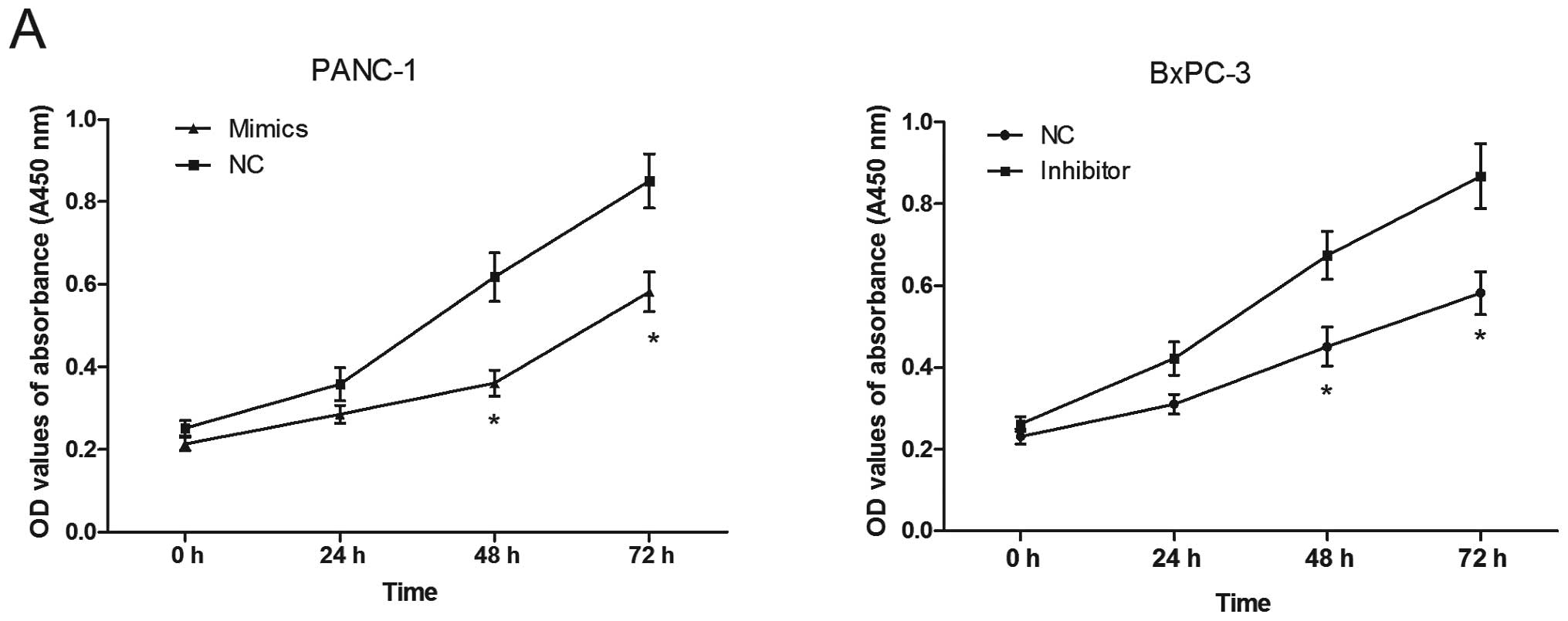
Using the transfected cells, CCK8 assays were
performed to examine the effects of miR-375 on PC cell
proliferation. As shown in Fig.
5A, the PANC-1 cells transfected with the miR-375 mimics
displayed significant growth inhibition compared with the cells
transfected with miR-NC at 48 and 72 h (both P<0.05). On the
contrary, the BxPC-3 cells transfected with the miR-375 inhibitor
showed an enhanced proliferation at 48 and 72 h (P<0.05).
We further assessed the effects of miR-375 on cell
apoptosis by flow cytometry and TUNEL assays. The apoptotic rate in
the PANC-1 cells transfected with the miR-375 mimics was
significantly higher than that of the cells transfected with miR-NC
(Fig. 5B; P<0.05). In the
BxPC-3 cells, the cells transfected with the miR-375 inhibitor
showed a decreased rate of apoptosis compared with the
miR-NC-transfected group (P<0.05). As shown in Fig. 5C, TUNEL assays demonstrated that
the number of apoptotic cells in the miR-375 mimic-transfected
group was greater than that in the miR-NC-transfected group
(miR-mimic-transfected cells, 120.16±10.04 cells per field;
miR-NC-transfected cells, 31.06±4.57 cells per field; P<0.05).
The downregulation of miR-375 induced by the inhibitor led to a
decrease in the number of apoptotic cells (miR-375
inhibitor-transfected cells, 42.56±7.11 cells per field;
miR-NC-transfected cells, 131.01±12.32 cells per field;
P<0.05).
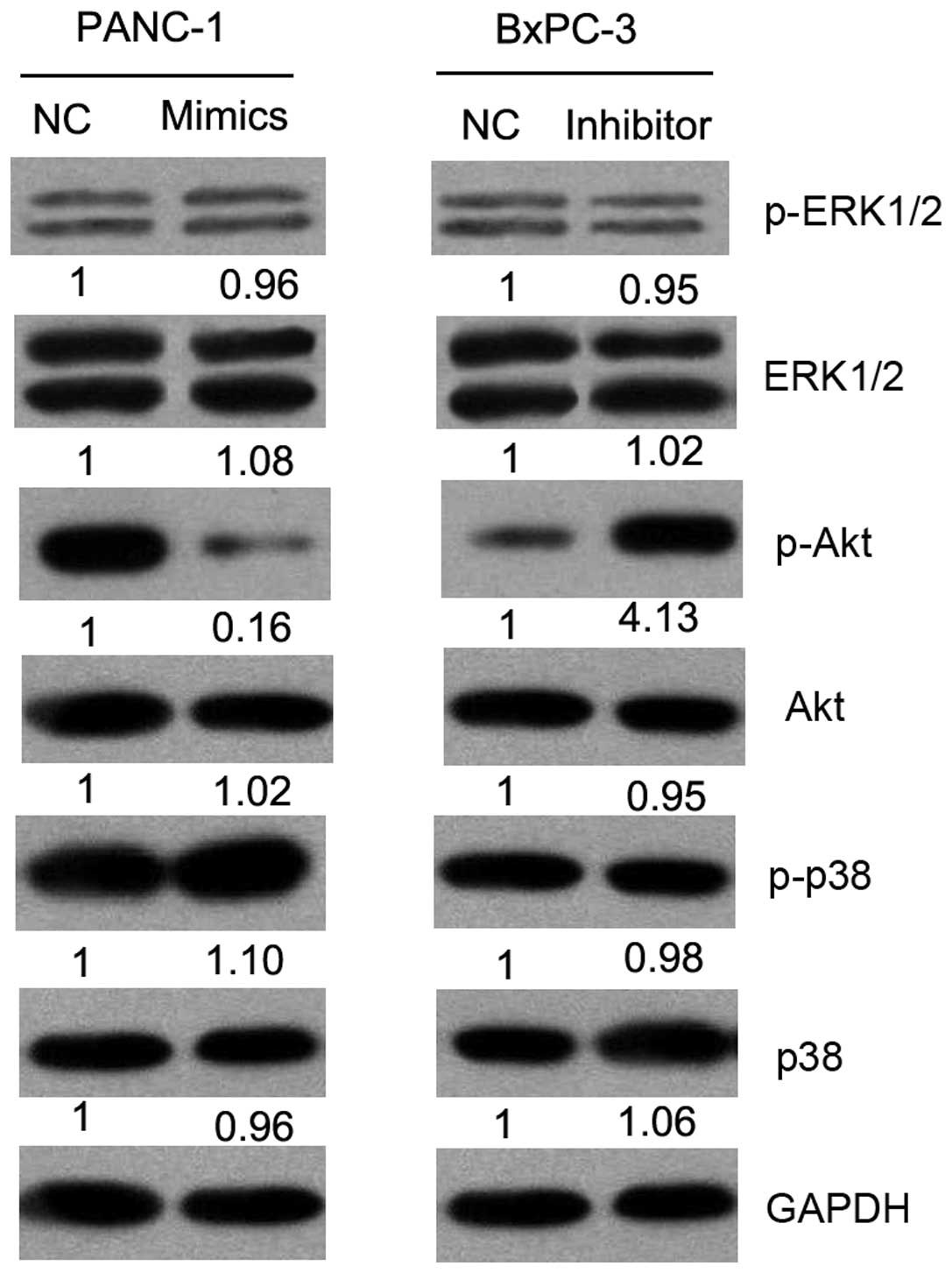
Effect of miR-375 on mitogen-activated
protein kinase (MAPK) and Akt signaling pathways in PC cells
PDK1 is an oncogene involved in the regulation of
cell cycle progression, apoptosis and invasion through the MAPK or
Akt signaling pathways. Thus, we investigated the effects of
miR-375 on the activation status of MAPK and Akt pathways by
western blot analysis (Fig. 6).
Our results revealed the decreased expression of phosphorylated
(p)-Akt in the miR-375 mimic-transfected cells and an increased
expression of p-Akt in the miR-375 inhibitor-transfected group as
compared with the controls (NC). There was no change observed in
the expression of ERK1/2, p-ERK1/2, p38, p-p38 and Akt. These
results suggest that miR-375 suppresses the malignant behavior of
PC cells through the Akt signaling pathway.
Discussion
In the present study, we investigated the role of
miR-375 in PC and observed a tendency towards the downregulation of
miR-375 expression in PC tissues compared to normal (non-tumor)
ones. We induced either the up- or downregulation of miR-375
expression by transfecting PC cells with miR-375 mimics or miR-375
inhibitor and observed that miR-375 suppressed tumor cell
proliferation and induced apoptosis. Furthermore, our results
demonstrated that the 3′UTR of human PDK1 contained a putative
target of miR-375 and that miR-375 regulated PDK1 expression at the
protein level in PC cells.
miRNAs are small non-coding RNA molecules that play
key roles in cell proliferation, apoptosis and differentiation in
cancer initiation and progression (15). A great deal of evidence has
suggested that miRNAs act either as oncogenes or tumor suppressor
genes in various types of cancer (5,7).
In this study, our results revealed that the upregulation of
miR-375 in PC cells suppressed their malignant behavior and that
the downregulation of miR-375 with the inhibitor had the opposite
effect. These data suggest that miR-375 acts as a tumor suppressor
gene in PC. Although miR-375 was initially shown to be expressed in
pancreatic islets (9), it is
worth mentioning that the aberrant expression of miR-375 has also
been reported in other malignancies (10–12). In the majority of studies, miR-375
has been shown to suppress cell growth and proliferation, such as
in HCC (16) and gastric cancer
(17). However, the
overexpression of miR-375 in cervical cancer cells has been shown
to decrease sensitivity to paclitaxel (18). These data from other studies,
together with our findings indicate that the function of miR-375 in
cancer is cel type-specific.
miRNAs exert their their regulatory effects on gene
expression through partial complementary elements in the 3′UTR of
their target mRNA, resulting in transcript degradation or
translational arrest (19). Since
a single miRNA can target multitudinous genes, the important
targets of miR-375 include yes-associated protein (YAP), PDK1 and
Janus kinase 2 (JAK2) (20–22). These targets play diverse roles in
different cellular systems. For instance, YAP1 is regulated by
miR-375 and inhibits cell growth in lung cancer (23). miR-375 targets p53 in gastric
cancer cells and abrogates cell cycle arrest and apoptosis
(24). Our results demonstrated
that miR-375 suppressed PDK1 expression by binding directly to the
3′UTR of PDK1 and an inverse correlation was observed between
miR-375 and PDK1 expression in the PC clinical specimens. PDK1 is a
key component of the PI3K/Akt signaling pathway and its
overexpression has been associated with cell growth and
proliferation in PC. Thus, the identification of miR-375 as a
regulator of PDK1 may have potential therapeutic implications in PC
and other malignancies.
ERK1/2and p38, members of MAPKs, are important
cellular protein kinases. Akt, also known as protein kinase B, is a
serine/threonine protein kinase. The activation of Akt, ERK1/2 and
p38 through phosphorylated forms plays a crucial role in the
modulation of cell survival and apoptosis (25,26). Previous studies have demonstrated
that miR-375 is involved in regulating target genes in the MAPK
signal pathway (27,28). In this study, our data
demonstrated that miR-375 modulated the phosphorylation of Akt in
PC cells, while no change was observed in p-ERK1/2 and p-p38
expression. To the best of our knowledge, our observations that
miR-375 effectively influenced PC cell proliferation and apoptosis
through the Akt signaling pathway have not been previously
reported. Therefore, our findings provide a more comprehensive
understanding of miR-375 in the development and progression of
PC.
In conclusion, this study demonstrates that miR-375
negatively regulates the oncogene, PDK1, and modulates PC cell
proliferation and apoptosis through the Akt signaling pathway. Our
findings indicate that targeting miR-375 through a genetic approach
may provide a novel strategy for the treatment of PC.
Acknowledgements
This study was supported by grants from the National
Natural Science Foundation of China (81201905), the China
Postdoctoral Science Foundation (2013M540374), the Shanghai
Postdoctoral Scientific Program of China (13R21415200) and Natural
Science Research Grants from the University of Jiangsu Province of
China (12KJB320009).
References
|
1
|
Chen X, Ma S and Zhang Z: Analysis of
clinical characteristics of pancreatic carcinoma in northern China.
Pancreas. 39:1116–1118. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar
|
|
3
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2013. CA Cancer J Clin. 63:11–30. 2013. View Article : Google Scholar
|
|
4
|
Vincent A, Herman J, Schulick R, Hruban RH
and Goggins M: Pancreatic cancer. Lancet. 378:607–620. 2011.
View Article : Google Scholar
|
|
5
|
Ruan K, Fang X and Ouyang G: microRNAs:
novel regulators in the hallmarks of human cancer. Cancer Lett.
285:116–126. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hrašovec S and Glavač D: microRNAs as
Novel Biomarkers in Colorectal Cancer. Front Genet. 3:1802012.
|
|
7
|
Chen PS, Su JL and Hung MC: Dysregulation
of microRNAs in cancer. J Biomed Sci. 19:902012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Liu H: microRNAs in breast cancer
initiation and progression. Cell Mol Life Sci. 69:3587–3599. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Poy MN, Eliasson L, Krutzfeldt J, et al: A
pancreatic islet-specific microRNA regulates insulin secretion.
Nature. 432:226–230. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang WH, Gui JH, Wang CZ, et al: The
identification of miR-375 as a potential biomarker in distal
gastric adenocarcinoma. Oncol Res. 20:139–147. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chang Y, Yan W, He X, et al: miR-375
inhibits autophagy and reduces viability of hepatocellular
carcinoma cells under hypoxic conditions. Gastroenterology.
143:177–187.8. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dai X, Chiang Y, Wang Z, et al: Expression
levels of microRNA-375 in colorectal carcinoma. Mol Med Rep.
5:1299–1304. 2012.PubMed/NCBI
|
|
13
|
Raimondi C and Falasca M: Targeting PDK1
in cancer. Curr Med Chem. 18:2763–2769. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Westmoreland JJ, Wang Q, Bouzaffour M,
Baker SJ and Sosa-Pineda B: Pdk1 activity controls proliferation,
survival, and growth of developing pancreatic cells. Dev Biol.
334:285–298. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nikitina EG, Urazova LN and Stegny VN:
microRNAs and human cancer. Exp Oncol. 34:2–8. 2012.
|
|
16
|
He XX, Chang Y, Meng FY, et al:
microRNA-375 targets AEG-1 in hepatocellular carcinoma and
suppresses liver cancer cell growth in vitro and in vivo. Oncogene.
31:3357–3369. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tsukamoto Y, Nakada C, Noguchi T, et al:
microRNA-375 is downregulated in gastric carcinomas and regulates
cell survival by targeting PDK1 and 14-3-3zeta. Cancer Res.
70:2339–2349. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shen Y, Wang P, Li Y, et al: miR-375 is
upregulated in acquired paclitaxel resistance in cervical cancer.
Br J Cancer. 109:92–99. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Garzon R, Calin GA and Croce CM: microRNAs
in Cancer. Annu Rev Med. 60:167–179. 2009. View Article : Google Scholar
|
|
20
|
Liu AM, Poon RT and Luk JM: microRNA-375
targets Hippo-signaling effector YAP in liver cancer and inhibits
tumor properties. Biochem Biophys Res Commun. 394:623–627. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li X, Lin R and Li J: Epigenetic silencing
of microRNA-375 regulates PDK1 expression in esophageal cancer. Dig
Dis Sci. 56:2849–2856. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ding L, Xu Y, Zhang W, et al: MiR-375
frequently downregulated in gastric cancer inhibits cell
proliferation by targeting JAK2. Cell Res. 20:784–793. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Nishikawa E, Osada H, Okazaki Y, et al:
miR-375 is activated by ASH1 and inhibits YAP1 in a
lineage-dependent manner in lung cancer. Cancer Res. 71:6165–6173.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liu Y, Xing R, Zhang X, et al: miR-375
targets the p53 gene to regulate cellular response to ionizing
radiation and etoposide in gastric cancer cells. DNA Repair (Amst).
12:741–750. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sheppard K, Kinross KM, Solomon B, Pearson
RB and Phillips WA: Targeting PI3 kinase/AKT/mTOR signaling in
cancer. Crit Rev Oncog. 17:69–95. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Dhanasekaran DN and Johnson GL: MAPKs:
function, regulation, role in cancer and therapeutic targeting.
Oncogene. 26:3097–3099. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang X, Yan Z, Zhang J, et al:
Combination of hsa-miR-375 and hsa-miR-142-5p as a predictor for
recurrence risk in gastric cancer patients following surgical
resection. Ann Oncol. 22:2257–2266. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Song J, Kim D, Chun CH and Jin EJ:
microRNA-375, a new regulator of cadherin-7, suppresses the
migration of chondrogenic progenitors. Cell Signal. 25:698–706.
2013. View Article : Google Scholar : PubMed/NCBI
|