1. Overview of programmed necrosis
(necroptosis)
Over the past 40 years, apoptosis has been generally
thought as the only prototype of programmed cell death. By
contrast, another cell demise termed ‘necrosis’ is simply described
as a passive and unwanted cell death in response to the
overexposure of chemical, physical or radioactive stress. The third
cell death mode, autophagy, is not discussed in this review as
there is still some controversy as to its role in cell survival and
cell death. Based on morphology, biochemistry and physiology,
necrotic cell death has unique characteristics, distinct from those
of apoptosis and autophagy. Some typical features discriminating
between apoptosis and necrosis are listed by criteria in Table I. Of note, the loss of membrane
integrity in cells undergoing necrosis induces the release of
intracellular debris, referred to as danger signals into the
microenvironmental niche, consequentially eliciting inflammatory
responses (1). Although little
attention has previously been paid to necrotic death, its
pathophysiological significance coupled to inflammatory response
has recently been emphasized.
 | Table IRepresentative characteristics
discriminating between apoptosis and necroptosis. |
Table I
Representative characteristics
discriminating between apoptosis and necroptosis.
| Cell death
modes/characteristicsa | Apoptosis | Programmed
necrosis |
|---|
| Membrane
integrity | Retained | Disintegrated |
| Caspase
requirement | Yes | No |
| Nuclear
morphology | Shrinkage | Swelling |
| DNA cleavage
pattern | Laddering | Smearing |
| RIP1 involvement | No | Yes |
| ROS generation | No | Yes |
Apart form apoptotic stress in nature, there exist a
variety of physical, chemical and biological stimuli causing
necrosis. These include high energy irradiation, DNA alkylating
agents and cytokines (2–4). Of the death stimuli listed above,
tumor necrosis factor (TNF)-α is a pleiotropic inflammatory
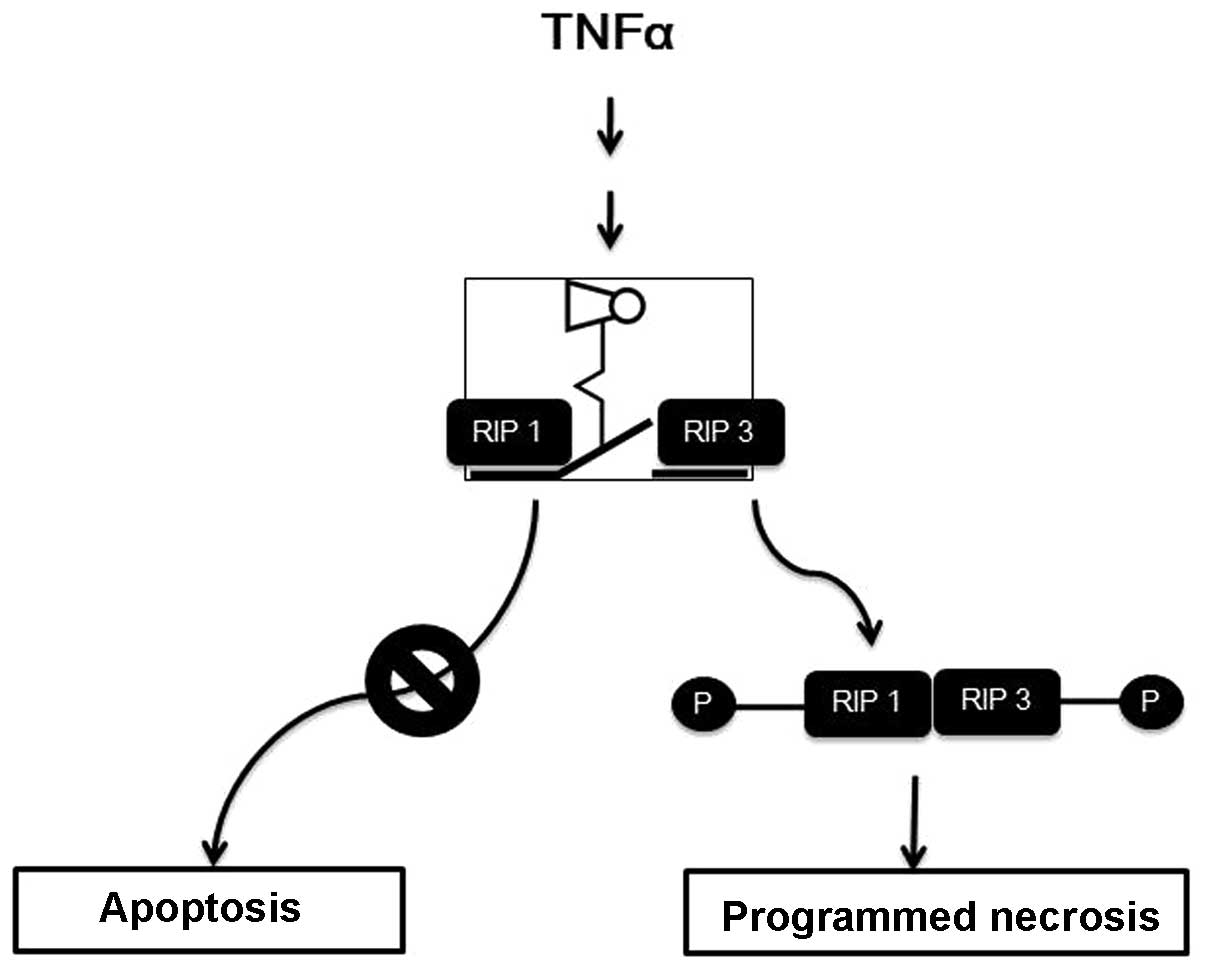
cytokine, and it initiates survival or programmed cell death,
apoptosis through the TNF-α receptor and a cascade of downstream
executioners (5). However, under
specific conditions in which the apoptotic machinery is blocked by
a pan-caspase inhibitor (zVAD peptide) or viral infection, the
cells themselves redirect the apoptotic cell demise into an
alternative cell death (Fig. 1).
Since a unified nomenclature on such a backup cell death program
has not been withdrawn, it is therefore referred to as necroptosis,
programmed necrosis, or caspase-independent cell death. In spite of
ambiguous nomenclature, there is a growing body of evidence
indicating that programmed necrosis is a backup cell death program
that is activated when caspase-driven cell death is blocked
(4,6). More precisely, programmed necrosis
contributes to N-methyl-D-aspartate (NMDA)-induced excitotoxicity
in neurons, as well as heavy metal poisoning and chemical-induced
toxicity (7–9). The precise manipulation of cell
death modes makes it possible to consequently define a new paradigm
of another cell death mode by analyzing the biochemical and
molecular parameters. As a result, Hitomi et al demonstrated
that a variety of proteins were related to necroptosis through a
genome-wide analysis (10).
Thereafter, proteins responsible specifically for programmed
necrosis have been extensively investigated and consequently, some
promising therapeutic proteins are discussed in this review. The
identification of some specific programmed necrotic proteins and
the development of small molecules specifically targeting receptor
interacting protein 1 (RIP1) make it conceivable that necrotic cell
death is not only an independent and specialized form of cell
death, but that it is also a part of an orchestrated signaling
network.
2. Therapeutic target molecules identified
for mediating necroptosis
Since necrosis has long been thought to be a passive
and unwanted cell response to devastating external stresses, the
identification of novel proteins responsible for necrotic death has
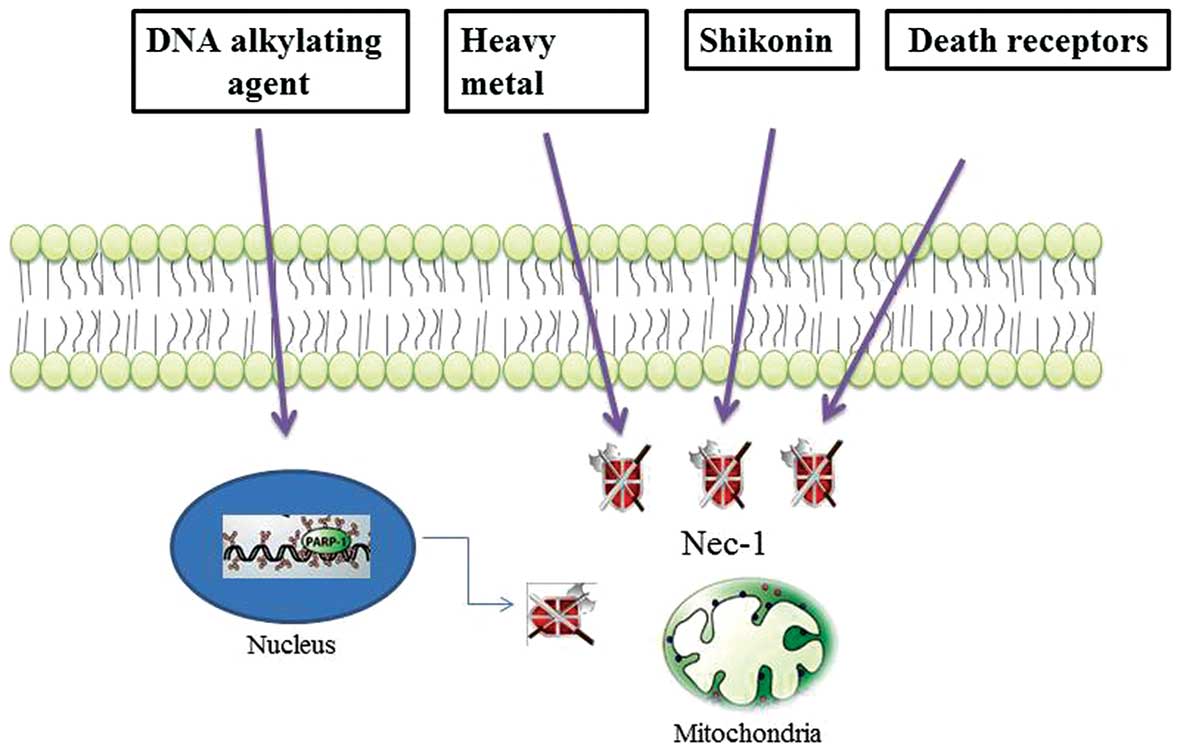
not been completed. However, accumulating evidence indicates that
DNA alkylating agents, shikonin and heavy metals induce programmed
necrosis-like cell death, distinct from necrosis or apoptosis
(Fig. 2) (11–13). Specifically, DNA
alkylation-induced DNA damage is repaired through the activation of
poly(ADP-ribose)polymerase-1 (PARP-1), which is a nuclear enzyme
that catalyzes the covalent linkage of long branched chains of PAR
to a variety of nuclear DNA-binding proteins, including PARP-1
(14,15). However, massive and intolerable
DNA damage to cells mediates necrosis through he excessive
activation of PARP-1, which depletes the ATP energy supply of the
cells supply and subsequently results in metabolic catastrophe
(16).
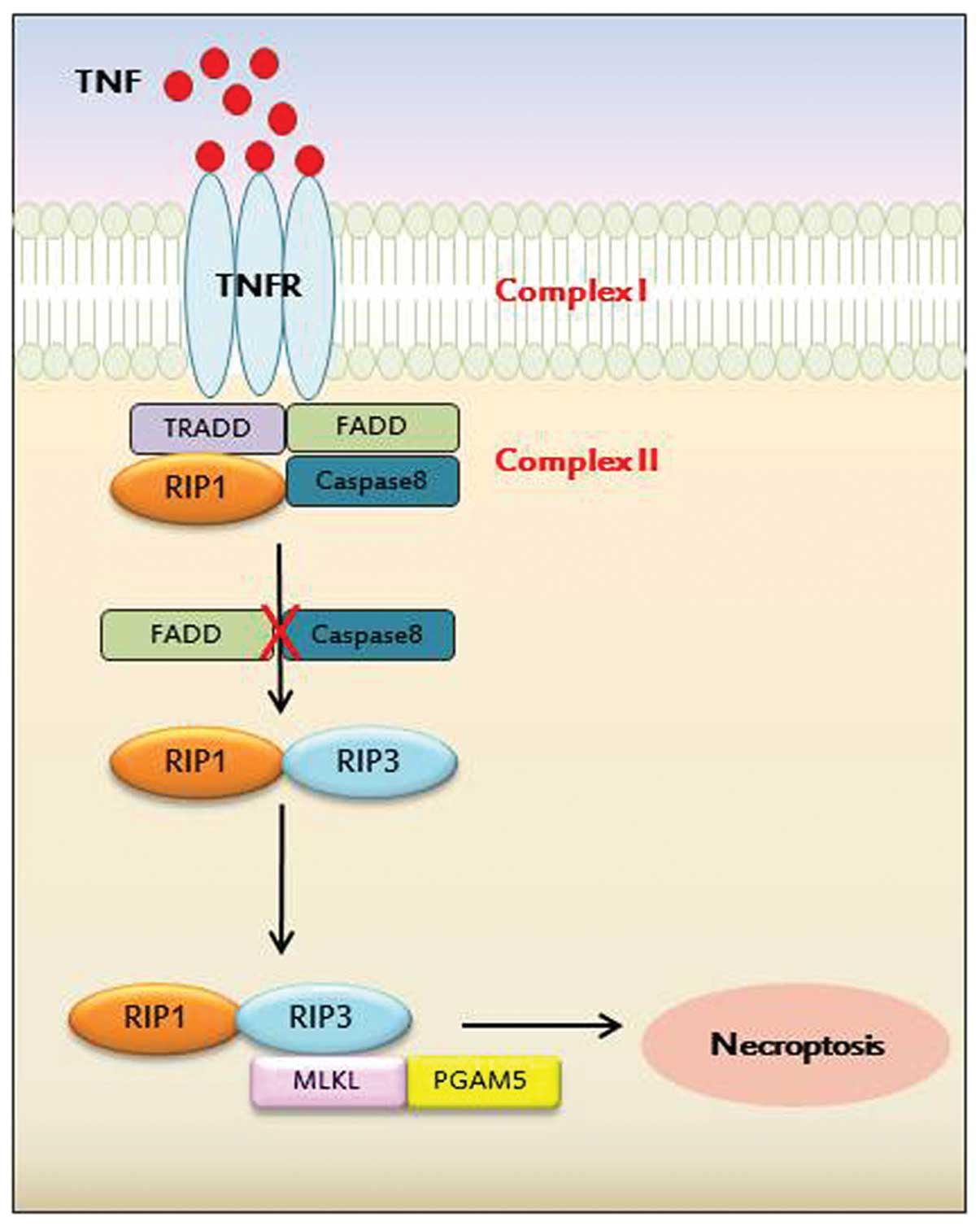
RIP1 was first identified as a regulator of
programmed necrosis upon TNFR stimulation (17). The activity of the RIP1 death
domain kinase is required for death receptor- and zVAD.fmk-mediated
necroptosis in murine and human cells. Later, three individual
research groups identified another novel protein, RIP3, as a
critical protein for the activation of programmed necrosis when
default cell death (apoptosis) is hindered (18–20). Furthermore, it has been
demonstrated that the pronecrotic complex formation between RIP1
and RIP3 is required for programmed necrosis, indicating that
downstream or upstream signaling networks of the RIP1-RIP3 complex
are plausible and can be tightly regulated by a cascade of
proteins. It has also been published that the NAD-dependent
deacetylase, SIR2, is involved in the regulation of TNF-mediated
programmed necrosis (21). It not
only recruits RIP3, but also catalyzes the deacetylation of RIP1 to
allow it to be in a stable conformation, forming a necrotic
complex.
Recently, the mixed lineage kinase domain-like
protein (MLKL) was identified as a RIP3 substrate, as well as a
biological target of the hit compound against necroptosis by a
combined approach of chemical biology and biochemistry (22). Unlike both RIP1 and RIP3 proteins,
MLKL does not possess kinase activity due to its absence of a
phosphate-binding loop and key amino acids for kinase. However, its
binding to RIP3 through kinase-like domain leads to an increased
RIP3 kinase activity through the formation of a stable complex.
Subsequently, MLKL is phosphorylated as a RIP3 substrate to form a
necrosis-inducing signaling complex, termed the necrosome, which
includes RIP1, RIP3 and MLKL. In addition, other kinases and
metabolism-related proteins have been disclosed by using
interference RNAs (18), and are
putatively expected to be involved in creating the signaling
network of programmed necrosis.
Apart from cytosolic proteins described above,
mitochondrial proteins have been suggested to be putative
candidates for mediating necroptosis. Pro-death Bcl2 proteins,
which have been documented to play a decisive role in the intrinsic
apoptotic pathway, have also been suggested to be invovled in
necrotic death (23,24). For instance, Bax, Bmf, BNIP3 and
Nix are candidate mitochondrial proteins responsible for specific
necrotic death. In light of mitochondrial function, cyclophilin-D
(CyP-D) and mitochondrial permeability transition (MPT) pore have
been gaining attention as the emerging targets to modulate necrosis
effectively. Of note, RIP3, being activated by a cascade of events
following TNFR ligation, has been suggested to interact with the
mitochondrial protein, glutamate dehydrogenase 1 (GLUD1), therefore
linking the signaling pathway from extracellular stimulation,
intracellular events, to mitochondria. Recently, the signaling
downstream of RIP1/RIP3 complex has been extensively explored.
Accordingly, the interaction of MLKL with the RIP1/RIP3 complex
recruits the mitochondrial protein phosphatase, PGAM5, functioning
as the convergent point for multiple necrosis pathways (25).
3. Small molecules that protect cells from
programmed necrosis, but not apoptosis
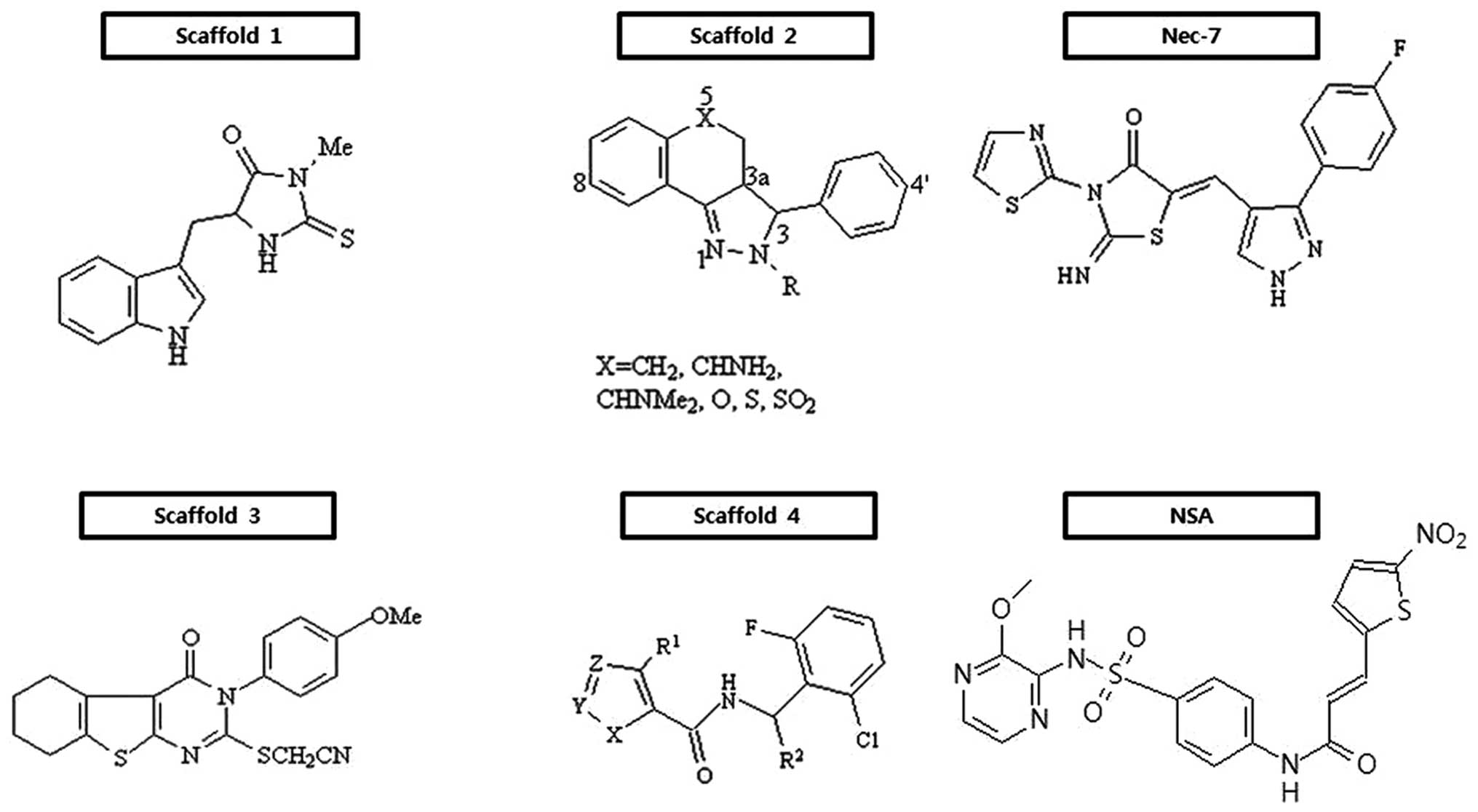
Small molecules that protect cells from undergoing
programmed necrosis are listed by scaffold in Fig. 3. Since the introduction of the
necroptosis concept to cell demise, the discovery and optimization
of small molecules with potent inhibitory activity against it have
been pursued for therapeutic use. The first successful outcome is a
series of hydantoin compounds containing indole derivatives
(Fig. 3, scaffold 1) (25,26), which are potent necrostatins. A
structure-activity relationship (SAR) analysis indicated that
several positions of the indole moiety were very vulnerable to
chemical modification, apart from electron-donating or -withdrawing
substituents at the 7-position, and that the hydantoin ring was
also very sensitive to structural modifications. In fact, the
substitution of the amide nitrogen and removal of a carbonyl group
led to a complete loss of activity. Also, steric bulk and extension
of the linker between the indole and hydantoin ring are found to be
detrimental for their inhibitory activity against necroptosis. Out
of this class bearing scaffold 1, a chemical necrostatin-1 (Nec-1)
has so modest pharmacokinetic profiles to be delivered to the
central nervous system (CNS) following intravenous administration
(27). Subsequently, an extensive
exploration for target molecules of this scaffold resulted in the
identification of RIP1 as an interacting molecule of Nec-1
(28). Mechanistically, Nec-1
inhibits RIP1 in an ATP-competitive manner.
Since then, the discovery of a series of tricyclic
derivatives (Fig. 3, scaffold 2)
(29) and substituted
3H-thieno[2,3-d]pyrimidin-4-ones (Fig. 3, scaffold 3) (30) were ensued. SAR of scaffold 2
demonstrates that the (3R, 3aR)-rel-diastereomers are
more potent than the corresponding (3R,
3aS)-rel-diastereomers. The replacement of fluorine or
methoxy at the 8-position of the tricyclic ring enhances the
protective activity, whereas that at the 6-, 7- and 9-positions is
fatal. Also, the introduction of a methoxy group to the 4-position
of the phenyl ring improves activity, while the location of the
methoxy at the 2-position of it deteriorates its potency; the
placement of amides at the 2-position in the tricyclic ring part
shows the best activity. Notably, in contrast to a hydantoin-indole
necrostatin, these derivatives do not protect cells from
zVAD-induced programmed necrosis in L929 cells, suggesting that
there is a mechanistic distinction between the two series of
compounds (29). In scaffold 3
(Fig. 3), the thioethylcyanide
moiety on the α-position of fused pyrimidone-4 part is required for
the inhibition of necroptosis. The presence of the -OMe group in
the para-position of the benzene ring bonded to pyrimidone nitrogen
is found to be critical for its protective activity. The
introduction of aliphatic rings, such as cyclopentyl, cycloheptyl
or benzene at the position of thiophene ring exhibits some variable
activities. It is apparent that derivatives with the methyl group
at the α- and β-position of the thiophene ring exhibit significant
activities. With increasing size of the aliphatic ring, their
inhibitory activities are detrimental. By contrast, substitution of
the phenyl ring for the cyclohexane ring keeps its active.
Furthermore, [1,2,3]thiadiazole derivatives (Fig. 3, scaffold 4) drawn through high
throughput screening have been found to effectively protect cells
from necroptosis (31). Through
SAR analysis, it has been demonstrated that secondary 2,6-dihalo
substituted benzyl amides are required for their antagonizing
effects on necroptosis. When the methyl group is located in the
benzylic position, the (S)-enantiomeric configuration has
its own ability to interfere with necroptosis. Small branched or
cyclic alkyl groups are favorable in the 4-position of [1,2,3]
thiadiazole. Of note, the replacement of [1,2,3] thiadiazole with a
variety of thiophene derivatives is tolerable. Through the
extensive optimization of necrostatins, a novel necrostatin, Nec-7,
bearing thiazole exerts differential biological activity from
structurally diverse necrostatins, such as Nec-1, Nec-3, Nec-4 and
Nec-5 (32). A series of Nec-7
derivatives suppresses TNF-α-induced necroptosis in the
FADD-deficient variant of human Jurkat T cells, but have no RIP1
inhibitory activity, suggesting that they may target other
necroptosis proteins. SAR analysis showed that various substituents
at the phenyl 4-position are essential, that the para-position of
the phenyl ring is tolerable to substituents and that the pyrazole
ring is susceptible to structural modification (32).
With the discovery of new targets, a hit compound
has been identified through the screening a library of 200,000
compounds for chemicals that protect necrosis. The compound,
(E)-N-{4-[N-(3-methoxypyrazin-2-yl)sulfamoyl]phenyl}-3-(5-nitrothiophene-2-yl)
acrylamide, is commonly referred to as necrosulfonamide (NSA) and
has been reported to be more potent than Nec-1, with an
IC50 of >1 μM under the necrosis- inducing context.
In an effort to reveal its target, MLKL has finally been proven to
be a specific target of NSA which can modify covalently the Cys 86
residue within the N-terminal CC domain of MLKL and consequently
interfere with the induction of necrosis (22).
Among those scaffolds listed above, scaffold 1 has
shown in vivo activity in some mouse models, such as middle
artery occlusion (MCAO) (27),
ischemic/reperfusion heart injury (33) and traumatic brain injury (TBI)
(34). Apart from efforts on the
development of necroptosis inhibitors, very little attempts have
been made to develop therapeutic drug targeting unregulated cell
death, literally necrosis. Recently, LG Life Sciences, Inc. (Seoul,
Korea) identified a series of necrosis inhibitors, referred to as
NecroX™, which has been of interest for therapeutic candidates of
liver diseases and fibrosis, ischemia and neurodegenerative
diseases (35). However, it acts
specifically as a scavenger of mitochondrial ROS, so that its
action mechanism is totally different from that of necroptosis
inhibitors. In a study from my group (unpublished data), NecroX™
was not effective against TNF-α-mediated necrosis, thus suggesting
that death-causing ROS are differentially derived from death
modes.
4. Control of diseases related to programmed
necrosis
Physiological outcomes of programmed necrosis during
viral infection are of significance as an innate immune defense
mechanism. In such a case that viruses or intracellular bacteria
encode caspase inhibitors, host cells themselves fail to operate
the quality control death program (apoptosis) enough to get rid of
infected cells, leading to the propagation of infectious agents. In
light of the pathophysiological aspects, however, an alternative
cell death to apoptosis may rather induce serious damage to
tissues, such as ischemic brain and heart tissue. Nerve cell death
occurs in neurodegenerative disorders, with a continuum of
apoptosis and necrosis being central to acute and chronic
degenerative events (36). In
addition, it has been known that chemical-induced pancreatitis is
associated with programmed necrosis (37). Clinically, parenchymal necrosis is
a key complication of pancreatitis, the severity of which depends
on the cell death modes. Conversely, caspase induction protects
from necrotizing pancreatitis (38). Sepsis is also thought to be
derived from cell death caused by acute uncontrolled microbial
infection. For instance, the pore-forming α-toxin from C.
septicum triggers a multifaceted necrotic cell death response
that is distinctively found in myonecrosis and sepsis (39). However, the detailed process by
which cell death is linked to pathogenic outcomes remains elusive;
programmed necrosis but not apoptosis may provide some insight into
its pathogenesis. Decisively, Nec-1 can significantly delay the
brain necrotic lesions induced by arterial occlusion, implying
clearly that there occurs necrotic cell death controlled by
RIP1-specific inhibitor in an ischemic setting. There is a growing
need that a series of novel small molecules be developed to treat
inflammatory-related diseases mentioned above. This may rather be
based on the programmed necrosis-targeting strategy beyond an
effective suppression of apoptosis. In fact, under an ischemic
condition, the penumbra of the injured brain are the battle ground
for stroke treatment, and immediate action should be taken for
treatment by restoring perfusion to the ischemic area (40). Accordingly, there is at least an
urgent need to develop safe and potent neuroprotective drugs that
can mitigate the damage of cells in the penumbra shortly after
onset and prior to hospital arrival.
A new paradigm of cell death makes it difficult to
delineate cell damage inflicted by extracellular stimuli, whether
it may be derived from microbial infection or chemotherapy.
Although it is not generally admitted, imatinib has been suggested
to induce cell death through a mixture of necrotic and apoptotic
death. Thus, it may be meaningful to address which type of cell
death will have a physiological effect over a treatment period.
5. Conclusions
In this review, the backup form of cell death to
apoptosis has on its own significant meaning in biology. Cells cope
actively with TNF-α-mediated cell death and consequently divert an
apoptotic force into an alternative one. More significantly, the
switch of death modes such as this have differential effects on
physiological outcomes according to the stimuli and tissue niche.
In this review, inflammatory diseases associated with programmed
necrosis were briefly discussed to demonstrate the identification
of small molecules against necroptosis. A variety of efforts in the
search for chemical entities have been made since the first
identification of RIP1. Presently, Nec-1 is the only small molecule
being developed for targeting a specific molecule, RIP1.
Thereafter, some necrotic proteins have been extensively explored
and signaling networks between molecules have been partly unveiled.
The identification of a novel programmed necrosis regulator, RIP3,
and elucidation of its signaling pathway will set out to adopt new
strategies, such as the suppression of RIP3 kinase activity or the
dissociation of the pronecrotic RIP1-RIP3 complex (Fig. 4). Therefore, the discovery of
novel small molecules which specifically and selectively inhibit
RIP1 or RIP3 will not only clearly elucidate its molecular
mechanisms, but may further translate into drug development
pipelines. Taken together, this review provides insight into the
molecular entity of small molecules against therapeutic target
proteins governing programmed necrosis.
Acknowledgements
This research was supported by the college of
pharmacy-specialized research fund (the Institute for New Drug
Development) of Keimyung University in 2012, and the Basic Science
Research Program through the National Research Foundation of Korea
(NRF) funded by the Ministry of Education (2013R1A1A2010212).
References
|
1
|
Iyer SS, Pulskens WP, Sadler JJ, et al:
Necrotic cells trigger a sterile inflammatory response through the
Nlrp3 inflammasome. Proc Natl Acad Sci USA. 106:20388–20393. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Xu Y, Huang S, Liu ZG and Han J:
Poly(ADP-ribose) polymerase-1 signaling to mitochondria in necrotic
cell death requires RIP1/TRAF2-mediated JNK1 activation. J Biol
Chem. 281:8788–8795. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Petit F, Arnoult D, Viollet L and
Estaquier J: Intrinsic and extrinsic pathways signaling during
HIV-1 mediated cell death. Biochimie. 85:795–811. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cho YS, Park SY, Shin HS and Chan FK:
Physiological consequences of programmed necrosis, an alternative
form of cell demise. Mol Cells. 29:327–332. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rangamani P and Sirovich L: Survival and
apoptotic pathways initiated by TNF-α: modeling and predictions.
Biotechnol Bioeng. 97:1216–1229. 2007.
|
|
6
|
Lamkanfi M, Festjens N, Declercq W, Vanden
Berghe T and Vandenabeele P: Caspases in cell survival,
proliferation and differentiation. Cell Death Differ. 14:44–55.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li Y, Yang X, Ma C, Qiao J and Zhang C:
Necroptosis contributes to the NMDA-induced excitotoxicity in rat’s
cultured cortical neurons. Neurosci Lett. 447:120–123.
2008.PubMed/NCBI
|
|
8
|
Scholz C, Wieder T, Stärck L, et al:
Arsenic trioxide triggers a regulated form of caspase-independent
necrotic cell death via the mitochondrial death pathway. Oncogene.
24:1904–1913. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Krumschnabel G, Ebner HL, Hess MW and
Villunger A: Apoptosis and necroptosis are induced in rainbow trout
cell lines exposed to cadmium. Aquat Toxicol. 99:73–85. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hitomi J, Christofferson DE, Ng A, et al:
Identification of a molecular signaling network that regulates a
cellular necrotic cell death pathway. Cell. 135:1311–1323. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Baritaud M, Cabon L, Delavallée L, et al:
AIF-mediated caspase-independent necroptosis requires ATM and
DNA-PK-induced histone H2AX Ser139 phosphorylation. Cell Death Dis.
3:e3902012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Park S, Shin H and Cho Y: Shikonin induces
programmed necrosis-like cell death through the formation of
receptor interacting protein 1 and 3 complex. Food Chem Toxicol.
55:36–41. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hsu TS, Yang PM, Tsai JS and Lin LY:
Attenuation of cadmium-induced necrotic cell death by
necrostatin-1: potential necrostatin-1 acting sites. Toxicol Appl
Pharmacol. 235:153–162. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
De Murcia G, Schreiber V, Molinete M, et
al: Structure and function of poly(ADP-ribose) polymerase. Mol Cell
Biochem. 138:15–24. 1994.
|
|
15
|
Lautier D, Lagueux J, Thibodeau J, Ménard
L and Poirier GG: Molecular and biochemical features of poly
(ADP-ribose) metabolism. Mol Cell Biochem. 122:171–193. 1993.
View Article : Google Scholar
|
|
16
|
Gobeil S, Boucher CC, Nadeau D and Poirier
GG: Characterization of the necrotic cleavage of poly(ADP-ribose)
polymerase (PARP-1): implication of lysosomal proteases. Cell Death
Differ. 8:588–594. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Holler N, Zaru R, Micheau O, et al: Fas
triggers an alternative, caspase-8-independent cell death pathway
using the kinase RIP as effector molecule. Nat Immunol. 1:489–495.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cho YS, Challa S, Moquin D, et al:
Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates
programmed necrosis and virus-induced inflammation. Cell.
137:1112–1123. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
He S, Wang L, Miao L, et al: Receptor
interacting protein kinase-3 determines cellular necrotic response
to TNF-α. Cell. 137:1100–1111. 2009.PubMed/NCBI
|
|
20
|
Zhang DW, Shao J, Lin J, et al: RIP3, an
energy metabolism regulator that switches TNF-induced cell death
from apoptosis to necrosis. Science. 325:332–336. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Narayan N, Lee IH, Borenstein R, et al:
The NAD-dependent deacetylase SIRT2 is required for programmed
necrosis. Nature. 492:199–204. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sun L, Wang H, Wang Z, et al: Mixed
lineage kinase domain-like protein mediates necrosis signaling
downstream of RIP3 kinase. Cell. 148:213–227. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Galluzzi L and Kroemer G: Necroptosis: a
specialized pathway of programmed necrosis. Cell. 135:1161–1163.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Baines CP: Role of the mitochondrion in
programmed necrosis. Front Physiol. 1:1562010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang Z, Jiang H, Chen S, Du F and Wang X:
The mitochondrial phosphatase PGAM5 functions at the convergence
point of multiple necrotic death pathways. Cell. 148:228–243. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Teng X, Degterev A, Jagtap P, et al:
Structure-activity relationship study of novel necroptosis
inhibitors. Bioorg Med Chem Lett. 15:5039–5044. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Degterev A, Huang Z, Boyce M, et al:
Chemical inhibitor of nonapoptotic cell death with therapeutic
potential for ischemic brain injury. Nat Chem Biol. 1:112–119.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Degterev A, Hitomi J, Germscheid M, et al:
Identification of RIP1 kinase as a specific cellular target of
necrostatins. Nat Chem Biol. 4:313–321. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jagtap PG, Degterev A, Choi S, Keys H,
Yuan J and Cuny GD: Structure-activity relationship study of
tricyclic necroptosis inhibitors. J Med Chem. 50:1886–1895. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wang K, Li J, Degterev A, Hsu E, Yuan J
and Yuan C: Structure-activity relationship analysis of a novel
necroptosis inhibitor, necrostatin-5. Bioorg Med Chem Lett.
17:1455–1465. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Teng X, Keys H, Jeevanandam A, et al:
Structure-activity relationship study of [1,2,3]thiadiazole
necroptosis inhibitors. Bioorg Med Chem Lett. 17:6836–6840.
2007.
|
|
32
|
Zheng W, Degterev A, Hsu E, Yuan J and
Yuan C: Structure-activity relationship study of a novel
necroptosis inhibitor, necrostatin-7. Bioorg Med Chem Lett.
18:4932–4935. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Smith CC, Davidson SM, Lim SY, Simpkin JC,
Hothersall JS and Yellon DM: Necrostatin: a potentially novel
cardioprotective agent? Cardiovasc Drugs Ther. 21:227–233. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
You Z, Yang J, Takahashi K, et al: Reduced
tissue damage and improved recovery of motor function after
traumatic brain injury in mice deficient in complement component
C4. J Cereb Blood Flow Metab. 27:1954–1964. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Choi JM, Park KM, Kim SH, et al: Effect of
necrosis modulator necrox-7 on hepatic ischemia-reperfusion injury
in beagle dogs. Transplant Proc. 42:3414–3421. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Gorman AM: Neuronal cell death in
neurodegenerative diseases: recurring themes around protein
handling. J Cell Mol Med. 12:2263–2280. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Gukovskaya AS, Mareninova OA, Odinokova
IV, et al: Cell death in pancreatitis: effects of alcohol. J
Gastroenterol Hepatol. 21(Suppl 3): S10–S13. 2006. View Article : Google Scholar
|
|
38
|
Mareninova OA, Sung KF, Hong P, et al:
Cell death in pancreatitis: caspases protect from necrotizing
pancreatitis. J Biol Chem. 281:3370–3381. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kennedy CL, Smith DJ, Lyras D, Chakravorty
A and Rood JI: Programmed cellular necrosis mediated by the
pore-forming α-toxin from clostridium septicum. PLoS Pathog.
5:e10005162009.
|
|
40
|
Yuan J: Neuroprotective strategies
targeting apoptotic and necrotic cell death for stroke. Apoptosis.
14:469–477. 2009. View Article : Google Scholar : PubMed/NCBI
|