Introduction
Cataract is the most common cause of visual
impairment in the elderly worldwide, particularly in developing
countries (1). Anterior
subcapsular cataract (ASC) and posterior capsule opacification
(PCO) are different types of cataract that share similar cellular
and molecular features (2,3).
PCO, also known as a secondary cataract, is the most common
long-term complication of modern cataract surgery. In the past few
decades, although advances in surgical techniques, intraocular lens
materials and designs have reduced the PCO rate, the incidence of
PCO is still ~20–40% in adults and 100% in children (4,5).
At present, cataract surgery and Nd:YAG laser capsulotomy are the
only effective treatments for ASC and PCO, however, they are likely
to induce many other complications and risks. Therefore, a better
understanding of the pathogenesis of these diseases is critical for
the development of new pharmacologic treatments.
Accumulating evidence has shown that the
epithelial-mesenchymal transition (EMT) of lens epithelial cells
(LECs) is a key pathological mechanism involved in the development
of ASC (6,7) and PCO (8,9).
PCO is caused by a wound healing response of residual LECs
following cataract surgery. After surgery, the levels of various
cytokines and growth factors increase in the aqueous humor and
stimulate the residual LECs to proliferate and undergo EMT
(10). Transforming growth factor
β (TGFβ), especially TGFβ2, the major isoform in the aqueous humor
of the eye, plays a central role in the cell biology of PCO
(11). During the process of EMT,
LECs undergo cytoskeletal rearrangement and loss of epithelial
phenotype, then migrate away from the original location onto the
posterior capsule, with the addition of a large amount of
extracellular matrix proteins (collagen and fibronectin)
deposition, and finally contribute to the development of PCO
(7,10). Unlike PCO, ASC is a primary
cataract that is mainly caused by ocular trauma, inflammation or
irritation (12). The
proliferation and EMT of LECs in situ lead to the formation
of subcapsular plaques just beneath the lens anterior capsule,
similar to the transdifferentiated cells in PCO (2). Thus, inhibition of the proliferation
of LECs and EMT may be a promising strategy to prevent ASC and
PCO.
Several signaling pathways are involved in the
process of LECs EMT in ASC and PCO development. Among these,
canonical TGFβ/Smad signaling has been identified to occupy a
crucial position in the signaling networks that control EMT of
LECs. TGFβ/Smad signaling transmits signals by binding to the
related transmembrane type I and II receptors, which subsequently
phosphorylate receptor-regulated Smad2 and Smad3 (13). The phosphorylated Smad2/3 bind to
the common mediator Smad4 to form a stable hetero-oligomeric
complex, and then the complex translocates to the nucleus where the
target gene expression is regulated (13). Recent studies have demonstrated
that the blockade of TGFβ2/Smad2/3 efficiently prevents the effect
of TGFβ2 on LECs migration, extracellular matrix production and EMT
(14,15). In addition to the canonical Smad
signaling, extracellular signal-regulated kinase (ERK) signaling is
involved in TGFβ-induced EMT in different types of cells (16–19). The activation of ERK1/2 signaling
enhances TGFβ-induced EMT, accompanied by morphological changes,
the upregulation of EMT markers and extracellular matrix
components. Blocking the function of ERK1/2 using a special
inhibitor results in the inhibition of TGFβ-induced EMT (17,20). In LECs EMT, it has been previously
reported that ERK1/2 is rapidly activated by TGFβ, and the specific
inhibitor of ERK1/2 blocks the morphologic change of LECs and the
upregulation of Slug induced by TGFβ (19).
Although the role of ERK1/2 signaling in EMT during
cancer progression and some fibrotic disorders has been studied,
the interaction of ERK1/2 with the canonical TGFβ/Smad signaling
pathway and other signaling pathways in fibrotic diseases is poorly
understood. In this study, we demonstrated that the TGFβ2-induced
activation of ERK1/2 is independent of TGFβ/Smad signaling in human
LECs, while the blockade of ERK1/2 signaling with the inhibitor
U0126 completely prevents TGFβ2-induced EMT. Moreover, blockade of
ERK1/2 signaling inhibits the canonical Smad signaling pathway, as
well as the Jagged/Notch pathway. We also found that non-canonical
TGFβ/ERK1/2 signaling can also be mediated by the Notch pathway.
Taken together, these results suggested that ERK1/2 signaling
cross-interacts with the TGFβ/Smad and the Jagged/Notch signaling
pathways, thus mediating EMT in LECs.
Materials and methods
Reagents and antibodies
U0126 (a selective inhibitor of MEK1 and MEK2) and
recombinant human TGFβ2 were purchased from Cell Signaling
Technology, Inc. (Danvers, MA, USA). SB431542 (a specific inhibitor
for TGFβ receptor type I/ALK5 kinase that phosphorylates Smad2/3)
and DAPT (an inhibitor of Notch receptor cleavage) were purchased
from Sigma-Aldrich (St. Louis, MO, USA). Antibodies against ERK1/2,
p-ERK1/2, Jagged-1, Notch-1, Notch-2, p-Smad2, p-Smad3, goat
anti-rabbit and horse anti-mouse horseradish peroxidase
(HRP)-conjugated secondary antibodies were purchased from Cell
Signaling Technology Inc. Antibodies against β-actin, α-SMA,
collagen type I (Col I), collagen type IV (Col IV), and fibronectin
(FN) were purchased from Abcam (Cambridge, UK).
Cell culture and treatment
The SRA01/04 human LEC line was kindly provided by
Professor Fu Shang at the Laboratory for Nutrition and Vision
Research (Boston, MA, USA), and cultured in Dulbecco’s modified
Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS). The
cells were grown at 37°C in a humidified atmosphere containing 5%
CO2 and dissociated with 0.25% trypsin-0.02%
ethylenediaminetetraacetic acid (EDTA) solution.
For TGFβ2 and U0126 treatments, the cells were
seeded in 6-well plates and treated with 10 ng/ml recombinant human
TGFβ2 and different concentrations of U0126 for different
time-points.
Quantitative PCR analysis for gene
expression
Total RNA was isolated from LECs using TRIzol
reagent (Invitrogen, Carlsbad, CA, USA), and the RNA was then
treated with DNase I (Sigma-Aldrich) to remove genomic DNA
contamination. The concentration of total RNA was quantified by
spectrophotometry and cDNA was synthesized with a reverse
transcription kit (Takara Bio Inc., Otsu, Japan). For quantitative
analysis of mRNA expression, the SYBR PrimeScript RT-PCR kit
(Takara Bio Inc.) was used to amplify the target genes, and the
reactions were performed with the ABI Prism 7000 sequence detection
system (Applied Biosystems, Foster City, CA, USA) according to the
manufacturer’s protocol. Glyceraldehyde 3-phosphate dehydrogenase
(GAPDH) was used as an internal control.
Western blot analysis for protein
expression
The cells were washed twice with PBS, and then lysed
in 100 μl of RIPA buffer with protease inhibitor cocktail for total
protein extraction. Protein was collected after centrifugation and
mixed with 5X SDS sample buffer. The samples were separated by 10%
SDS-PAGE, and then transferred to PVDF membranes. The membranes
were blocked with 5% non-fat milk for 1 h and the membranes were
subsequently incubated with different primary antibodies at 4°C
overnight. The membranes were washed with 1X PBS containing 0.1%
Tween-20 (PBST) three times, and incubated with HRP-conjugated
secondary antibodies for 1 h at room temperature. The protein bands
were detected with chemiluminescence detection reagents. β-actin
was used as the loading control. Densitometric analysis was
conducted by ImageJ software 1.41 (National Institutes of Health,
Bethesda, MD, USA).
Statistical analysis
Experiments presented in the figures are
representative of three or more different repetitions. Data were
presented as mean ± standard error of the mean (SEM) and analyzed
with SPSS 15.0 software (SPSS, Inc., Chicago, IL, USA). A standard
Student’s t-test was used for statistical analysis. P<0.05 was
considered to indicate statistical significance.
Results
Blockade of ERK1/2 signaling by U0126
prevents TGFβ2- induced EMT in LECs
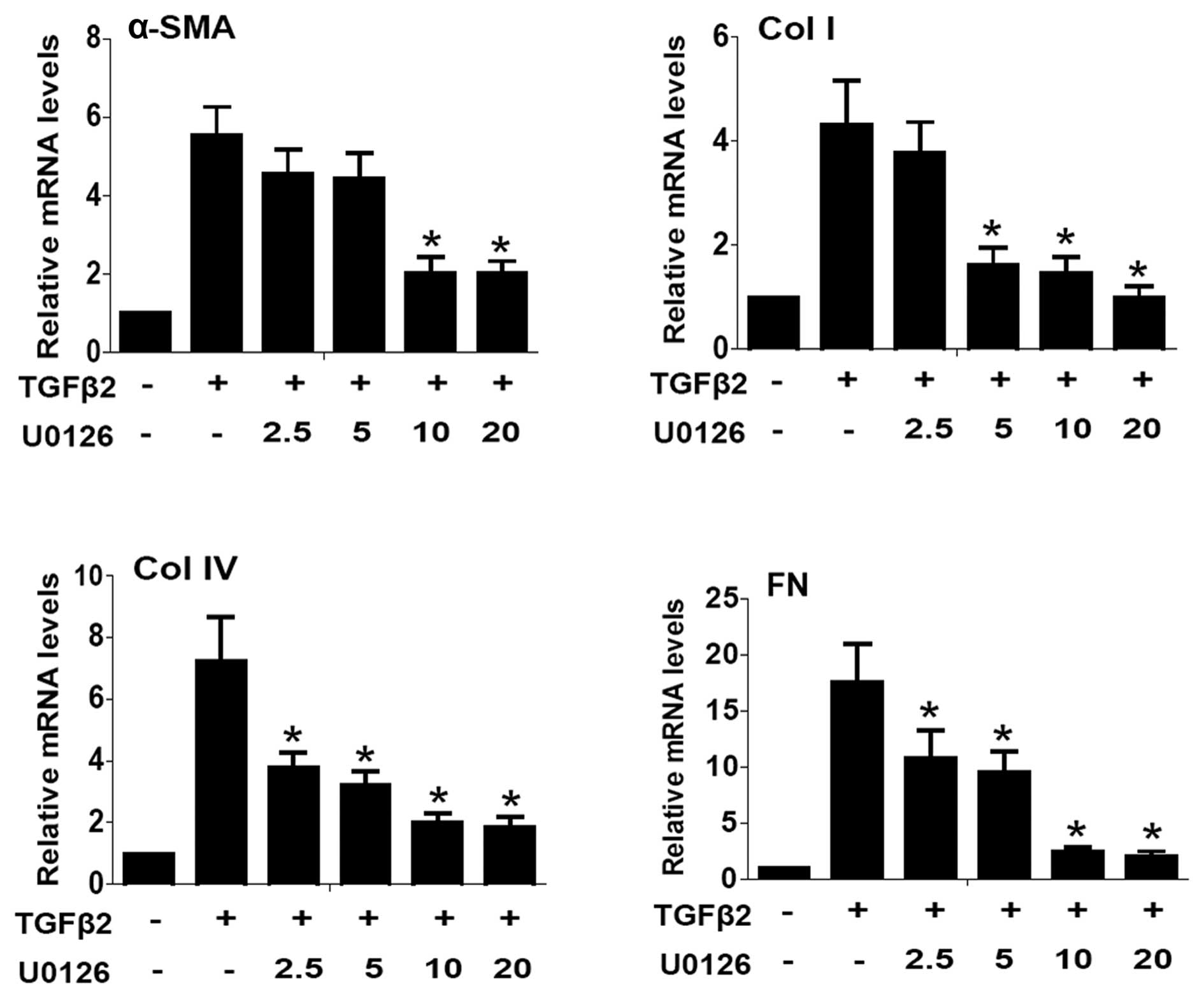
To examine whether the blockade of ERK1/2 signaling
prevented TGFβ2-induced EMT in LECs, U0126 (a selective inhibitor
of MEK1 and MEK2) was used. EMT markers such as α-SMA, Col I, Col
IV and FN were investigated at mRNA and protein levels by
quantitative PCR and western blot analysis, respectively. As shown
in Fig. 1, quantitative PCR
results showed that the mRNA expression of α-SMA, Co1 I, Col IV and
FN were upregulated ~5.5- 4.3- 7.2- and 17.7-fold in TGFβ2-induced
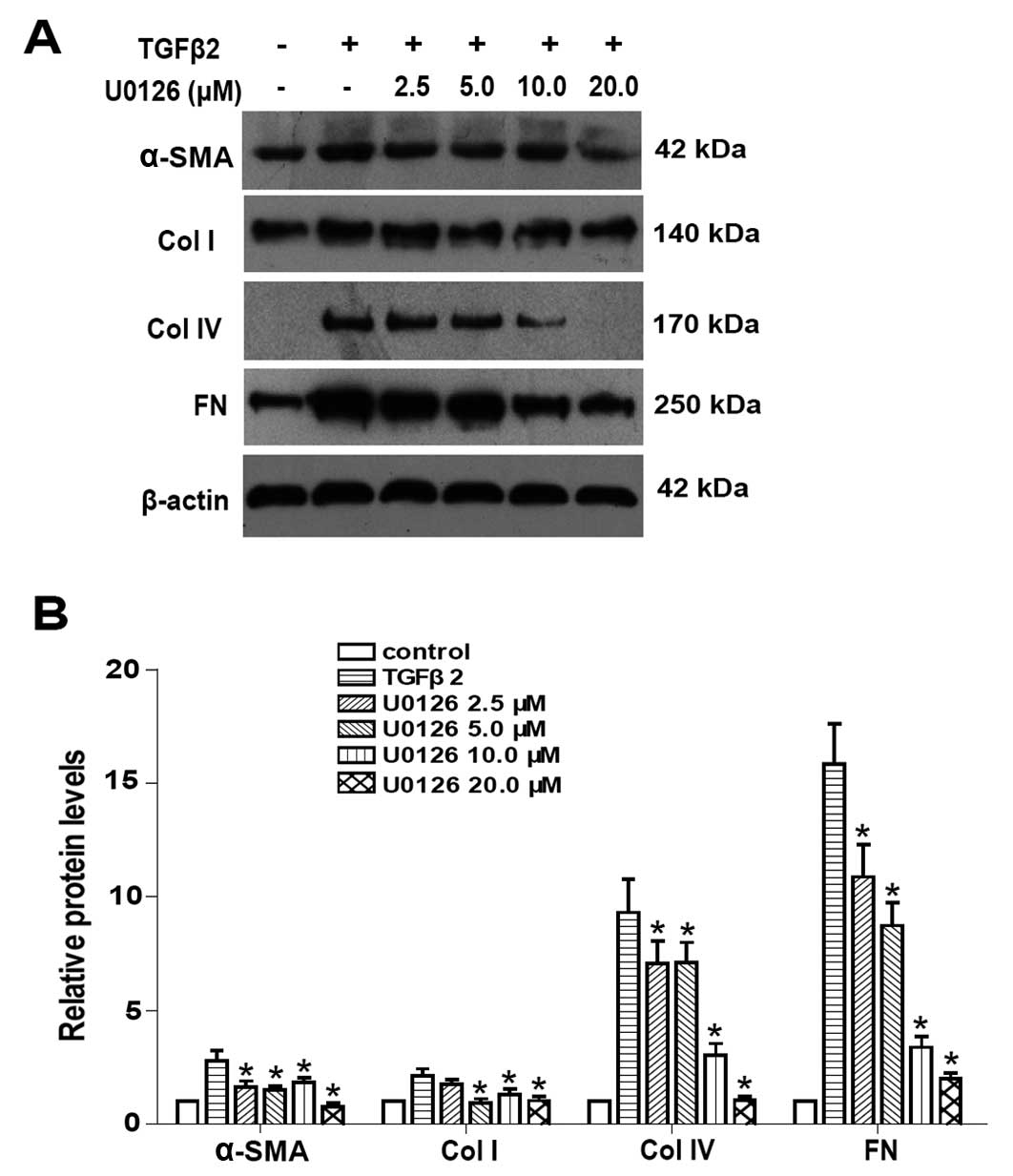
LECs for 24 h. In addition, western blot analysis results showed
that TGFβ2 significantly increased the protein expression of α-SMA,
Col I, Col IV and FN in LECs (Fig.
2). Co-treatment with U0126 markedly abrogated the upregulation
of α-SMA, Col I, Col IV and FN induced by TGFβ2 at the mRNA and
protein levels (Figs. 1 and
2: P<0.05 vs. TGFβ2 treated
with DMSO group). Maximum effect of U0126 was observed at a
concentration of 20.0 μM, however, there was no obvious difference
between 10.0 and 20.0 μM at mRNA level. These data suggested that
the blockade of ERK1/2 pathway by U0126 effectively attenuated
TGFβ2-induced EMT in LECs.
TGFβ2-induced ERK1/2 activation is
independent of the canonical TGFβ/Smad pathway
To determine whether the canonical TGFβ/Smad
signaling is required for the activation of ERK1/2 pathway by
TGFβ2, SB431542 (a specific inhibitor for TGFβ receptor type I/ALK5
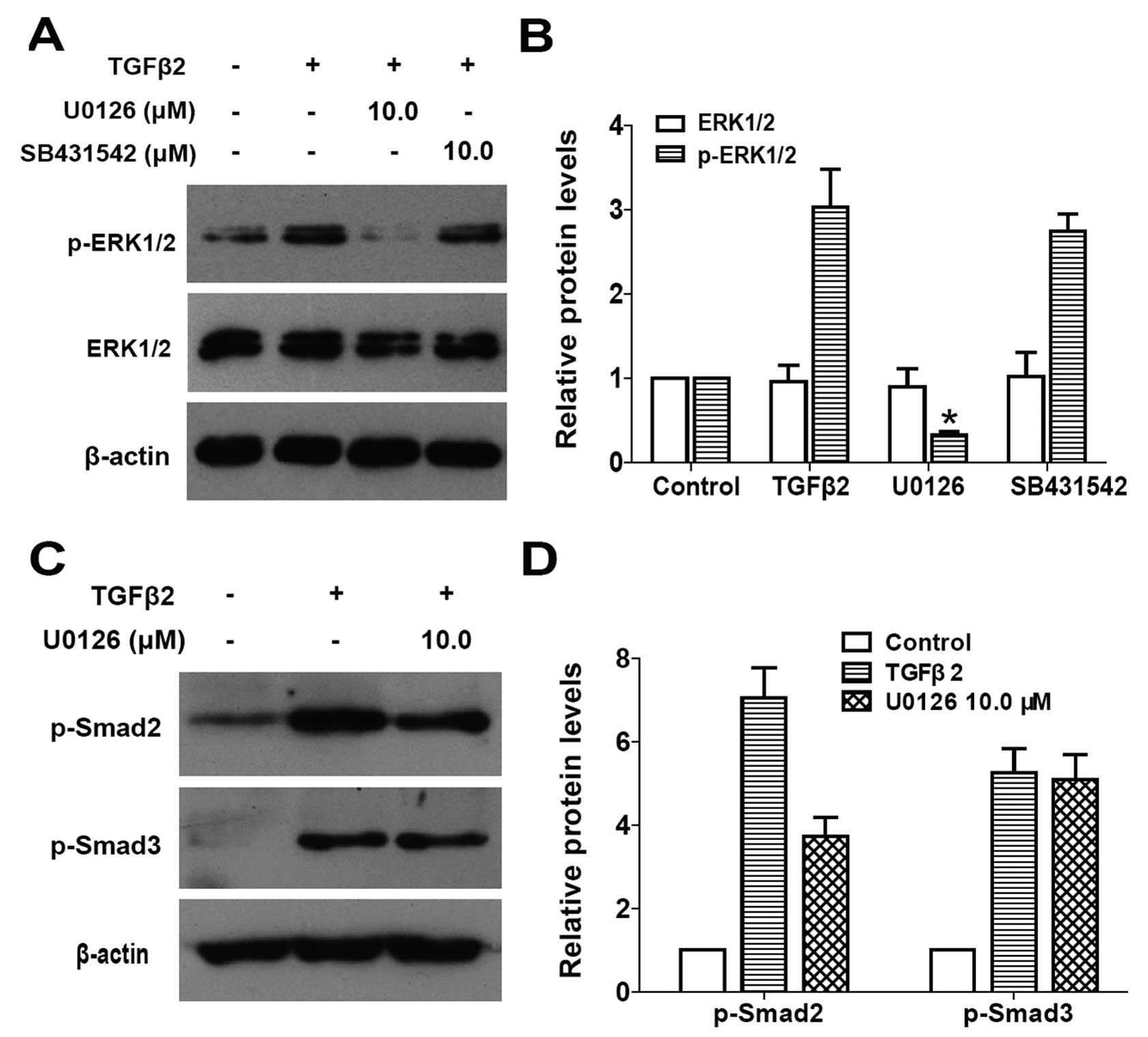
kinase that phosphorylates Smad2/3) was used. As shown in Fig. 3A and B, when LECs were stimulated
by TGFβ2 for 30 min, ERK1/2 was activated via phosphorylation but
with an unchanged total protein level, while U0126 treatment
completely inhibited the TGFβ2-induced activation of ERK1/2.
However, SB431542 treatment had no effect on the phosphorylation of
ERK1/2 (Fig. 3A and B: P<0.05
vs. TGFβ2 treated with DMSO group). These results indicated that
TGFβ2-induced ERK1/2 activation is independent of the canonical
TGFβ/Smad pathway in LECs.
U0126 mediates canonical TGFβ/Smad
signaling by inhibiting the phosphorylation of Smad2
To examine whether there is a crosstalk between the
ERK1/2 signaling and the canonical TGFβ/Smad pathway, the effect of
U0126 on the activation of receptor-regulated Smad proteins Smad2
and Smad3 was examined. As shown in Fig. 3C and D, TGFβ2 alone clearly
induced apparent phosphorylation of Smad2 and Smad3 following
60-min treatment, whereas co-treatment with U0126 inhibited the
phosphorylation of Smad2, but had no effect on the phosphorylation
of Smad3 in LECs (Fig. 3C and D:
P<0.05 vs. TGFβ2 treated with DMSO group). Collectively, these
data suggested that U0126 inhibits the canonical TGFβ2/Smad
signaling transduction by inhibiting the phosphorylation of Smad2.
Thus, there is a crosstalk between ERK1/2 signaling and the
canonical TGFβ2/Smad signaling pathway in LECs.
U0126 prevents TGFβ2-induced EMT partly
by inhibiting the Jagged/Notch pathway
Accumulating evidence suggests that the Notch
signaling pathway is a vital regulator in the induction of EMT
during embryonic development, cancer metastasis and various
fibrotic diseases (21). Results
of a previous study also found that the Jagged/Notch pathway is
activated through canonical TGFβ2/Smad signaling during EMT in
human LECs, while blockade of the Notch pathway with the specific
inhibitor DAPT strongly inhibited TGFβ2-induced EMT (unpublished
data). Therefore, we investigated whether inactivation of ERK1/2
signaling with U0126 inhibited Notch signaling activated by TGFβ2,
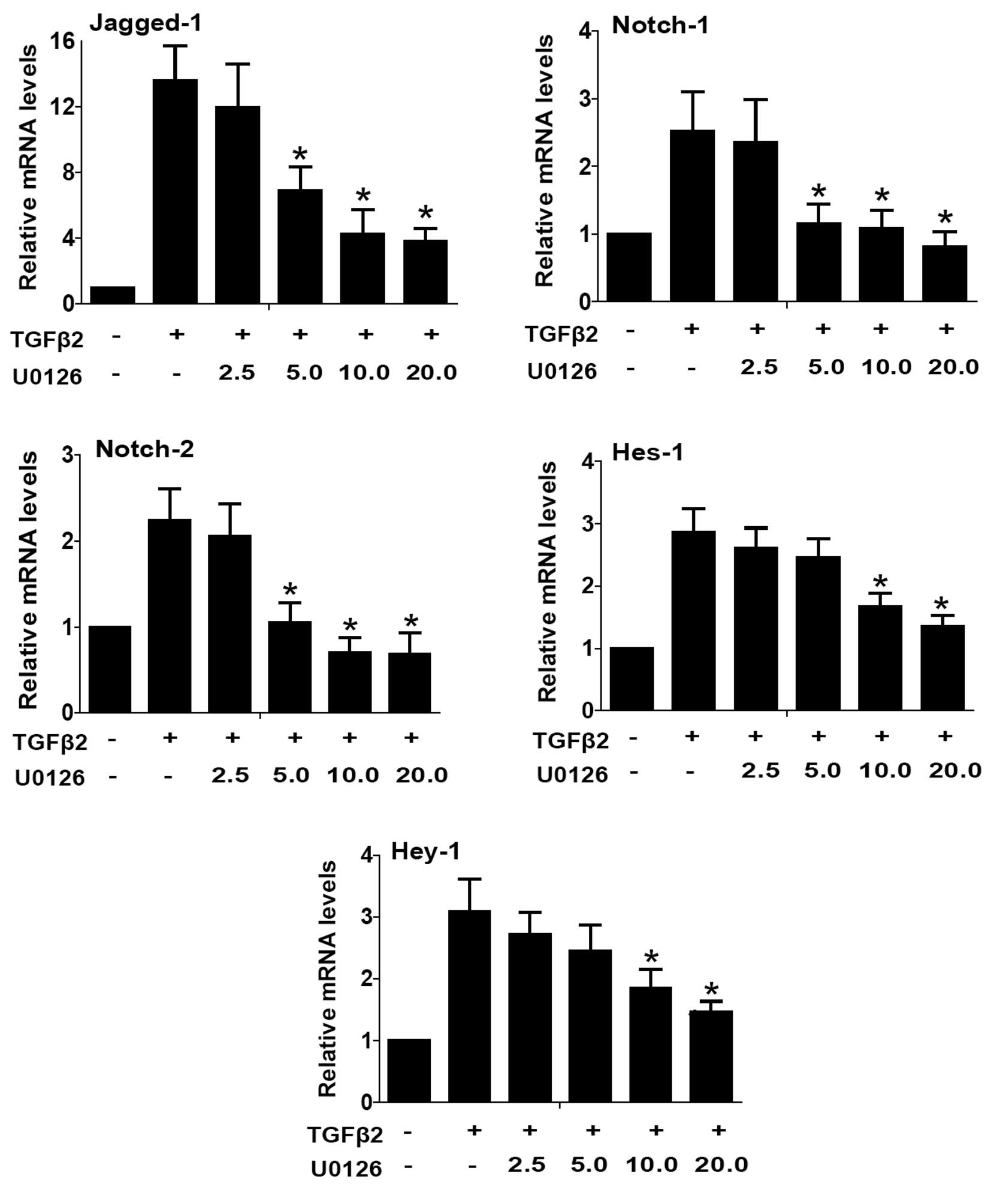
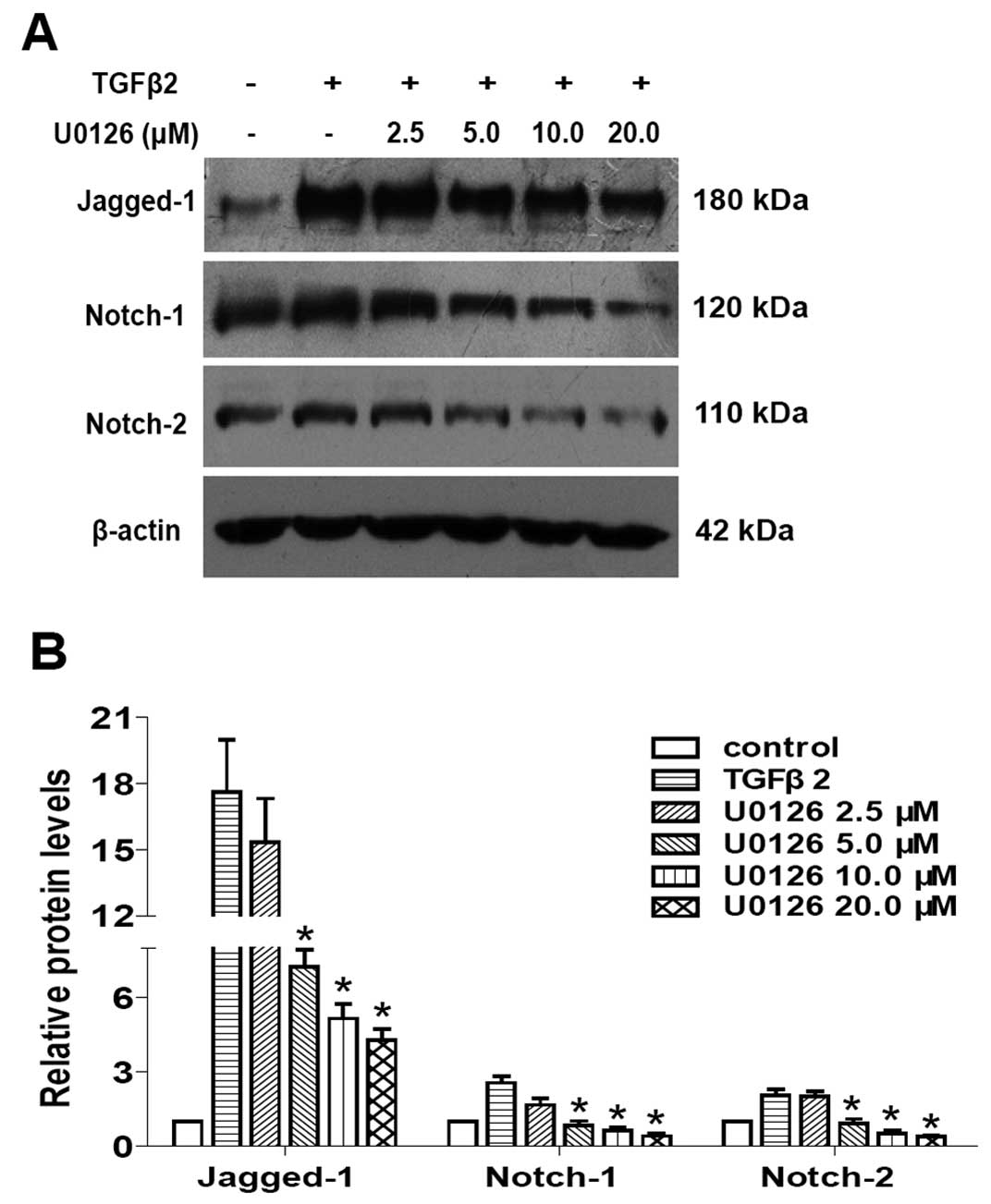
and subsequently inhibited LECs EMT. As shown in Figs. 4 and 5, TGFβ2 treatment alone significantly
increased the expression of Jagged-1, Notch-1 and Notch-2 at mRNA
and protein levels, while U0126 completely attenuated the
TGFβ2-induced upregulation of Jagged-1, Notch-1 and Notch-2
(Figs. 4 and 5: P<0.05 vs. TGFβ2 treated with DMSO
group). In addition, U0126 treatment attenuated TGFβ2-induced Notch
target genes Hes-1 and Hey-1 expression (Fig. 4: P<0.05 vs. TGFβ2 treated with
DMSO group). These results suggested that U0126 prevents
TGFβ2-induced EMT partly by downregulating the Jagged/Notch
pathway. Thus, non-canonical ERK1/2 signaling also contributes to
the TGFβ2-induced activation of the Notch pathway in LECs.
Non-canonical TGFβ/ERK1/2 signaling can
be mediated by the Notch pathway
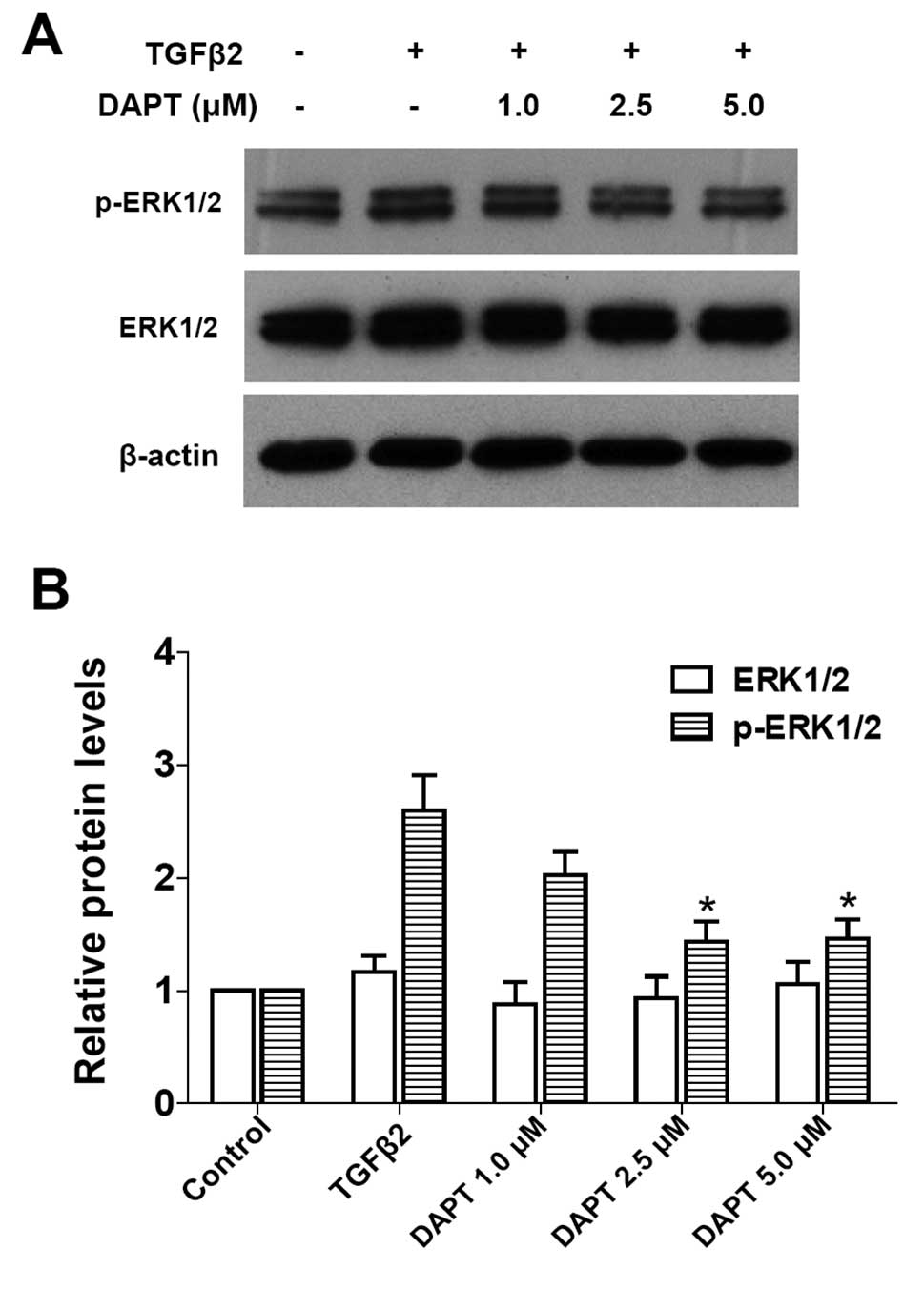
It is unclear whether blockade of Notch signaling is
able to modulate ERK1/2 signaling pathway activated by TGFβ2. As
expected, blockade of the Notch pathway by DAPT clearly inhibited
the TGFβ2-induced activation of ERK1/2 pathway in LECs (Fig. 6: P<0.05 vs. TGFβ2 treated with
DMSO group). These results suggested that the non-canonical
TGFβ/ERK1/2 signaling can be mediated by the Notch pathway
conversely in LECs. This finding also indicated that there is a
crosstalk between the ERK1/2 signaling and Notch pathways.
Discussion
A growing number of studies have proven that the
development of ASC and PCO largely attributes to the EMT of LECs in
response to a variety of cytokines, typically TGFβ2. Activation of
ERK1/2 pathway plays a critical role in carcinogenesis, cancer
metastasis, and various fibrotic diseases, including PCO (19,22–24). In this study, we investigated the
role of ERK1/2 signaling in TGFβ2-induced EMT in human LECs, with a
focus on the interaction of ERK1/2 signaling with the canonical
TGFβ2/Smad and the Jagged/Notch pathways. We found that the
activation of ERK1/2 signaling by TGFβ2 is independent of canonical
TGFβ2/Smad signaling in LECs, while the blockade of ERK1/2
signaling with U0126 markedly prevented TGFβ2-induced EMT.
Furthermore, the blockade of ERK1/2 signaling inhibits the
canonical Smad signaling pathway, as well as the Jagged/Notch
pathway. By contrast, we demonstrated that non-canonical
TGFβ/ERK1/2 signaling can also be mediated by the Notch pathway.
Therefore, our data suggest that ERK1/2 signaling cross-interacts
with the canonical TGFβ/Smad and Jagged/Notch signaling pathways,
thus regulating EMT in LECs.
TGFβ signaling occupies a key position in the
signaling networks that regulates EMT. It includes canonical Smad
signaling and non-canonical Smad independent signaling pathways.
Previous studies have reported that ERK1/2 signaling is involved in
TGFβ-induced EMT in LECs and other types of cells (16–19). The activation of ERK1/2 signaling
promotes TGFβ-induced EMT and ECM components deposition, whereas
the inactivation of ERK1/2 inhibits TGFβ-induced EMT effectively
(19,20). In the present study, we found that
ERK1/2 is rapidly activated by TGFβ2 stimulation, and that MEK1/2
inhibitor U0126 blocks this response completely. Nevertheless,
SB431542, a specific inhibitor for the canonical TGFβ/Smad2/3
signaling transduction, has no effect on the activation of ERK1/2
induced by TGFβ2. These data indicate that TGFβ2-induced ERK1/2
activation is independent of the TGFβ/Smad pathway in LECs. In
addition, inactivation of ERK1/2 signaling strongly prevents the
upregulation of EMT markers induced by TGFβ2. These results suggest
that ERK1/2 signaling pathway is a critical mediator for TGFβ
induction of EMT in LECs, and ERK1/2 inhibitor can be useful for
abrogating EMT phenotype.
It has been reported that non-canonical Smad
signaling, such as the p38MAPK and PI3K/AKT pathways, can crosstalk
and integrate with the canonical TGFβ/Smad signaling, thereby
contributing to EMT (25). To
examine whether there is a crosstalk between the non-canonical
TGFβ/ERK1/2 signaling and the canonical TGFβ/Smad signaling, the
effect of U0126 on the activation of receptor-regulated Smad2 and
Smad3 induced by TGFβ2 was investigated. We found that U0126
inhibits the phosphorylation of Smad2 induced by TGFβ2, but cannot
inhibit the phosphorylation of Smad3 in LECs. These results suggest
that U0126 mediates the canonical TGFβ/Smad signaling by inhibiting
the phosphorylation of Smad2. Therefore, there is a crosstalk
between the non-canonical TGFβ/ERK1/2 and the canonical TGFβ/Smad
signaling in LECs EMT.
Evidence suggests that the Notch signaling pathway
is a vital regulator in the induction of EMT during embryonic
development, cancer metastasis and various fibrotic diseases
(21). Activated Jagged/Notch
signaling has been confirmed in a large range of fibrotic diseases
developed in the kidney, liver and lung (26). Moreover, our former study found
that the Notch signaling pathway is upregulated via canonical
TGFβ2/Smad signaling in LECs EMT, while blockade of the Notch
pathway with DAPT markedly reverses TGFβ2-induced EMT. In this
study, we have shown that U0126 attenuates the TGFβ2-induced
upregulation of Jagged-1, Notch-1 and Notch-2, as well as
TGFβ2-induced Notch target genes Hes-1 and Hey-1 expression. These
results suggest that non-canonical ERK1/2 signaling also
contributes to the TGFβ2-induced activation of the Notch pathway in
LECs. Inactivation of ERK1/2 with U0126 abrogates TGFβ2-induced EMT
partly by suppressing the Jagged/Notch pathway. Furthermore, we
observed that blockade of the Notch pathway by DAPT inhibits the
TGFβ2-induced activation of the ERK1/2 pathway. This means
non-canonical TGFβ/ERK1/2 signaling can be mediated by the Notch
pathway inversely in LECs. Collectively, these data indicate that
there is a crosstalk between the ERK1/2 signaling and the Notch
pathway in LECs EMT.
In summary, our results provide evidence that the
TGFβ2-induced activation of ERK1/2 is independent of canonical
TGFβ/Smad signaling in human LECs. Inactivation of ERK1/2 signaling
with U0126 completely inhibits TGFβ2-induced EMT in LECs. In
addition, the blockade of ERK1/2 signaling inhibits the canonical
Smad signaling pathway, as well as the Jagged/Notch pathway. We
also found that non-canonical TGFβ/ERK1/2 signaling can be mediated
by the Notch pathway conversely. Thus, findings of this study
suggest that ERK1/2 signaling cross-interacts with the canonical
TGFβ/Smad and the Jagged/Notch signaling pathways, thus mediating
EMT in LECs. Therefore, ERK inhibitor may have therapeutic value in
the prevention and treatment of ASC and PCO.
Acknowledgements
We would like to thank Professor Fu Shang for kindly
providing the SRA01/04 human LEC line for this study. The study was
funded by the grant from the Guangdong Natural Science Foundation
(S2012020010878).
References
|
1
|
McCarty CA and Taylor HR: Recent
developments in vision research: light damage in cataract. Invest
Ophthalmol Vis Sci. 37:1720–1723. 1996.PubMed/NCBI
|
|
2
|
Nathu Z, Dwivedi DJ, Reddan JR, Sheardown
H, Margetts PJ and West-Mays JA: Temporal changes in MMP mRNA
expression in the lens epithelium during anterior subcapsular
cataract formation. Exp Eye Res. 88:323–330. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Shin EH, Basson MA, Robinson ML, McAvoy JW
and Lovicu FJ: Sprouty is a negative regulator of transforming
growth factor β-induced epithelial-to-mesenchymal transition and
cataract. Mol Med. 18:861–873. 2012.PubMed/NCBI
|
|
4
|
Apple DJ, Solomon KD, Tetz MR, et al:
Posterior capsule opacification. Surv Ophthalmol. 37:73–116. 1992.
View Article : Google Scholar
|
|
5
|
Hodge WG: Posterior capsule opacification
after cataract surgery. Ophthalmology. 105:943–944. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Srinivasan Y, Lovicu FJ and Overbeek PA:
Lens-specific expression of transforming growth factor beta1 in
transgenic mice causes anterior subcapsular cataracts. J Clin
Invest. 101:625–634. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
de Iongh RU, Wederell E, Lovicu FJ and
McAvoy JW: Transforming growth factor-beta-induced
epithelial-mesenchymal transition in the lens: a model for cataract
formation. Cells Tissues Organs. 179:43–55. 2005.PubMed/NCBI
|
|
8
|
Wallentin N, Wickström K and Lundberg C:
Effect of cataract surgery on aqueous TGF-beta and lens epithelial
cell proliferation. Invest Ophthalmol Vis Sci. 39:1410–1418.
1998.PubMed/NCBI
|
|
9
|
Meacock WR, Spalton DJ and Stanford MR:
Role of cytokines in the pathogenesis of posterior capsule
opacification. Br J Ophthalmol. 84:332–336. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Awasthi N, Guo S and Wagner BJ: Posterior
capsular opacification: a problem reduced but not yet eradicated.
Arch Ophthalmol. 127:555–562. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Allen JB, Davidson MG, Nasisse MP,
Fleisher LN and McGahan MC: The lens influences aqueous humor
levels of transforming growth factor-beta 2. Graefes Arch Clin Exp
Ophthalmol. 236:305–311. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Eldred JA, Dawes LJ and Wormstone IM: The
lens as a model for fibrotic disease. Philos Trans R Soc Lond B
Biol Sci. 366:1301–1319. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Akhurst RJ and Hata A: Targeting the TGFβ
signalling pathway in disease. Nat Rev Drug Discov. 11:790–811.
2012.
|
|
14
|
Li J, Tang X and Chen X: Comparative
effects of TGF-β2/Smad2 and TGF-β2/Smad3 signaling pathways on
proliferation, migration, and extracellular matrix production in a
human lens cell line. Exp Eye Res. 92:173–179. 2011.
|
|
15
|
Dawes LJ, Sleeman MA, Anderson IK, Reddan
JR and Wormstone IM: TGFbeta/Smad4-dependent and -independent
regulation of human lens epithelial cells. Invest Ophthalmol Vis
Sci. 50:5318–5327. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chung EJ, Chun JN, Jung SA, Cho JW and Lee
JH: TGF-β-stimulated aberrant expression of class III β-tubulin via
the ERK signaling pathway in cultured retinal pigment epithelial
cells. Biochem Biophys Res Commun. 415:367–372. 2011.
|
|
17
|
Chen XF, Zhang HJ, Wang HB, et al:
Transforming growth factor-β1 induces epithelial-to-mesenchymal
transition in human lung cancer cells via PI3K/Akt and MEK/Erk1/2
signaling pathways. Mol Biol Rep. 39:3549–3556. 2012.
|
|
18
|
Aomatsu K, Arao T, Sugioka K, et al: TGF-β
induces sustained upregulation of SNAI1 and SNAI2 through Smad and
non-Smad pathways in a human corneal epithelial cell line. Invest
Ophthalmol Vis Sci. 52:2437–2443. 2011.
|
|
19
|
Choi J, Park SY and Joo CK: Transforming
growth factor-beta1 represses E-cadherin production via slug
expression in lens epithelial cells. Invest Ophthalmol Vis Sci.
48:2708–2718. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Xie L, Law BK, Chytil AM, Brown KA, Aakre
ME and Moses HL: Activation of the Erk pathway is required for
TGF-beta1-induced EMT in vitro. Neoplasia. 6:603–610. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang Z, Li Y, Kong D and Sarkar FH: The
role of Notch signaling pathway in epithelial-mesenchymal
transition (EMT) during development and tumor aggressiveness. Curr
Drug Targets. 11:745–751. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Neuzillet C, Tijeras-Raballand A, de
Mestier L, Cros J, Faivre S and Raymond E: MEK in cancer and cancer
therapy. Pharmacol Ther. 141:160–171. 2013. View Article : Google Scholar
|
|
23
|
Tanahashi T, Osada S, Yamada A, et al:
Extracellular signal-regulated kinase and Akt activation play a
critical role in the process of hepatocyte growth factor-induced
epithelial-mesenchymal transition. Int J Oncol. 42:556–564.
2013.
|
|
24
|
Pacheco-Domínguez RL, Palma-Nicolas JP,
López E and López-Colomé AM: The activation of MEK-ERK1/2 by
glutamate receptor-stimulation is involved in the regulation of RPE
proliferation and morphologic transformation. Exp Eye Res.
86:207–219. 2008.PubMed/NCBI
|
|
25
|
Zhang YE: Non-Smad pathways in TGF-beta
signaling. Cell Res. 19:128–139. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Leask A: Targeting the jagged/notch
pathway: a new treatment for fibrosis? J Cell Commun Signal.
4:197–198. 2010. View Article : Google Scholar : PubMed/NCBI
|