Introduction
Acute kidney injury (AKI) is a life-threatening
condition with high morbidity and mortality, even in patients who
have received medical intervention. As there is a lack of effective
remedies for AKI other than dialysis, experimental efforts are
being made to explore the pathogenesis of AKI and to seek materials
with therapeutic potency. Cell cycle arrest is known to be
beneficial for repairing of damaged DNA, thereby reducing the
severity and teratogenicity of the injury (1). For this reason, cyclin-dependent
kinase inhibitors (CDKIs) have been much investigated and have
proven to possess cell protective properties in AKI (2–10).
It is therefore believed that cell cycle regulation is a potential
remedy for the treatment of AKI.
Based on sequence and the inhibitory effects they
exert on cyclin-dependent kinase (CDK), the seven CDKIs are divided
into two families. The CIP/KIP family includes p21, p27 and p57,
which act with multiple CDK and extensively inhibit the cell cycle
(11). The INK4 family includes
p16, p15, p18 and p19, which only interact with CDK4/6 and
specifically arrest the cell cycle in early G1 phase (12). CDKIs studies in AKI have mainly
focused on the CIP/KIP family, especially p21, which has been
identified as the protective factor in AKI (5–10).
Compared to CIP/KIP family members, the role of INK4 members in AKI
has yet to be determined. However, previous studies have reported
that some novel additional biological functions are present in INK4
family members, such as p16 and p19. P16 controlled apoptosis
induced by ultraviolet light and cisplatin through the intrinsic
mitochondrial cell death pathway (13–14). Overexpression of p19 conferred
resistance to cells exposed to UV irradiation (15–16).
Therefore, we hypothesized the beneficial behaviors
of p18 and investigated its role in cisplatin-induced AKI using
p18−/− mice. As oxidative stress is important factor in
cisplatin-induced AKI, we also investigated the effect of p18 on
cisplatin-induced endoplasmic reticulum stress (ERS) in an attempt
to elucidate the possible mechanism involved in p18 actions.
Materials and methods
Animals
P18+/−mice in a C57BL/6 and 129/Sv background were
kind gifts from Professor Tao Cheng of the laboratory of Cancer
Research Center at Pittsburgh University. P18−/− or p18+/+ mice
were generated from p18+/− breeding pairs. The mice were genotyped
by a PCR approach, using tail DNA as previously described (17).
The primer sequences for genotype identification
included: p18 WT forward, 5′-AGCCATCAAATTTATTCATGTTGCAGG-3′; p18
MG-47 reverse, 5′-CCTCCATCAGGCTAATGACC-3′; and PGKNEO reverse,
5′-CCAGCCTCTGAGCCCAGAAAGCGAAGG-3′.
The detailed characteristics of the p18−/− mice were
previously described by Franklin et al (18). Briefly, p18−/− mice grew and
developed to become larger in body size than their p18+/+
littermates. Accordingly, the heart, liver and kidneys of the
p18−/− mice exhibited proportional organomegaly; however, no
abnormal structures, such as hepatic hypertrophy, glomerular
sclerosis, diffuse kidney tubular atrophy, or dermal abnormalities,
were detected in p18−/− mice.
Littermates or age-matched male mice (8–12 weeks)
were used in our experiments. The animals were housed in a specific
pathogen-free facility with access to water and food ad
libitum at the Second Military Medical University Animal
Center. All procedures were approved by the Ethics Committee of the
Experimental Animals Center of the University.
Animal experiments
AKI was induced by a single intraperitoneal
injection of cisplatin (Sigma, St. Louis, MO, USA) at a dose of
12.5 mg/kg in p18−/− (n=35) and p18+/+ (n=35) mice, while the
controls (n=15) were injected with isovolumic saline.
After cisplatin injection, the 28-day survival of
p18−/− (n=15) and p18+/+ (n=15) mice was determined, while the
other animals were dealt with at day 3 after the injection. Blood
samples were collected using the method of eye enucleation. Kidneys
were collected after the animals were sacrificed by cervical
dislocation. Kidneys and blood were collected at day 3 after
cisplatin injection for morphological and renal function analysis.
Serum creatinine (SCr) and urea nitrogen were determined by
enzymatic colorimetric assay. Kidney tissues were stained with
hematoxylin and eosin (H&E) and morphological assessment was
determined under light microscopy by the same experienced
histologist. Tubular necrosis, brush border loss and cast formation
were used as the main damage parameters. Scoring was performed
according to the percentage of damaged tubuli in the kidney as
follows: I, 0–25%; II, 25–50%; III, 50–75%; IV, >75%.
Terminal deoxynucleotidyl-transferase-mediated dUTP
nick end-labeling (TUNEL) and analysis of ERS signal proteins by
quantitative PCR (qPCR) and western blot analysis were performed at
day 3 after cisplatin injection for the p18−/− and p18+/+
kidneys.
TUNEL
A commercial kit (Fuzhou Maixin Biotechnology
Development Co., Ltd., Fuzhou, China) was used to detect apoptotic
cells for in situ kidneys. Briefly, paraffin-embedded
sections were deparaffinized in xylene and rehydrated through
graded concentrations of ethanol. After being washed with PBS, the
sections were treated with 0.5% pepsin at 37°C for 8 min, and 0.3%
Triton X-100 for 10 min at room temperature. To inactivate
endogenous peroxidase, the sections were incubated in 3%
H2O2 at 37°C for 15 min and then incubated
with terminal deoxynucleotidyl transferase (TdT) in a humid chamber
at 37°C for 1 h. The signals were detected with a horseradish
peroxidase-conjugated sheep anti-alkaline phosphatase antibody.
Quantitative measurement of apoptotic cells was performed by
examining 10 randomly selected fields under a light microscope
(magnification, ×400) in the cortex. Twelve sections from at least
six animals of each group were counted, and the data were presented
as the mean number of apoptotic cells in each HPF field.
Differences were considered statistically significant if
P<0.05.
qPCR
Total RNA was extracted from kidney tissues (renal
cortex) by means of the TRIzol reagent (Invitrogen Life
Technologies, Carlsbad, CA, USA), and an RT kit (Takara Bio, Inc.,
Shiga, Japan) was used to synthesize cDNA. The expression of signal
proteins in ERS was determined by qPCR using the ABI PRISM 7000
Sequence Detection System, and PCR reactions were performed using
the SYBR-Green real-time PCR Master mix (Toyobo Co., Ltd., Osaka,
Japan). The ribosomal gene 18S (18S rRNA) was selected as an
endogenous reference and the samples were assayed in triplicate.
Based on the analysis by the ΔΔCt method, the expression of the
target genes was determined. The primer sequences used for qPCR
were: 18S rRNA forward, 5′-AGGAGTGGGCCTGCGGCTTA-3′ and reverse,
5′-GCCGGGTGAGGTTTCCCGTG-3′; Grp78 forward,
5′-AGACATTTGCCCCAGAAGAA-3′ and reverse, 5′-ATCTTTGGTTGCTTGTCGCT-3′;
Grp94 forward, 5′-TGAAGGAGAAGCAGGACAAAA-3′ and reverse,
5′-AGTCGCTCAACAAAGGGAGA-3′; and CCAAT/enhancer-binding
protein-homologous protein (CHOP) forward,
5′-TATCTCATCCCCAGGAAACG-3′ and reverse,
5′-GGACGCAGGGTCAAGAGTAG-3′.
Western blot analysis
Protein was extracted from the renal cortex using a
lysis buffer containing 50 mM Tris-HCl (pH 8.0), 50 mM NaCl, 50 mM
sodium fluoride, 0.1% Nonidet P-40, 5 mM EDTA, 2 mM
phenylmethylsulfonyl fluoride, 1 mM dithiothreitol, 10 μg/ml
leupeptin, 2 μg/ml pepstatin, and 1 μg/ml aprotinin. After a 30-min
incubation on ice, the lysates were heated at 100°C for 15 min and
centrifuged at 12,000 × g for 15 min at 4°C. Lysates containing
equal amounts of proteins (100 μg) were dissolved in an SDS sample
buffer, separated on 12% SDS slab gels and transferred
electrophoretically onto polyvinylidene difluoride (PVDF)
membranes. Equal protein loading and protein transfer was confirmed
by Ponceau S staining. After blocking with 5% non-fat dry milk in
TBST, the membrane was incubated at 4°C overnight with the
following primary antibodies: mouse anti-GAPDH (1:5,000 dilution;
Kangcheng Biotechnology, Shanghai, China), rabbit anti-p18 (1:200
dilution; Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA),
rabbit anti-grp78 (1:1,000 dilution), rabbit anti-phosphorylation
of pancreatic endoplasmic reticulum (ER) eukaryotic translation
initiation factor 2α (eIF2α) kinase (PERK) (1:1,000 dilution),
rabbit anti-phospho-PERK (1:1,000 dilution), rabbit anti-eIF2α
(1:1,000 dilution) and rabbit anti-phospho-eIF2α (1:1,000 dilution)
(all from Cell Signaling Technology, Beverly, MA, USA). After
washing, a horseradish peroxidase-conjugated secondary antibody was
applied. Proteins that bound to the secondary antibody were
visualized using ECL (Amersham Pharmacia Biotech, Amersham,
UK).
Statistical analysis
Data are presented as mean ± SD and were analyzed
for significance using an ANOVA model. Comparisons between the two
groups were made using the t-test or Wilcoxon-Mann-Whitney test.
Differences were considered tatistically significant if
P<0.05.
Results
Deletion of p18 aggravated
cisplatin-induced AKI
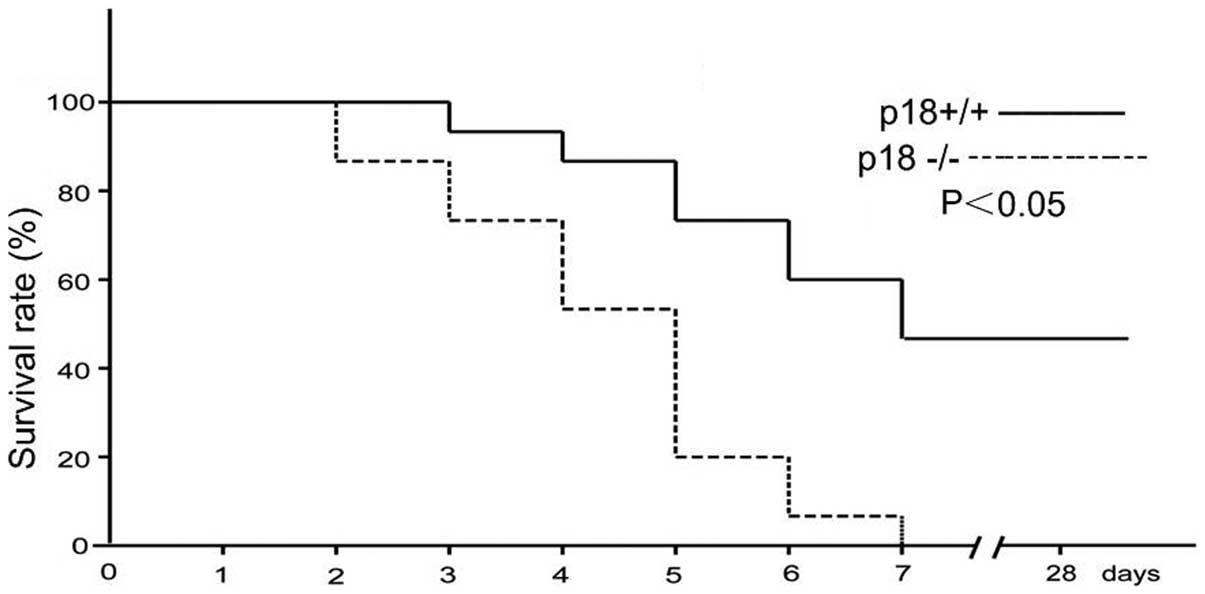
As shown in Fig.
1, the 28-day survival of p18−/− mice was significantly worse
than that of their p18+/+ counterparts. All 15 p18−/− mice died at
day 7 after cisplatin injection, while the 7-day survival rate for
the p18+/+ mice was 53.3%, with no deaths occurring in p18+/+ mice
from day 8 to 28. A significant difference was observed between the
survival curves of p18−/− and p18+/+ mice after cisplatin injection
(P<0.05 for the log-rank test).
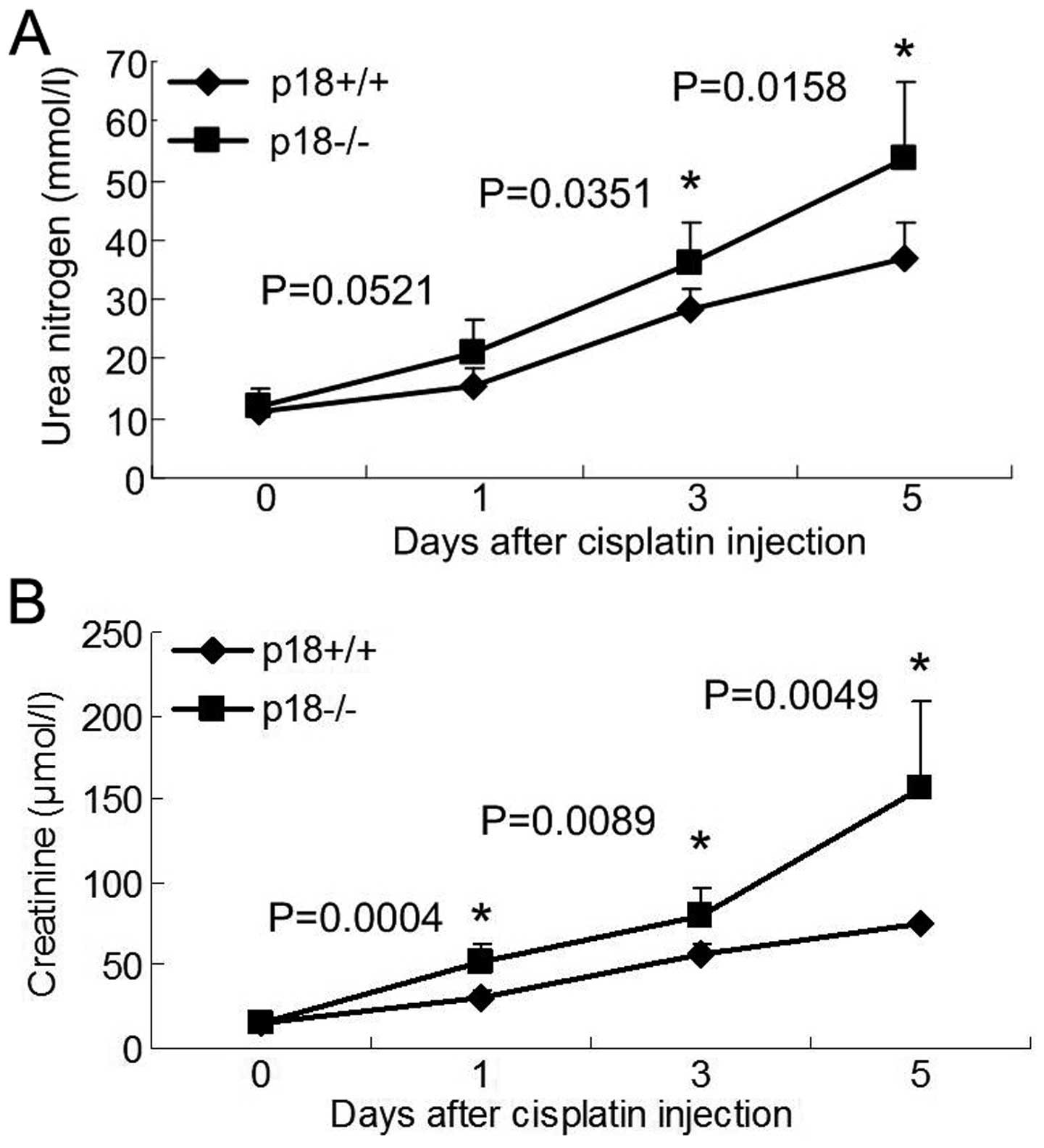
Compared to p18+/+ mice, aggravated urea nitrogen
(Fig. 2A) and creatinine
(Fig. 2B) of p18−/− mice was
demonstrated at day 3 after cisplatin injection.
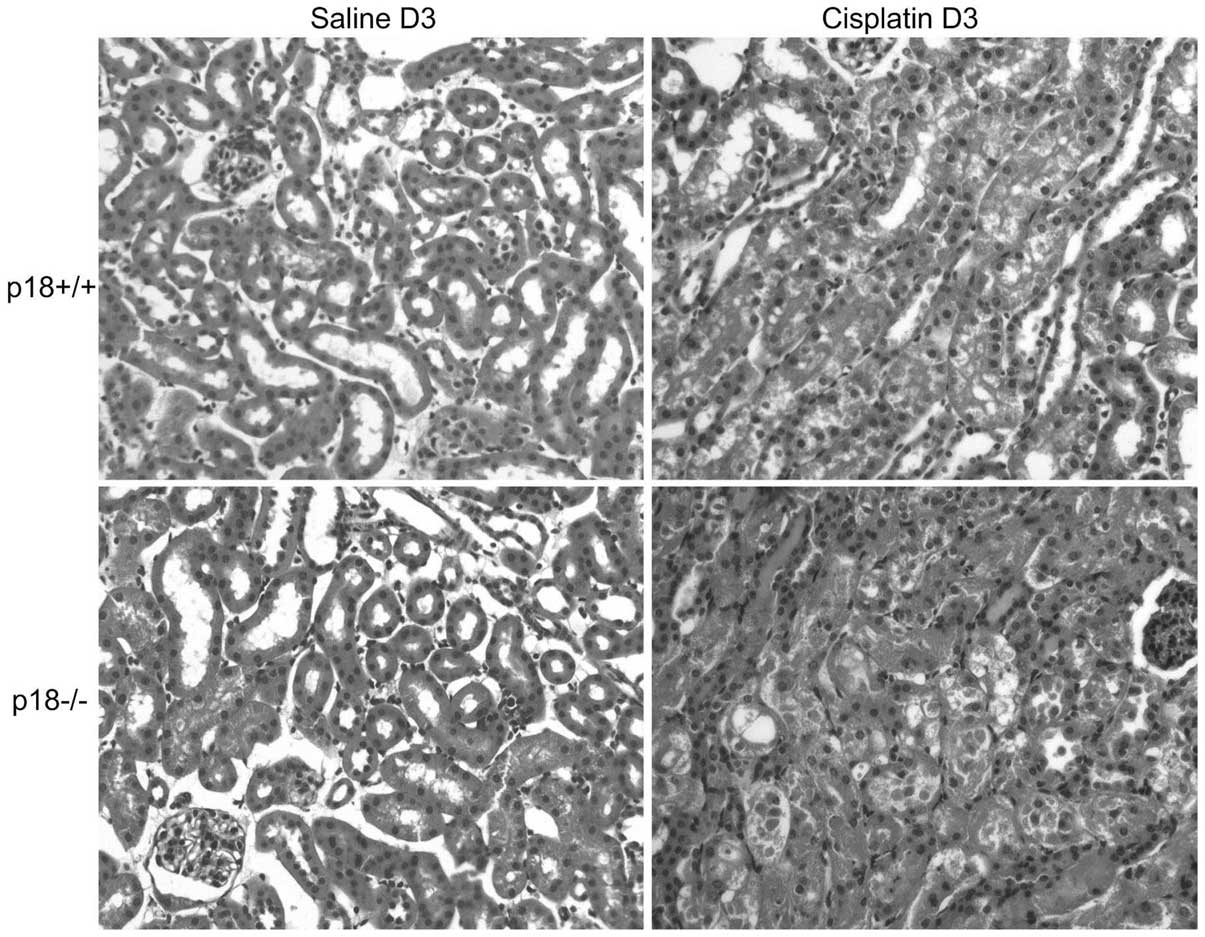
Aggravated morphological changes were present in
p18−/− mice at day 3 after cisplatin injection as demonstrated by
H&E staining (Fig. 3 and
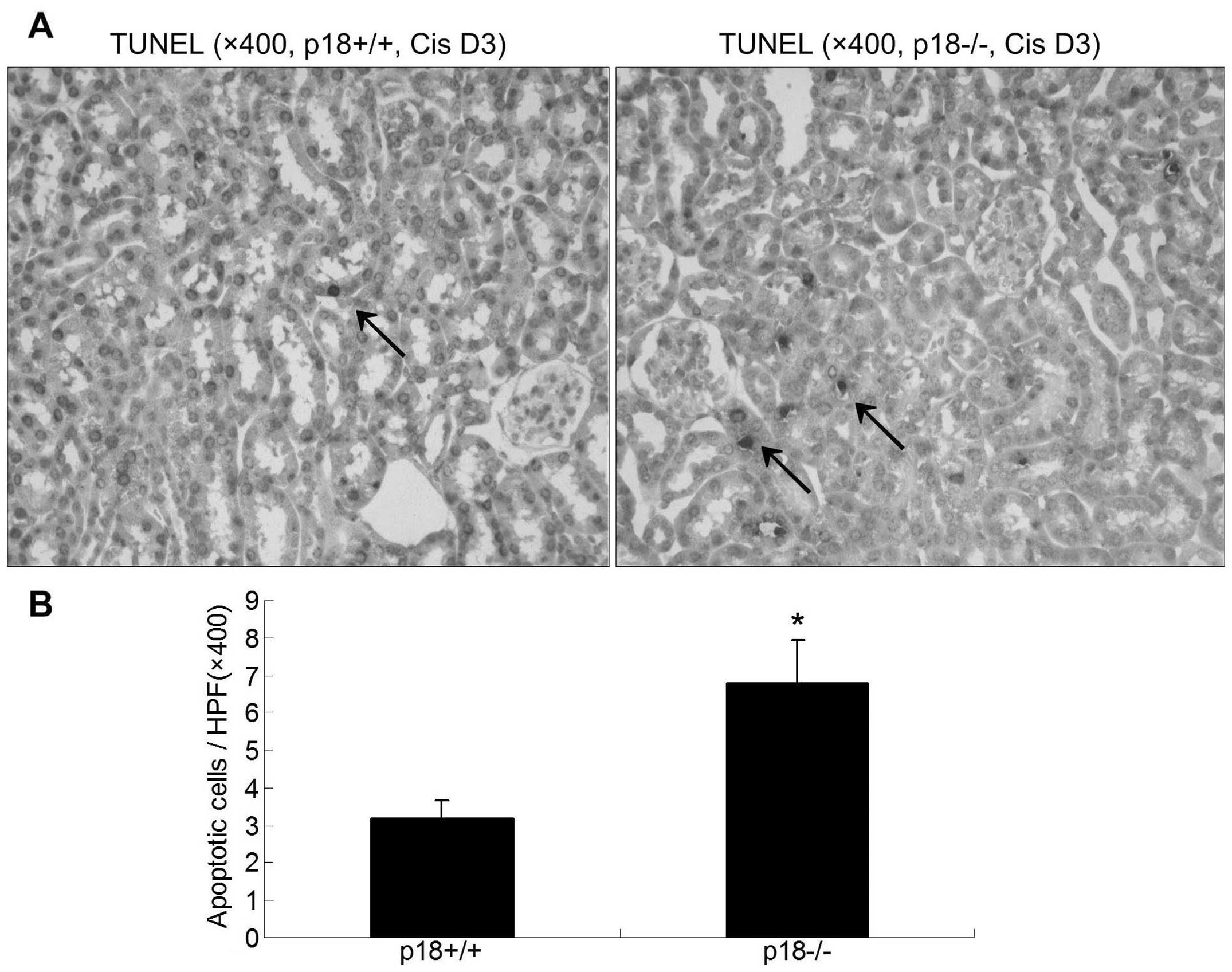
Table I) and TUNEL assessment
(Fig. 4). A higher degree of
kidney damage and a higher percentage of apoptotic cells were
present in p18−/− kidneys as compared to p18+/+ kidneys at day 3
after cisplatin injection.
 | Table IHistological assessment of p18−/− and
p18+/+ mice at day 3 after cisplatin (12.5 mg/kg, i.p)
injection. |
Table I
Histological assessment of p18−/− and
p18+/+ mice at day 3 after cisplatin (12.5 mg/kg, i.p)
injection.
| No. of mice in each
grade |
|---|
|
|
|---|
| Groups | I | II | III | IV |
|---|
| p18−/− (n=15) | 0 | 2 | 6 | 7 |
| p18+/+ (n=15) | 0 | 4 | 8 | 3 |
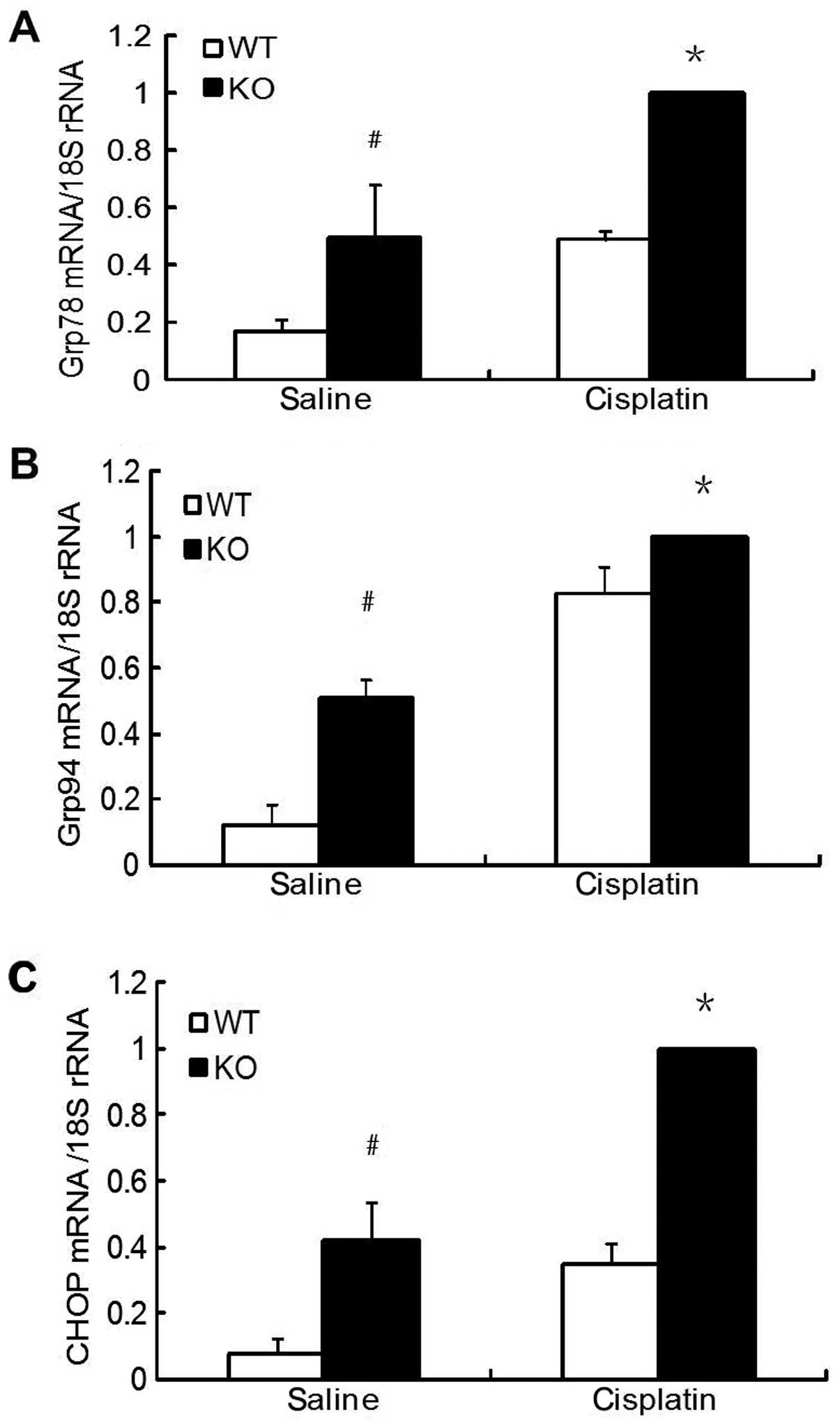
Deletion of p18 aggravated
cisplatin-induced ERS
As shown in Fig.
5, the expression of molecular chaperones grp78 and grp94 mRNAs
was upregulated in kidneys of animals with AKI at day 3 after
cisplatin injection. However, compared to p18+/+ mice, the basal
and inducible expression of grp78 and grp94 mRNAs was significantly
higher in the p18−/− mice. Similar results were observed in the
analysis of CHOP mRNA, a particular transcription factor activated
by ERS.
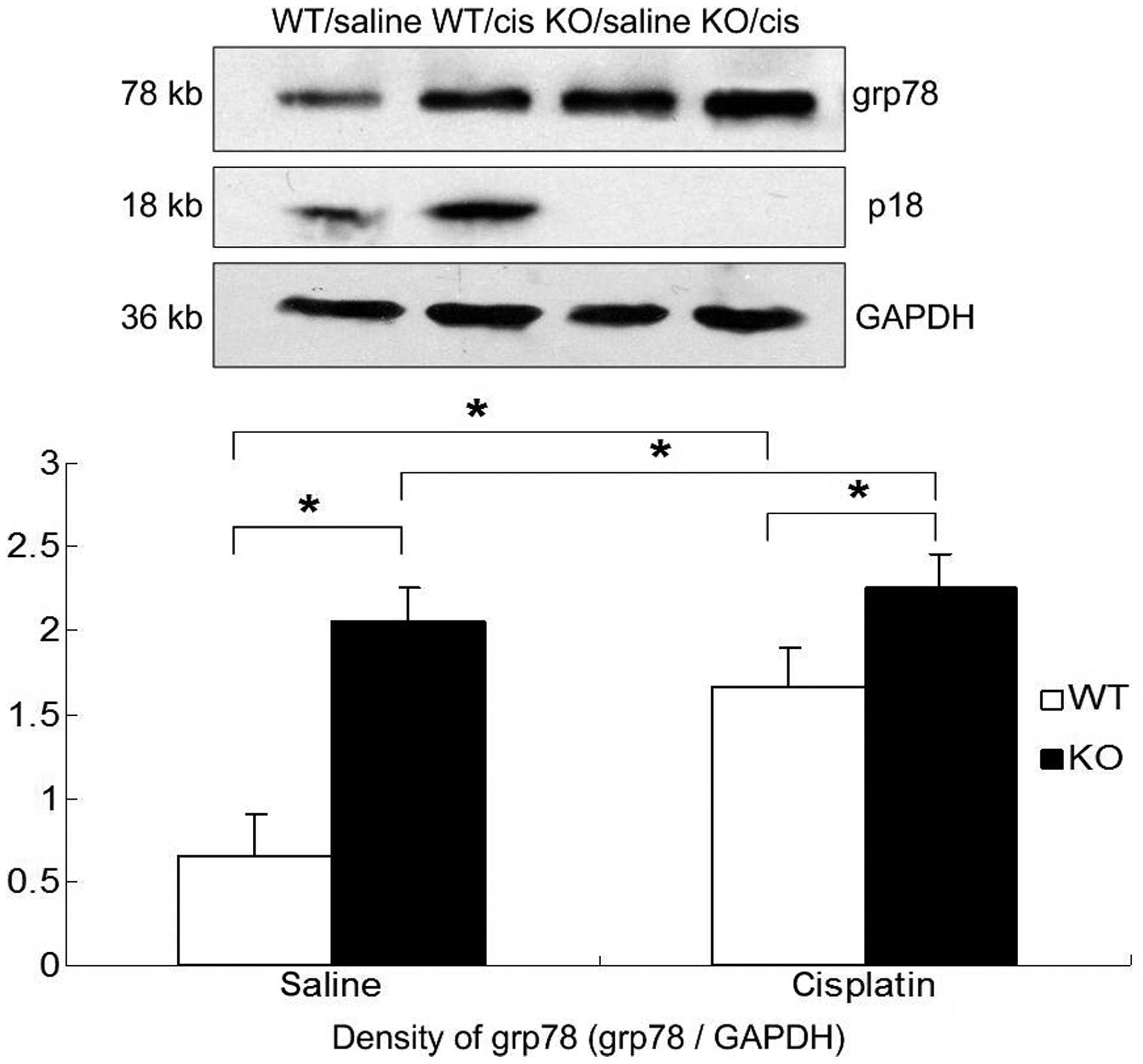
Results were confirmed by western blot analysis
(Fig. 6). The renal expression of
grp78 protein was upregulated after cisplatin injection in p18−/−
and p18+/+ mice. Compared to p18+/+ mice, the basal and inducible
renal expression of grp78 protein was significantly higher in
p18−/− mice.
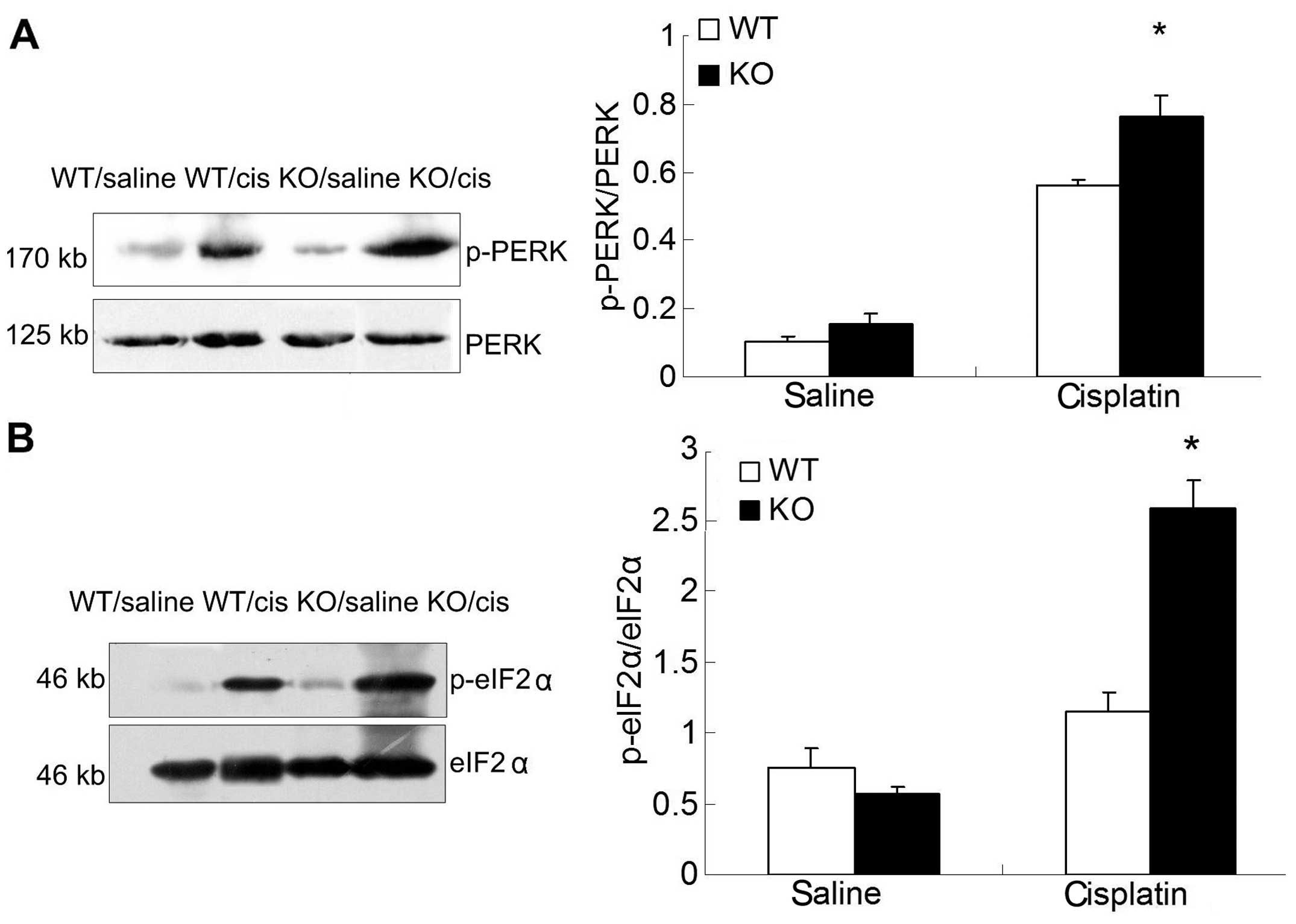
As a rapid response to ERS, PERK was also analyzed
by western blot analysis. The degree of PERK/eIF2α phosphorylation
was higher in p18−/− mice as compared to that of p18+/+ mice after
cisplatin injection (Fig. 7).
Discussion
Clinical use of cisplatin is largely limited due to
drug resistance and nephrotoxicity (19–20). Since the mechanism by which
cisplatin produces its nephrotoxic effect is similar to human AKI
(21–22), AKI animals were administered
cisplatin.
As the manifestation of cisplatin nephrotoxicity is
apoptosis and/or necrosis, cell death pathways were involved in the
mechanism of cisplatin nephrotoxicity. Previous studies have
confirmed that in addition to the classical death-receptor and
mitochondrial pathways (23–26), the ERS pathway is activated in
cisplatin-induced kidney injury in vitro and in vivo
(27–30). Therefore, the effect of p18
deletion on the ERS pathway was investigated to elucidate the
actions of p18 in cisplatin-induced AKI. In ERS, the unfolded
protein response (UPR) was identified and considered to interpret
the mechanism of ERS-induced apoptosis (31–35). Three transmembrane proteins are
activated in UPR: inositol-requiring enzyme-1 (IRE1), PERK and
activating translation factor-6 (ATF6). In this study, upregulation
of the molecular chaperones and CHOP and activation of the
PERK/eIF2α pathway were analyzed to evaluate ERS severity in
cisplatin-induced AKI.
It was found that p18 exerted protective actions in
cisplatin-induced AKI. Compared to p18+/+ mice, p18−/− mice
exhibited a higher degree of kidney damage, accompanied with
aggravated renal function and worse survival after cisplatin
injection. Deletion of p18 also aggravated cisplatin-induced ERS.
Compared to p18+/+ mice, the basal and inducible expression of the
molecular chaperones (grp78 and grp94) and transcription factor
(CHOP) in kidney were significantly higher in the p18−/− mice. The
degree of PERK/eIF2α phosphorylation was also higher in p18−/− mice
kidneys compared to p18+/+ mice after cisplatin injection. These
results indicate that the effect of p18 on cell death pathways,
such as the ERS pathway, may be the facet of its protective
mechanism in cisplatin-induced AKI. However, other classical death
pathways affected by p18 cannot be excluded as they were not the
focus of our investigation.
P18, as a member of the INK4 family, is different
from p21, whose protection in cisplatin-induced AKI has been
demonstrated in previous studies (2–10).
Protection of p21 occurs mainly due to the inhibitory effect on
CDK2 activity, which has been demonstrated as an important factor
in the promotion of apoptosis in cisplatin-induced AKI (36–37). However, the INK4 family members
only interact with CDK4/6 and arrest the cell cycle in the early G1
phase, with no direct interaction with CDK2 (12,38). INK4 family members are also
considered to be involved more in cell differentiation than CIP/KIP
family members as they are often mutant or deleted in a number of
tumors (39). No abnormality or
defect exists in p21 gene knockout mice, whereas p18 gene knockout
mice acquire pituitary tumors with age (18,40). These limitations may be the
possible reasons for INK4 members rarely being investigated in AKI.
However, some studies have reported the involvement of INK4 family
members in the cell response to genotoxic agents, such as p16 and
p19 (13–16), as well as p18. These observations
suggest that INK4 family members also exert protection and are
involved in organ injury, despite the differences between the INK4
and CIP/KIP family members.
In conclusion, protection of p18 was demonstrated in
cisplatin-induced AKI by using p18 gene knockout mice in this
study. The main results of this study are the finding that p18
regulates cell death pathways, such as the ERS pathway, against
cisplatin-induced AKI, although the exact signal transduction
pathways connecting p18 to cell death pathways remain to be
investigated in detail.
Acknowledgements
We would like to thank Professor Tao Cheng for
kindly providing the p18 gene knockout mice and Professor Jun Gu
for his helpful suggestions and careful review of our
manuscript.
References
|
1
|
Weinert T: DNA damage and checkpoint
pathways: molecular anatomy and interactions with repair. Cell.
94:555–558. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Megyesi J, Andrade L, Vieira JM Jr,
Safirstein RL and Price PM: Coordination of the cell cycle is an
important determinant of the syndrome of acute renal failure. Am J
Physiol Renal Physiol. 283:F810–F816. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhou H, Kato A, Yasuda H, et al: The
induction of cell cycle regulatory and DNA repair proteins in
cisplatin-induced acute renal failure. Toxicol Appl Pharmacol.
200:111–120. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Price PM, Safirstein RL and Megyesi J:
Protection of renal cells from cisplatin toxicity by cell cycle
inhibitors. Am J Physiol Renal Physiol. 286:F378–F384. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Megyesi J, Safirstein RL and Price PM:
Induction of p21WAF1/CIP1/SDI1 in kidney tubule cells affects the
course of cisplatin-induced acute renal failure. J Clin Invest.
101:777–782. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhou H, Fujigaki Y, Kato A, et al:
Inhibition of p21 modifies the response of cortical proximal
tubules to cisplatin in rats. Am J Physiol Renal Physiol.
291:F225–F235. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nowak G, Price PM and Schnellmann RG: Lack
of a functional p21WAF1/CIP1 gene accelerates caspase-independent
apoptosis induced by cisplatin in renal cells. Am J Physiol Renal
Physiol. 285:F440–F450. 2003.PubMed/NCBI
|
|
8
|
Yu F, Megyesi J, Safirstein RL and Price
PM: Identification of the functional domain of p21(WAF1/CIP1) that
protects cells from cisplatin cytotoxicity. Am J Physiol Renal
Physiol. 289:F514–F520. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Miyaji T, Kato A, Yasuda H, Fujigaki Y and
Hishida A: Role of the increase in p21 in cisplatin-induced acute
renal failure in rats. J Am Soc Nephrol. 12:900–908.
2001.PubMed/NCBI
|
|
10
|
Nath KA: Provenance of the protective
property of p21. Am J Physiol Renal Physiol. 289:F512–F513. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hengst L and Reed SI: Inhibitors of the
Cip/Kip family. Curr Top Microbiol Immunol. 227:25–41. 1998.
|
|
12
|
Sherr CJ and Roberts JM: Inhibitors of
mammalian G1 cyclin-dependent kinases. Genes Dev. 9:1149–1163.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Al-Mohanna MA, Manogaran PS, Al-Mukhalafi
ZK, Al-Hussein AK and Aboussekhra A: The tumor suppressor
p16(INK4a) gene is a regulator of apoptosis induced by ultraviolet
light and cisplatin. Oncogene. 23:201–212. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Le HV, Minn AJ and Massagué J:
Cyclin-dependent kinase inhibitors uncouple cell cycle progression
from mitochondrial apoptotic functions in DNA-damaged cancer cells.
J Biol Chem. 280:32018–32025. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Scassa ME, Marazita MC, Ceruti JM, et al:
Cell cycle inhibitor, p19INK4d, promotes cell survival and
decreases chromosomal aberrations after genotoxic insult due to
enhanced DNA repair. DNA Repair (Amst). 6:626–638. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tavera-Mendoza LE, Wang TT and White JH:
p19INK4D and cell death. Cell Cycle. 5:596–598. 2006. View Article : Google Scholar
|
|
17
|
Yuan Y, Shen H, Franklin DS, Scadden DT
and Cheng T: In vivo self-renewing divisions of haematopoietic stem
cells are increased in the absence of the early G1-phase inhibitor,
p18INK4C. Nat Cell Biol. 6:436–442. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Franklin DS, Godfrey VL, Lee H, et al: CDK
inhibitors p18(INK4c) and p27(Kip1) mediate two separate pathways
to collaboratively suppress pituitary tumorigenesis. Genes Dev.
12:2899–2911. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang D and Lippard SJ: Cellular processing
of platinum anticancer drugs. Nat Rev Drug Discov. 4:307–320. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Siddik ZH: Cisplatin: mode of cytotoxic
action and molecular basis of resistance. Oncogene. 22:7265–7279.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pabla N and Dong Z: Cisplatin
nephrotoxicity: mechanisms and renoprotective strategies. Kidney
Int. 73:994–1007. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Heyman SN, Lieberthal W, Rogiers R and
Bonventre JV: Animal models of acute tubular necrosis. Curr Opin
Crit Care. 8:526–534. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lieberthal W, Triaca V and Levine J:
Mechanisms of death induced by cisplatin in proximal tubular
epithelial cells: apoptosis vs. necrosis Am J Physiol.
270:F700–F708. 1996.PubMed/NCBI
|
|
24
|
Razzaque MS, Koji T, Kumatori A and
Taguchi T: Cisplatin-induced apoptosis in human proximal tubular
epithelial cells is associated with the activation of the Fas/Fas
ligand system. Histochem Cell Biol. 111:359–365. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Seth R, Yang C, Kaushal V, Shah SV and
Kaushal GP: p53-dependent caspase-2 activiation in mitochondrial
release of apoptosis-inducing factor and its role in renal tubular
epithelial cell injury. J Biol Chem. 280:31230–31239. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Park MS, De Leon M and Devarajan P:
Cisplatin induces apoptosis in LLC-PK1 cells via activation of
mitochondrial pathways. J Am Soc Nephrol. 13:858–865.
2002.PubMed/NCBI
|
|
27
|
Muruganandan S and Cribb AE:
Calpain-induced endoplasmic reticulum stress and cell death
following cytotoxic damage to renal cells. Toxicol Sci. 94:118–128.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cribb AE, Peyrou M, Muruganandan S and
Schneider L: The endoplasmic reticulum in xenobiotic toxicity. Drug
Metab Rev. 37:405–442. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Peyrou M, Hanna PE and Cribb AE:
Cisplatin, gentamicin, and p-aminophenol induce markers of
endoplasmic reticulum stress in the rat kidneys. Toxicol Sci.
99:346–353. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Liu H and Baliga R: Endoplasmic reticulum
stress-associated caspase 12 mediates cisplatin-induced LLC-PK1
cell apoptosis. J Am Soc Nephrol. 16:1985–1992. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bernales S, Papa FR and Walter P:
Intracellular signaling by the unfolded protein response. Annu Rev
Cell Dev Biol. 22:487–508. 2006. View Article : Google Scholar
|
|
32
|
Malhotra JD and Kaufman RJ: The
endoplasmic reticulum and the unfolded protein response. Semin Cell
Dev Biol. 18:716–731. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Mori K: Tripartite management of unfolded
proteins in the endoplasmic reticulum. Cell. 101:451–454. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kleizen B and Braakman L: Protein folding
and quality control in the endoplasmic reticulum. Curr Opin Cell
Biol. 16:343–349. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Szegezdi E, Logue SE, Gorman AM and Samali
A: Mediators of endoplasmic reticulum stress-induced apoptosis.
EMBO Rep. 7:880–885. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Price PM, Yu F, Kaldis P, et al:
Dependence of cisplatin-induced cell death in vitro and in vivo on
cyclin-dependent kinase 2. J Am Soc Nephrol. 17:2434–2442. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yu F, Megyesi J and Price PM: Cytoplasmic
initiation of cisplatin cytotoxicity. Am J Physiol Renal Physiol.
295:F44–F52. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hirai H, Roussel MF, Kato JY, Ashmun RA
and Sherr CJ: Novel INK4 proteins, p19 and p18, are specific
inhibitors of the cyclin D-dependent kinases CDK4 and CDK6. Mol
Cell Biol. 15:2672–2681. 1995.PubMed/NCBI
|
|
39
|
Cordon-Cardo C: Mutation of cell cycle
regulators. Biological and clinical implaications for human
neoplasia. Am J Pathol. 147:545–560. 1995.PubMed/NCBI
|
|
40
|
Shankland SJ and Wolf G: Cell cycle
regulatory proteins in renal disease: role in hypertrophy,
proliferation, and apoptosis. Am J Physiol Renal Physiol.
278:F515–F529. 2000.PubMed/NCBI
|