Soft tissue sarcomas are a complex group of rare
mesenchymal lesions, many of which are distinguishable from the
others only through careful histological and ultramicroscopic
investigations. Their diagnosis is problematic due to their rarity;
15–20% of these sarcomas are poorly differentiated, with a wide
cellular variety that makes their classification difficult. In
addition, histological subtypes which are morphologically similar
present cytogenetic and molecular differences that influence the
prognosis.
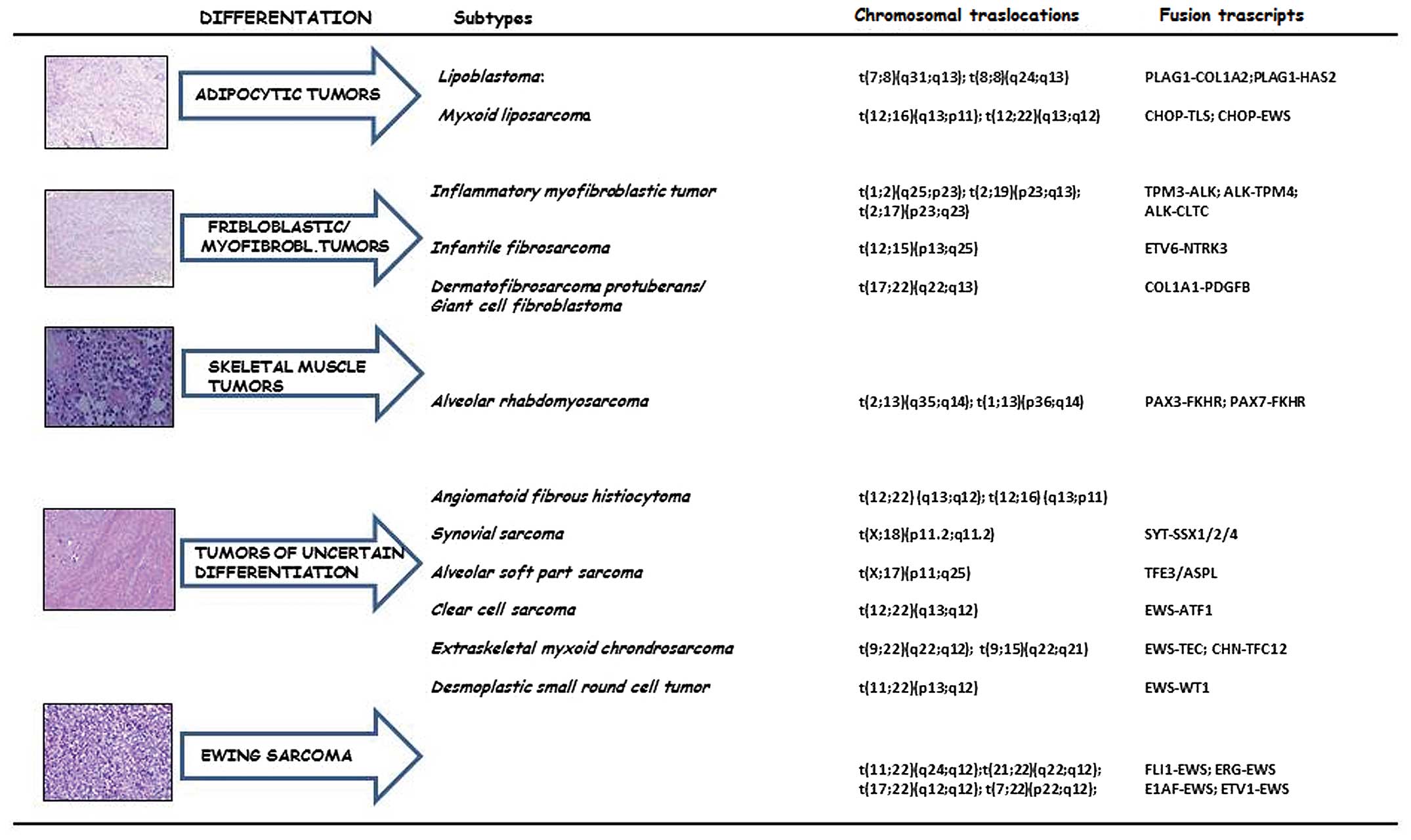
Sarcomas are generally classified on the basis of
tumor cell line differentiation rather than on the type of tissue
from which they arise. A number of histotypes of differentiated
tumor cells have been identified: adipocyte differentiation,
fibroblast/myofibroblast differentiation, fibrohistiocytic
differentiation, smooth/skeletal muscle differentation, tumors of
uncertain differentation and a separate group which includes
Ewing’s sarcoma (ES). However, not all histological types described
present specific chromosomal alterations. For this reason, they are
often grouped into ‘sarcomas with specific genetic alterations’ and
‘sarcomas with no specific genetic alterations’ (Fig. 1). In pleomorphic sarcomas, only
cytogenetic and molecular biology allow a correct diagnosis,
identifying specific chromosomal and molecular rearrangements
(1).
Thus, the combination of morphological and molecular
techniques represents an important progress, not only for a more
adequate diagnostic definition, but also for the prognostic and
therapeutic indications of these malignancies. Comparative
genomics, in situ hybridization and gene array analysis
allowed the rapid acquisition of the fundamentals of the
biology/genetics of sarcomas (2).
However, these techniques are not affordable for all laboratories.
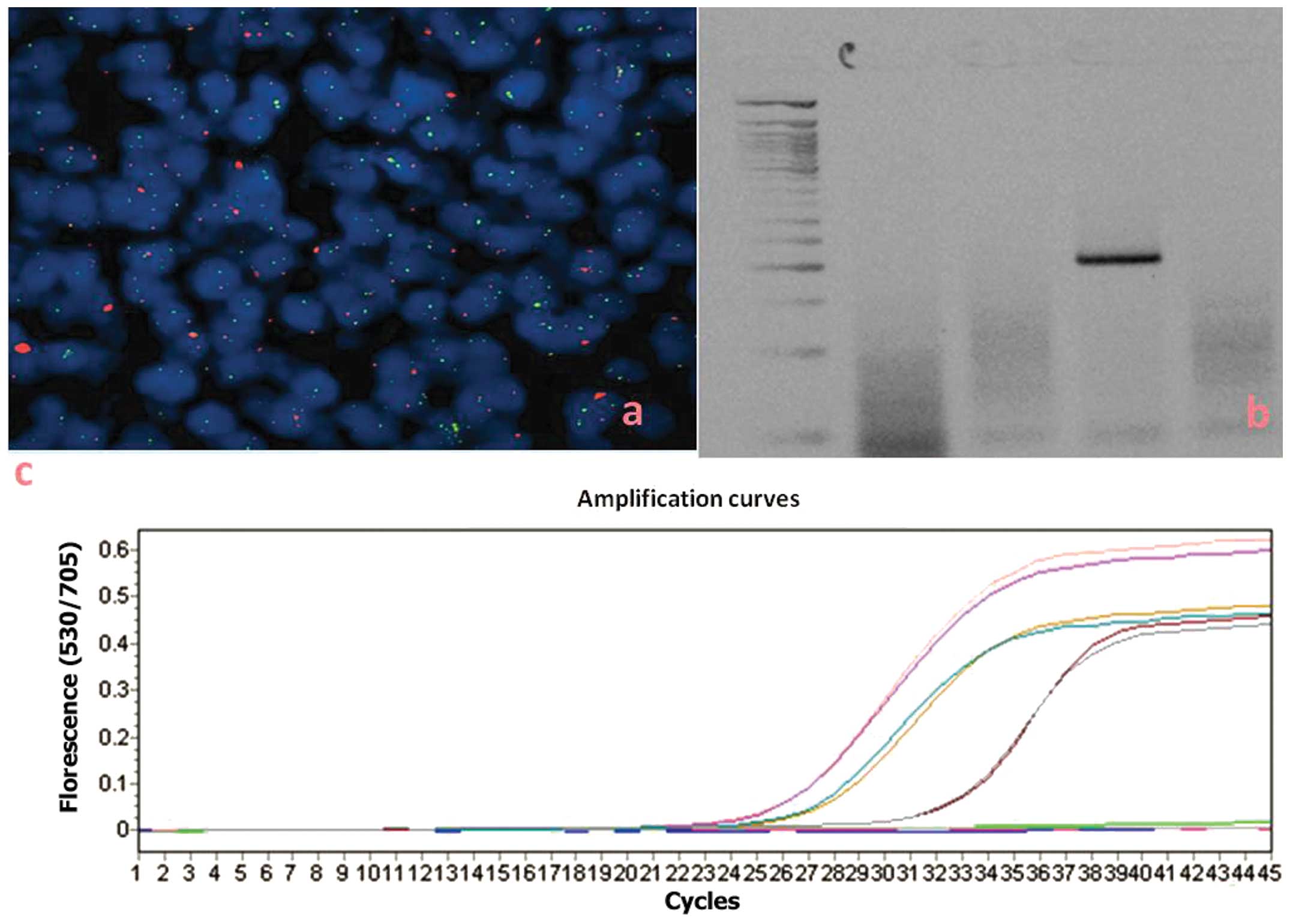
The cytogenetic techniques (Fig.
2a) and those associated with polymerase chain reaction (PCR)
technology (Fig. 2b and c) have
instead allowed us to extend the possibility to identify gene
translocations/amplifications specific for these tumors in all
diagnostic pathology laboratories, making the costs and methods
more affordable for all.
In this review, we summarize not only all known
chromosomal aberrations associated with soft tissue tumors, but
also the different methods that help identify them and characterize
the fusion transcripts produced.
Lipogenic tumors represent a heterogeneous group of
lesions, mainly represented by liposarcomas. Among the principal
histological subtypes of liposarcoma, myxoid liposarcoma (MLS) is
the second most common, followed by well-differentiated liposarcoma
(3), that have not been
implicated in such chromosomal translocations or fusion genes.
Lipoblastomas are pediatric neoplasms, typically
benign lesions, composed of adipose cells in different stages of
maturation within a variably myxoid matrix, and they contain clonal
rearrangements of chromosome band 8q12. In lipoblastomas, several
chromosomal rearrangements have been described, involving the
pleiomorphic adenoma gene 1 (PLAG1) oncogene. In particular, it was
shown that the hyaluronic acid synthase 2 (HAS2) or collagen 1 α2
(COL1A2) gene promoter regions are fused to the entire PLAG1 coding
sequence (4). The PLAG1 status
was investigated through in situ hybridization techniques,
particularly fluorescence detection [fluorescence in situ
hybridization (FISH)] and chromogenic detection [chromogenic in
situ hybridization (CISH)] (5).
The non-random reciprocal translocation
t(12;16)(q13;p11) is a characteristic of MLS (6,7).
FISH is an alternative to ancestral cytogenetic methods, using
painting probes against the centromeres of chromosome 12 metaphases
and interphase nuclei (8–10). Early studies using FISH relied on
the use of cosmid probes derived from YAC clones, which map at the
CHOP locus (11,12). Currently, centralized laboratories
specialized in FISH for the diagnosis of sarcomas, use dual-color,
break-apart FISH probes spanning the genomic regions of FUS (16p11)
(Vysis Inc., Downers Grove, IL, USA) (13,14).
Using Southern blot techniques, in samples with
cytogenetic rearrangements in the region 12q13, it has been shown
that CHOP/DDIT3 t(12;16)(q13;p11) is the gene involved in
translocation (15). A chimeric
transcript between the CHOP gene, which encodes a transcription
factor, and the gene TLS/FUS that localizes in the region 16p11, is
produced (16,17). The protein FUS/TLS interacts with
several nuclear receptors and specific transcription factors.
Through RT-PCR (18), different
variants of the fusion transcript FUS/CHOP can be detected. The
most frequent variants are the I and II variants, which are
generated by alternative splicing between exon 2 of CHOP and exons
5 and 7 of the FUS gene (19–22). The different expression profile of
the transcript FUS/CHOP has also been revealed by nested-PCR
(23,24) or nested-PCR and direct sequencing
(25,26) in frozen or paraffin-embedded
biopsies. Cloning studies have demonstrated that the different
variants have similar activities in transforming mesenchymal cells
using the same molecular pathways (27).
Real-time PCR is a more specific and sensitive
technique for both frozen and formalin-fixed, paraffin-embedded
(FFPE) tissues (28). The
TLS-CHOP chimeric product is also capable of promoting the
development of MLS and tumorigenesis through the repression of the
expression of a microRNA, miR-486, as demonstrated in studies on
cloned cell lines from NIH3T3 fibroblasts and MLS tissues (29).
MLS can also present the t(12; 22)(q13,q12)
aberration with its chimeric transcript EWS/CHOP fusion between
exon 7 of the EWS gene and exon 2 of CHOP. FISH is an excellent
method for detecting the presence of gene rearrangements in CHOP,
but RT-PCR is the only method able to detect fusion partners, FUS
or EWS (30,31). Patients who exhibit these fusion
products show a more favorable clinical history compared to other
patients positive for transcripts and different variants.
Therefore, a correct diagnosis also related to biomolecular data is
important, particularly in cases where the myxomatous change is
minimal compared to their dominant counterparts which resemble
pleomorphic malignant fibrous histiocytoma (32).
Fibrosarcoma is a primary malignant tumor composed
of immature fibroblasts. Several forms are recognized: infantile
fibrosarcoma, which presents at birth or in early childhood,
identical to the adult form apart from the clinical course and is
much more favorable, as well as dermatofibrosarcoma protuberans
(DFSP).
Infantile fibrosarcoma has the same characteristics
of benign lesions of childhood, such as infantile fibromatosis and
myofibromatosis (33). The
specific translocation t(12;15)(p13;q25) (34), which makes the differential
diagnosis possible, was identified by cytogenetic techniques and
cloning of chromosome break sequences. FISH is a specific method,
although more costly, and can be used to determine the status of
chromosomal rearrangements of the regions 12p13 and 15q25 (35). Early studies were carried out
using FISH probes or chromosome-specific bacterial artificial
chromosome (BAC) clone probes (36). These were then replaced by
commercial probes for dual-color fusion, the Ets variant 6 (ETV6)
gene (Abbott Molecular, Inc., Des Plaines, IL, USA) (37).
The translocation generates a fusion transcript
ETV6- neurotrophic tyrosine kinase, receptor, type 3 (NTRK3). The
ETV6 gene is localized on chromosome arm 12p13, while the NTRK3
gene is in the chromosomal region 15q25 (38). Techniques of cloning and
sequencing have shown that the fusion occurs between exon 5 of the
ETV6 gene and exon 13 of the NTRK3 gene (38,39). Reverse transcriptase PCR (RT-PCR)
is a specific method, fast and economical, for determining the
presence of the fusion gene, ETV6-NTRK3, in fresh tissue samples or
archived material (40–42).
DFSP is a rare variant that is derived from the
fibrous component of the dermis and grows slowly, forming ulcers on
the skin and subcutaneous tissues. The translocation
t(17;22)(q22;q13) is highly specific for DFSP and can generate a
chimeric transcript COL1A1/platelet-derived growth factor subunit B
(PDGFB) and a supernumerary ring chromosome, r(17,22) (43,44).
Early studies on the detection of chromosomal
translocations by FISH, were based on chromosome painting and
α-satellite probes. Later studies reported the use of a dual-color
dual-fusion BAC probe for COL1A1/PDGFB translocation, obtained by
cloning vectors, BACs, covering the PDGFB gene entirely. In this
manner, the different variants of the chimeric transcript were
identified (45,46). Currently, commercial FISH probes
are available, e.g., ZytoLight-SPEC COL1A1-PDGFB Dual Color Dual
Fusion Probe (ZytoVision GmbH, Bremerhaven, Germany) (47). Different methods, such as Southern
blot analysis, RT-PCR and FISH, based on frozen tissue specimens or
using archival FFPE tumor samples have shown that COL1A1/PDGFB
chimeric genes are present in all cases of DFSP (43,48,49).
Finally, to detect the copy number changes on the
chromosomal regions, 17q and 22q, comparative genomic hybridization
(CGH) studies have been performed, and have confirmed the
amplification of 17q, but not always that of 22q (50,51)1. The breakpoint is at the level of
exon 2 of PDGFB in the region 22q, but may involve several exons of
the COL1A1 gene in the region 17q, such as exons 8, 10, 22, 24, 27,
32, 34, 38, 40, 45, 46 and 47, as shown by RT-PCR followed by
sequencing analysis (52,53). The same methodology has allowed to
identify additional variants of chimeric transcript, such as the
one due to fusion at the level of exon 2 of PDGFB with COL1A1 gene
but at the level of exon 41 (54). All possible variants of the fusion
transcript can be detected using either multiplex RT-PCR (55) or FISH on paraffin-embedded tissues
(46).
Real-time PCR provides a more sensitive alternative
for analyzing the presence of the fusion transcript or the
amplification of the regions affected by the rearrangement in fresh
tissue or archived samples (56).
Real-time PCR also allows the quantification of mRNA transcripts
and the PDGFB chimeric gene, which is overexpressed compared to
benign counterparts (normal tissue or dermatofibroma) (57,58).
Rhabdomyosarcomas originate most often in striated
muscles at the level of the arms and legs and are more common in
children than in adults. Three main forms are known: polymorphous
rhabdomyosarcoma and alveolar rhabdomyosarcoma in adults, and
embryonal rhabdomyosarcoma, which is more common in children. The
tumor is ubiquitous and the most common sites are the arms, but
also the head and neck, the urogenital tract and retroperitoneum.
The progression is extremely aggressive, with a great tendency to
recurrence and metastasis. The alveolar variant tends to have a
worse prognosis than the embryonal variant.
Two chromosomal translocations, t(2;13)(q35;q14) and
t(1;13)(p36;q14), are present in approximately 80% of all alveolar
rhabdomyosarcoma cases (59–61).
The presence of t(2;13) in rhabdomyosarcoma cell
lines was demonstrated by ancillary approaches, such as classical
cytogenetics and FISH, using painting cosmid probes labeled with
digoxigenin and biotin on both metaphases and interphase nuclei
(62). t(2;13)(q35;q14) was
firstly detected by FISH on interphase nuclei in minimally invasive
biopsies of patients treated with neoadjuvant chemotherapy and then
on non-suitable tumor material. In early hybridization studies, two
cosmid clones were used in interphase cells with inserts of regions
proximal or distal to the 13q14 breakpoint and a yeast clone with
the inserted region distal to the 2q35 point (63). Present commercial
dual-split-signal color FISH probes (Abbott Molecular) are more
sensitive and specific to chimeric products, due to the two
translocations involving the 13q14 region (64,65).
Studies on cDNA cloning and sequencing have shown
that t(2;13)(q35;q14) produces a fusion transcript between the
paired box 3 (PAX3) gene and FKHR gene, respectively (66,67); the PAX7-FKHR fusion transcript
results from the t(1;13) translocation (68).
However, the survival and mortality rate in
metastatic patients depend on the rearrangement type: the 4-year
survival rate is 75% for patients with PAX7-FKHR vs. 8% for those
with PAX3-FKHR. If PAX3-FKHR is expressed, there is a significant
risk of death (P=0.019); besides, these patients may present bone
marrow involvement (69).
The technical related issues associated with FISH in
the diagnosis of alveolar rhabdomyosarcoma in children have been
avoided by using RT-PCR, although this method is less sensitive
than FISH (70,71); however, the chimeric transcript
PAX3/FKHR has been detected in cell lines and in fresh tissue
samples and FPPE samples using only very small amounts of tumor
tissue (72,73). With multiplex RT-PCR it is also
possible to calculate the residual disease (74) and allows the differential
diagnosis of alveolar rhabdomyosarcoma and ES (72).
The presence of both translocation t(2;13)(q35;q14)
and t(1;13)(p36;q14) products is detected with specific primers for
the regions flanking the breakpoints in 13q and 1p, as well as for
2q (75), with RNA extracted from
fresh or frozen tissue and formalin-fixed, paraffin-embedded tissue
samples (76). The quality of the
extracted RNA and the absence of specific primers for unusual
variants are the technique limitations, but certainly the problems
related to RNA quality will be reduced with fresh or frozen
meterial or with the increase in the number of neoplastic cells
after laser capture microdissection (77). The specificity of the test is very
high (94–100%), compared to electrophoresis on an agarose gel.
Discordant data have been obtained by analyzing the same samples by
Southern blot analysis (78). A
correct diagnosis also requires ancillary data, such as clinical
history, immunohistochemistry and histology, while the uncertain
cases may require further FISH investigation.
Multiplex fluorescent analysis of chromosomal
translocations (MFACT) is another alternative method which can be
used in place of conventional RT-PCR. This method has the advantage
of completely eliminating the manipulation of the PCR products and
thus it greatly reduces the risk of cross-contamination (79).
Real-time PCR using a hydrolysis probe, is a highly
sensitive and specific method that reveals and further quantifies
the chimeric transcripts PAX3-FKHR and PAX7-FKHR in the peripheral
blood of patients, yielding similar results to nested-PCR. Patients
positive for the fusion transcripts are at high risk of tumor
progression (80). An initial
screening for immunohistochemistry helps to select patients for
molecular investigations, since it has been shown that only
patients with alveolar rhabdomyosarcoma and not those with the
embryonic phenotype that present with >50% of cells
immunoreactive for myogenin, show the rearrangement of PAX
(81).
Tumors that escape histological classification, as
they lack a definite differentiation from a certain type of
mesenchymal tissue, are encompassed in this category. They mostly
occur between the ages of 15 and 40 years and are more common in
males.
Synovial sarcoma is the most common lesion in this
group. The peak of incidence is the third decade of life, and the
male/female incidence ratio is approximately 1.2:1. Synovial
sarcoma can be mono- or biphasic and the difference is at the
histopathological level: biphasic synovial sarcoma presents with
epithelial and spindle cells, while monophasic synovial sarcoma
almost always presents with spindle cells.
The t(x;18)(p11.2;q11.2) translocation has been
observed in patients with this neoplasm, often as a unique
cytogenetic abnormality (82),
for which the SYT gene on chromosome 18 is juxtaposed to one of the
two genes related, SSX1 or SSX2, but distinct on chromosome X
(83,84).
The first cytogenetics and FISH studies on
paraffin-embedded samples were performed using centromeric probes
together with whole chromosome painting probes for chromosomes X
and 18 (85). The translocation
(X;18)(p11;q11) breakpoint has been demonstrated by both Southern
blot analysis and FISH analysis using specific yeast artificial
chromosome (YAC) probes (86,87). Dual-color break-apart probes,
synthesized by cloning the breakpoint regions of interest in BAC
clones and labeled with fluorescein-12-dUTP and TexRed-5-dUTP, were
very useful in CISH and FISH (88). Currently, interphase FISH is
performed on fixed, paraffin-embedded tissues using a commercially
available LSI SS18 dual-color break-apart probe (Abbott
Molecular/Vysis Inc.), a more specific and sensitive probe than
non-commercial CISH probes (89,90).
The SS18-SSX chimeric product was detected even in
FFPE samples by ISH using biotinylated tyramide and probes labeled
with digoxigenin specific for the cDNA. The method produces signals
in epithelial cells of biphasic sarcomas, with mild or focal
positivity in monophasic tumors (91).
RT-PCR has a specificity of 100% with a sensitivity
of 96% for the detection of the fusion transcript in
paraffin-embedded lesions with the t(X;18) (SYT-SSX) translocation,
limited only by the use of particular fixatives that produce a poor
quality of extracted RNA (92,93). The molecular analysis evaluates
the incidence of molecular variants, using sets of specific primers
for SS18-SSX1 and SS18-SSX2; however, a statistically significant
association between histological subtype (monophasic vs. biphasic)
and SSX1 or SSX2 (94) has not
been found. New variants for both SS18-SSX1 and SS18-SSX2 have been
shown by sequencing the products of RT-PCR (95–97).
FISH and RT-PCR investigations are useful and
necessary in those cases where the differential diagnosis between
synovial and other spindle cell sarcomas (98) is difficult.
The fusion transcripts are detected and quantified
by real-time RT-PCR, more sensitive and rapid than RT-PCR, using
specific primers and TaqMan fluorescent probes complementary to the
breakpoints in the genes involved in the translocation (99–101). Multiplex real-time PCR analyzes
all variants of chimeric transcripts together, using appropriate
sets of primers and probes: SS18-SSX1 has been shown to be present
in both monophasic and biphasic synovial sarcomas more frequently
than the SS18-SSX2 variant. They are mutually exclusive (102).
CCS is a malignancy that can be morphologically
confused with non-cutaneous melanoma as it presents the same
immunophenotype. The differential diagnosis is possible thanks to
the translocation t(12;22)(q13;q12), for which the chimeric gene
EWSR1/ATF1 is formed in melanocytic tumors of soft tissues. Less
commonly, CCS can be marked by t(2;22)(q34;q12), which produces the
fusion transcript, EWSR1/CREB1, typical of gastrointestinal CCS,
but that can also characterize CCS of soft tissue (103).
In order to characterize the translocation and its
chimeric product, the first studies were conducted on cell lines,
such as KAO, obtained from a girl of 9 years, or the HS-MM.
melanoma cells, which were used as negative controls for t(12;22);
CCS cells alone have been shown to be positive for the
translocation and the EWS/ATF1 fusion gene, analyzed by FISH and
RT-PCR, respectively (104).
Nowdays, slides of FPPE samples and tumor microarray
(TMA) are routinely analyzed by interphase FISH with commercial
probes LSY EWS dual-color break-apart (Vysis Inc.) and the positive
cases can be analyzed by RT-PCR to determine the type of chimeric
transcript EWS ATF1 (105,106). All cases of melanoma are
negative to FISH for the same region, the 22q12 (107).
Four variants of the fusion transcript are detected
by RT-PCR and sequencing, due to different breakpoints in the
relevant gene regions: three subtypes are due to in-frame fusion
and are type 1, 2 and 3, due to the fusion-EWS exon 8 and ATF1-exon
4, EWS-ATF1-exon 7 and 5, and EWS-ATF1-exon 10 and 5, respectively;
the subtype 4 is due to the out-of-frame fusion of the region with
exon 7 of EWS and exon 7 of ATF1. In addition to these four main
transcripts, which may also occur together, there can be
out-of-frame fusion between exon 10 of EWS and 3 of ATF1, or
between exon 8 of EWS and 4 of ATF1, with insertion of nucleotides
at the junction point (108,109). RT-PCR is performed using
extracted RNA from either frozen or FFPE tissue (110).
Real-time PCR is much more sensitive than classical
RT-PCR, and is highly specific and very helpful in the differential
diagnosis of melanoma, on fresh or frozen or FPPE samples (111).
DSRCT is a rare and aggressive malignancy, with
typical localization to serosa of the abdominal-pelvic peritoneum,
with a male/female incidence ratio of 4:1 and occurs during
adolescence or early adulthood. In 40% of patients, it metastasizes
to the liver, lungs and lymph nodes.
The tumor shows epithelial and mesenchymal
properties and neural differentiation, and the cells present the
translocation t(11;22)(p13;q12), which juxtaposes the gene, EWSR1,
to the tumor suppressor gene, WT1 (112,113), whose identification in
specialist laboratories is very helpful for a correct differential
diagnosis, complicated by similarities with other small round cell
tumors. The fusion protein EWSR1/WT1 acts as a potent
transcriptional activator (114).
In clinical diagnostics of FFPE tissue sections,
interphase FISH is routinely performed with a commercially
available EWSR1 (22q12) dual color, break-apart rearrangement
probe, but the t(11;22) is also found in 90% of EWS/primitive
neuroectodermal tumor (PNET) and CCS cases (105).
The biological differences between DSRCT, ES and CCS
can be explained by the presence of the different partners in the
EWS gene translocation. Studies using Southern blot analysis,
multi-enzymatic digestion and northern blot analysis have
demonstrated that gene rearrangement in the region 22q12 produces
the fusion of the EWS gene on 11p13 with WT1, the gene involved in
Wilms tumor. RT-PCR, with the use of a primer for exon 7 of EWS and
primers for exons 8 or 9 of WT1, confirmed the data (115). The chimeric mRNA is due to an
in-frame fusion of the amino-terminal domain of EWS with the
zinc-finger DNA-binding domain of WT1, which can undergo
alternative splicing (116).
Chromosomal translocation and fusion with EWS affect two
independent biochemical functions of WT1, binding activity to DNA
and transcriptional regulation, a deregulation that influences
tumorigenesis in intra-abdominal DSRCT (114,117).
The variability in the breakpoint EWS produces
molecular variants of the fusion gene EWS-WT1, as happens for the
chimeric gene EWS-FLI1 in ES, such as an in-frame splicing of exon
9 of EWS to exon 8 of WT1, a variant found in a DSRCT unusually
arising on hand, or an in-frame junction of EWS to exons 8–10 of
WT1 (118).
To differentiate DSRCT from EWS/PNET, when the
genetic information is not available, immunohistochemistry is
recommended with an anti-WT1 antibody, highly specific and
sensitive, that is a reliable index for the presence of the EWS-WT1
chimeric product (119).
RT-PCR can detect all chimeric messages that are
formed by fusion between exons 1–7 of EWS and exons 8–10 of WT1
(120). Multiple in-frame cDNA
can be also detected. It is produced by large internal deletions,
insertions of small parts of heterologous DNA at the site of the
junction between the two exons EWS and WT1, or the loss of exon 6
of EWS or exon 9 of WT1. The molecular diversity and functionality
of these fusion transcripts may have significant biological
implications for their tumorigenic potential (121).
IMT is a mesenchymal tumor that presents with
fibroblastic and myofibroblastic spindle cell proliferation mixed
with lymphocytes, plasma cells and histiocytes (122). It can commonly occur in
children, teenagers and adults under the age of 40. It was
described for the first time in lungs and remains the most frequent
mesenchymal endobronchial tumor in childhood (123). IMT can localize in any
anatomical site, but it rarerly occurs in the liver. The lesion
often presents with ambiguous morphological, structural and
vascular properties, since it has both an inflammatory and
neoplastic nature and therefore, diagnosis can be difficult.
Cytogenetic analysis has revealed clonal chromosomal
abnormalities exhibiting the neoplastic nature of the disease.
Approximately half of inflammatory myofibroblastic tumors present
rearrangements of the locus of the anaplastic lymphoma kinase (ALK)
gene on chromosome 2p23, resulting in the aberrant expression of
ALK. FISH of interphase nuclei of FPPE samples were carried out
using a commercial dual-color (red and green) ALK probe (Vysis
Inc.), that labeled at telomeric region in SpectrumOrange and at
centromeric region in SpectrumGreen of chromosome 2 (124,125). FISH leads to a correct diagnosis
of IMT, even in cases in which the inflammatory component is
minimal and during prenatal life as well (126). It is also possible to perform
FISH on samples of fine needle aspiration (FNA) or endoscopic
ultrasound-guided FNA (EUS-FNA) (127,128).
The disease can also present morphological
characteristics of other similar lesions, such as child congenital
fibrosarcoma (CIFS) and hemangiopericytoma, but the identification
of the present transcript fusion by RT-PCR facilitates the correct
diagnosis (129,130).
ALK, which is normally downregulated in neural
tissues, is overexpressed in IMT cells with the 2p23 rearrangement,
in which the N-terminal domain of tropomyosin (TPM) is fused to the
C-terminus of ALK. Cloning studies have shown two fusion products,
TPM4-ALK and TPM3-ALK, which encode for oncoproteins with
constitutive kinase activity (131,132). RT-PCR seems the best method to
identify the ALK fusion transcripts (133,134).
ALK can be also fused with clathrin heavy chain
(CTLC), a gene localized to 17q23; however, patients that are
t(2;17) positive show other abnormal karyotypes as well (135).
RT-PCR with specific pairs of primers and direct
sequencing of the amplification products have also enabled the
identification of new partners of ALK, such as dynactin-1, when the
alteration der(2)
t(2;12)(p23;q11) is present (136), or when the SEC31L1/ALK fusion
gene, due to translocation t(2;4)(p23;q21), is present in two
variants of different lengths (137).
A partial response by the inhibitor of ALK,
crizotinib, has been reported in a patient with inflammatory
myofibroblastic tumor with ALK translocation (138,139).
ASPS represents approximately 0.5–1% of soft tissue
sarcomas and typically develops in adolescents and young adults; in
children the localization is typical on the head and neck (140,141). Although growth is indolent, up
to 79% of patients develop metastatic disease, since a significant
percentage of them is resistant to conventional chemotherapeutic
drugs. The development of chemoresistant metastases contributes to
the increase in the mortality rate.
The translocation is confirmed by dual-and
triple-color FISH on metaphases and interphase nuclei (145). Early studies of fluorescence
in situ were performed using YAC and cosmid probes from the
genomic regions of interest (146).
RT-PCR can be performed on frozen and FPPE tumor
tissue to detect the presence of the resulting ASPSCR1-TFE3 fusion
transcripts and its variants. The location of the fusion
transcript, if present, leads to the proper diagnosis of ASPS,
previously considered as a subgroup of RCC in children (147,148). The chimeric products of
t(X;17)(p11;25) are detected by nested RT-PCR, which is more
sensitive, also in circulating tumor cells in peripheral blood of
ASPS patients with distant metastases (149).
Preliminary clinical studies have shown that
patients with ASPS positive for der(17)t(X;17)(p11;25) respond to treatment
with trabectedin, the only currently available clinical drug which
has shown to be effective in the treatment of this disease
(150).
EMC presents strings of small acidophilic cells
similar to chondroblasts in a myxoid stroma, and occurs
particularly in the lower extremities, particularly in the fifth
decade of life with a male/female incidence ratio of 2:1 (151). Patients may have long-term
survival; however, local recurrences and metastases occur in
approximately half of the cases, commonly in the lungs (152–154). Unlike bone chondrosarcoma, EMC
behaves in a less aggressive manner. Therefore, it is deemed as two
prognostically distinct entities (152).
Previous cytogenetic data have included a
translocation t(9;22)(q22–31;q12), that produced the EWS/CHN
chimeric gene and showed complex karyotypes (155,156). The translocation
t(9;17)(q22;q11.2) is less frequent and combines with the CHN RBP56
gene, also known as TAF15, TAF2N or TAFII68 (157). A third translocation was also
identified, typical of EMC, but less common, affecting chromosomes
9 and 15 and forming the chimeric gene, CHN/TCF12. Gene TCF12, also
known as HTF4, presents different isoforms by alternative splicing
and the breakpoint affects the region of intron 5 (158,159). Further molecular analyses have
revealed additional chromosomal aberrations that can aid in the
diagnosis of EMC, identifying other chimeric variants, such as the
fused trascript TFG (TRK-fused gene)/CHN associated with
t(3;9)(q11-q12;q22) (156,160,161).
The translocation occurs due to different
breakpoints in various introns of the gene, EWS and CHN (termed
NR4A3, NOR1 or TEC) (162),
resulting in different variants of the chimeric products, EWS/CHN.
The most frequent are: type 1, for the fusion between exons 12 of
EWS and 3 of CHN, and type 5, between exons 13 of EWS and 3 of CHN.
The chimeric gene RBP56/CHN is always formed by fusion between
exons 6 of RBP56 and 3 of CHN. The mapping of the different regions
of breakpoints in the EWS and CHN genes has shown that there are no
sequence-specific recombinases or homology to explain the various
breakpoints, due to other associated events such as deletions,
duplications and inversions (163).
In FISH on formalin-fixed, FNA biopsy and
paraffin-embedded tissues, using commercial LSY EWSR1 (22q12)
dual-color, break-apart probe (Vysis Inc.), it is possible to
demonstrate the presence of the EWSR1 gene rearrangement (13,164,165). The translocation at gene NR4A3
has been shown by dual-color FISH using a custom probe, synthesized
by means of BAC and telomeric chromosome clones, labeled with
SpectrumGreen and SpectrumOrange (166). Variant translocations were also
detected by interphase FISH, such as t(9;15)(q22;q21) and
t(7;9;17)(q32;q22;q11), with satellite probes for chromosomes 7, 8
and 12, and telomeric probes for 1q and 19q (Vysis Inc.) (167).
RT-PCR on archival FPPE samples, using specific
pairs of primers, is a useful technique for detecting both the
chimeric products due to the main translocations, such as EWS-CHN
or RBP56-CHN (168), and
different transcripts from EWSR1/TAF15/TFG-NR4A3 fusion, such as
EWSR1-CREB1 fusion transcript which is present in cases of primary
pulmonary myxoid sarcoma (169).
ES is a bone cancer, the most frequent after
osteosarcoma, histologically characterized by sheets of small round
cells, blue staining with H&E, which can be confused with
lymphoma or embryonic rhabdomyosarcoma. It is rare in newborns and
after the age of 30, with a higher incidence at the age of 16. ES
and PNET not only have similar microscopic characteristics, but
also show the same genetic alteration, a translocation (170–172); thus, they are subsequently
grouped in a class of tumors defined as ‘Ewing’s sarcoma family
tumors’ (ESFT). The typical translocation affects the region of
chromosome 22 in which the family of ETS transcription factors are
mapped. In 90% of cases the chimeric gene has the region of
chromosome 11 as a partner of the gene EWS, producing the fusion
transcript, EWSR1-FLI1; chromosome 21 is the less frequent partner
and in particular the translocation forms the product gene,
EWSR1-ERG (173). The region of
chromosome 22 with EWSR1 can translocate into other chromosomal
regions, t(21;22), t(7;22), t(17;22) and t(2;22), producing
different chimeric transcripts according to the fusion partner
(ERG, ETV1, E1AF and FEV) (173–175).
Colorimetric or fluorescence studies have used YAC
probes, tested on paraffin-embedded tissue sections (176), while others have used and
validated constructed probes on cell lines (177). Using a triple-target FISH on
interphase nuclei, metaphase chromosomes and DNA fibers, it has
been proven how the transcript EWS/ERG, in particular, may occur.
The technique has been performed with co-hybridization of probes
cloned in cosmids, complementary to the telomeric and centromeric
regions of the region with the EWS breakpoint. FISH, in particular,
has shown that an inversion of the ERG gene or part of it may be
followed by an insertion in the EWS gene on der(22) (178). It is now of routine use to
investigate translocations involving the EWSR1 gene using a dual
interphase LSY color break-apart EWS-FISH (179), while the different variations of
the formed fusion transcripts have been investigated by RT-PCR with
specific pairs of primers (180).
RT-PCR amplifies RNAs extracted from fresh or
cryopreserved tissue samples; however, the results are specific and
confirmed on archivial FPPE material (181). The method is also sensitive to
detect minimal residual disease (182) and a simultaneous detection of
all chimeric products can be done using a mixture of primers. This
method can be very useful in clinical practice, to guarantee
diagnosis, to perform investigations of minimal metastatic and
residual disease and to evaluate the prognostic significance of the
subtypes of chimeric transcripts even when fresh tumor tissue is
not available (182).
The specificity of EWS transcripts with their
respective partners for ES was tested using nested RT-PCR on
different samples of FPPE tissue (183,184).
Combining the biomolecular investigations by nested
RT-PCR, more sensitive than conventional RT-PCR, with cytogenetic
analysis by FISH in FPPE samples, a clear diagnosis of ES/PNET is
possible. In particular, in situ hybridization of nuclear
extraction (NE-FISH) is more reliable than that of thin-section
(TS-FISH) in detecting the translocation of EWSR1 (185). The specificity of the
amplification product for nested RT-PCR is confirmed by the
subsequent digestion of the PCR fragments with different
restriction endonucleases, a rapid method to determine the
combination of exons present in a chimeric mRNA (186).
The use of western blotting to detect the fusion
protein of 68-kDa EWS/FLI1 in samples of surgical biopsies and in
fine needle aspirates of ES, also detected in cell lines of ES,
bypasses the problems related to the quality of mRNA extracted from
paraffin-embedded samples or the risk of contamination in
amplification techniques, such as RT-PCR (187).
It is possible to genotype the allelic
discrimination for single nucleotide polymorphisms (SNPs) in the
EWS gene using a TaqMan assay real-time-PCR. The analysis revealed
a higher incidence of the presence of homozygous TT in patients
with ES. The analysis also allowed the detection and identified the
region around the SNP, formed by a hexamer palindrome
(5′-GCTAGC-3′) and three nucleotides (GTC), very close to the
breakpoints in both the EWS and FLI1 genes. In patients homozygous
for this set of alleles, there a tendency to fracture doubles,
increasing the possibility of a translocation. SNP can be then a
candidate marker for susceptibility to ES (188). Moreover, investigations by
TaqMan real-time-PCR with a set of pairs of specific primers and
probes can quantify the different chimeric transcripts in ES
(EWS-FLI1, EWS-ERG, EWS-TV1, EWS-ETV4 and EWS-FEV) (189).
A number of studies have been undertaken to increase
knowledge on chromosomal aberrations and facilitate the diagnosis
of subsequent lesions, detecting specific translocations and
chimeric products (190–192). The described chromosomal
rearrangements not only aid in the diagnosis and classification of
soft tissue tumors, but are particularly useful in the differential
diagnosis of patients with an uncertain or dubious morphology
(193). Routine techniques, such
as FISH and RT-PCR, can be within the reach of pathology
laboratories, helping the pathologist in the diagnosis of such
neoplasms (194). Obviously, as
for all biomolecular methods, an essential condition is mandatory
for the correct development of these methods and to ensure useful
results for diagnosis; this includes all pre-analytical stages of
preparation of the biological sample. Appropriate sampling and all
stages, ranging from fixation/inclusion to the cutting of sections
destined for FISH and purification of nucleic acids, must be
conducted in the correct manner and by established standardized
procedures.
|
1
|
Dei Tos AP and Dal Cin P: The role of
cytogenetics in the classification of soft tissue tumours. Virchows
Arch. 431:83–94. 1997.PubMed/NCBI
|
|
2
|
Bennicelli JL and Barr FG: Genetics and
the biologic basis of sarcomas. Curr Opin Oncol. 11:267–274. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fletcher Christopher DM, Unni K Krishnan
and Mertens Fredrik: WHO: Pathology and Genetics of Tumours of Soft
Tissue and Bone. IARC Press; Lyon: 2002
|
|
4
|
Gisselsson D, Hibbard MK, Dal Cin P, Sciot
R, Hsi BL, Kozakewich HP and Fletcher JA: PLAG1 alterations in
lipoblastoma: involvement in varied mesenchymal cell types and
evidence for alternative oncogenic mechanisms. Am J Pathol.
159:955–962. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Martins C, Fonseca I, Roque L, Pereira T,
Ribeiro C, Bullerdiek J and Soares J: PLAG1 gene alterations in
salivary gland pleomorphic adenoma and carcinoma ex-pleomorphic
adenoma: a combined study using chromosome banding, in situ
hybridization and immunocytochemistry. Mod Pathol. 18:1048–1055.
2005. View Article : Google Scholar
|
|
6
|
Turc-Carel C, Limon J, Dal Cin P, Rao U,
Karakousis C and Sandberg AA: Cytogenetic studies of adipose tissue
tumors. II. Recurrent reciprocal translocation t(12;16)(q13;p11) in
myxoid liposarcomas. Cancer Genet Cytogenet. 23:291–309. 1986.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Paulien S, Turc-Carel C, Dal Cin P,
Jani-Sait S, Sreekantaiah C, Leong SP, Vogelstein B, Kinzler KW,
Sandberg AA and Gemmill RM: Myxoid liposarcoma with t(12;16)
(q13;p11) contains site-specific differences in methylation
patterns surrounding a zinc-finger gene mapped to the breakpoint
region on chromosome 12. Cancer Res. 50:7902–7907. 1990.
|
|
8
|
Mezzelani A, Sozzi G, Pierotti MA and
Pilotti S: Rapid differential diagnosis of myxoid liposarcoma by
fluorescence in situ hybridisation on cytological preparations.
Clin Mol Pathol. 49:308–309. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Aoki T, Hisaoka M, Kouho H, Hashimoto H
and Nakata H: Interphase cytogenetic analysis of myxoid soft tissue
tumors by fluorescence in situ hybridization and DNA flow cytometry
using paraffin-embedded tissue. Cancer. 79:284–293. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sozzi G, Minoletti F, Miozzo M, Sard L,
Musso K, Azzarelli A, Pierotti MA and Pilotti S: Relevance of
cytogenetic and fluorescent in situ hybridization analyses in the
clinical assessment of soft tissue sarcoma. Hum Pathol. 28:134–142.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Schoenmakers EF, Kools PF, Mols R,
Kazmierczak B, Bartnitzke S, Bullerdiek J, Dal Cin P, De Jong PJ,
Van den Berghe H and Van de Ven WJ: Physical mapping of chromosome
12q breakpoints in lipoma, pleomorphic salivary gland adenoma,
uterine leiomyoma, and myxoid liposarcoma. Genomics. 20:210–222.
1994. View Article : Google Scholar
|
|
12
|
Gisselsson D, Mandahl N, Pålsson E,
Gorunova L and Höglund M: Locus-specific multifluor FISH analysis
allows physical characterization of complex chromosome
abnormalities in neoplasia. Genes Chromosomes Cancer. 28:347–352.
2000. View Article : Google Scholar
|
|
13
|
Downs-Kelly E, Goldblum JR, Patel RM,
Weiss SW, Folpe AL, Mertens F, Hartke M, Tubbs RR and Skacel M: The
utility of fluorescence in situ hybridization (FISH) in the
diagnosis of myxoid soft tissue neoplasms. Am J Surg Pathol.
32:8–13. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Narendra S, Valente A, Tull J and Zhang S:
DDIT3 gene break-apart as a molecular marker for diagnosis of
myxoid liposarcoma - assay validation and clinical experience.
Diagn Mol Pathol. 20:218–224. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Aman P, Ron D, Mandahl N, Fioretos T, Heim
S, Arheden K, Willén H, Rydholm A and Mitelman F: Rearrangement of
the transcription factor gene CHOP in myxoid liposarcomas with
t(12;16)(q13;p11). Genes Chromosomes Cancer. 5:278–285. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Crozat A, Aman P, Mandahl N and Ron D:
Fusion of CHOP to a novel RNA-binding protein in human myxoid
liposarcoma. Nature. 363:640–644. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Knight JC, Renwick PJ, Dal Cin P, Van den
Berghe H and Fletcher CD: Translocation t(12;16)(q13;p11) in myxoid
liposarcoma and round cell liposarcoma: molecular and cytogenetic
analysis. Cancer Res. 55:24–27. 1995.PubMed/NCBI
|
|
18
|
Yang X, Nagasaki K, Egawa S, Maruyama K,
Futami H, Tsukada T, Yokoyama R, Beppu Y, Fukuma H, Shimoda T,
Mukai K, Yabe H, Hanaoka I, Yabe Y and Yamaguchi K: FUS/TLS-CHOP
chimeric transcripts in liposarcoma tissues. Jpn J Clin Oncol.
25:234–239. 1995.PubMed/NCBI
|
|
19
|
Panagopoulos I, Mandahl N, Mitelman F and
Aman P: Two distinct FUS breakpoint clusters in myxoid liposarcoma
and acute myeloid leukemia with the translocations t(12;16) and
t(16;21). Oncogene. 11:1133–1137. 1995.
|
|
20
|
Willeke F, Ridder R, Mechtersheimer G,
Schwarzbach M, Duwe A, Weitz J, Lehnert T, Herfarth C and von
Knebel Doeberitz M: Analysis of FUS-CHOP fusion transcripts in
different types of soft tissue liposarcoma and their diagnostic
implications. Clin Cancer Res. 4:1779–1784. 1998.PubMed/NCBI
|
|
21
|
Kanoe H, Nakayama T, Hosaka T, Murakami H,
Yamamoto H, Nakashima Y, Tsuboyama T, Nakamura T, Ron D, Sasaki MS
and Toguchida J: Characteristics of genomic breakpoints in TLS-CHOP
translocations in liposarcomas suggest the involvement of Translin
and topoisomerase II in the process of translocation. Oncogene.
18:721–729. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Huang HY and Antonescu CR: Molecular
variability of TLS-CHOP structure shows no significant impact on
the level of adipogenesis: a comparative ultrastructural and RT-PCR
analysis of 14 cases of myxoid/round cell liposarcomas. Ultrastruct
Pathol. 27:217–226. 2003. View Article : Google Scholar
|
|
23
|
Panagopoulos I, Aman P, Mertens F, Mandahl
N, Rydholm A, Bauer HF and Mitelman F: Genomic PCR detects tumor
cells in peripheral blood from patients with myxoid liposarcoma.
Genes Chromosomes Cancer. 17:102–107. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Rivero ER, Mesquita RA, de Sousa SC and
Nunes FD: Detection of TLS/FUS-CHOP fusion transcripts in a case of
oral liposarcoma. Ann Diagn Pathol. 10:36–38. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Panagopoulos I, Lassen C, Isaksson M,
Mitelman F, Mandahl N and Aman P: Characteristic sequence motifs at
the breakpoints of the hybrid genes FUS/CHOP, EWS/CHOP and FUS/ERG
in myxoid liposarcoma and acute myeloid leukemia. Oncogene.
15:1357–1362. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hisaoka M, Tsuji S, Morimitsu Y, Hashimoto
H, Shimajiri S, Komiya S and Ushijima M: Detection of TLS/FUS-CHOP
fusion transcripts in myxoid and round cell liposarcomas by nested
reverse transcription-polymerase chain reaction using archival
paraffin-embedded tissues. Diagn Mol Pathol. 7:96–101. 1998.
View Article : Google Scholar
|
|
27
|
Schwarzbach MH, Koesters R, Germann A,
Mechtersheimer G, Geisbill J and Winkler S: Comparable transforming
capacities and differential gene expression patterns of variant
FUS/CHOP fusion transcripts derived from soft tissue liposarcomas.
Oncogene. 23:6798–6805. 2004. View Article : Google Scholar
|
|
28
|
Patil N, Abba M, Hödl P, Schwarzbach M and
Allgayer H: A real time PCR based approach for the quantitative
detection of FUS-CHOP fusion transcripts in human liposarcoma. Adv
Med Sci. 57:37–45. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Borjigin N, Ohno S, Wu W, Tanaka M, Suzuki
R, Fujita K, Takanashi M, Oikawa K, Goto T, Motoi T, Kosaka T,
Yamamoto K and Kuroda M: TLS-CHOP represses miR-486 expression,
inducing upregulation of a metastasis regulator PAI-1 in human
myxoid liposarcoma. Biochem Biophys Res Commun. 427:355–360. 2012.
View Article : Google Scholar
|
|
30
|
Suzuki K, Matsui Y, Endo K, Kubo T,
Hasegawa T, Kimura T, Ohtani O and Yasui N: Myxoid liposarcoma with
EWS-CHOP type 1 fusion gene. Anticancer Res. 30:4679–4683.
2010.PubMed/NCBI
|
|
31
|
Powers MP, Wang WL, Hernandez VS, Patel
KS, Lev DC, Lazar AJ and López-Terrada DH: Detection of myxoid
liposarcoma-associated FUS-DDIT3 rearrangement variants including a
newly identified breakpoint using an optimized RT-PCR assay. Mod
Pathol. 23:1307–1315. 2010. View Article : Google Scholar
|
|
32
|
Suzuki K, Matsui Y, Hashimoto N, et al:
Variation in myxoid liposarcoma: Clinicopathological examination of
four cases with detectable TLS-CHOP or EWS-CHOP fusion transcripts
whose histopathological diagnosis was other than myxoid
liposarcoma. Oncol Lett. 3:293–296. 2012.
|
|
33
|
Bourgeois JM, Knezevich SR, Mathers JA and
Sorensen PH: Molecular detection of the ETV6-NTRK3 gene fusion
differentiates congenital fibrosarcoma from other childhood spindle
cell tumors. Am J Surg Pathol. 24:937–246. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Knezevich SR, McFadden DE, Tao W, Lim JF
and Sorensen PH: A novel ETV6-NTRK3 gene fusion in congenital
fibrosarcoma. Nat Genet. 18:184–187. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Adem C, Gisselsson D, Dal Cin P and
Nascimento AG: ETV6 rearrangements in patients with infantile
fibrosarcomas and congenital mesoblastic nephromas by fluorescence
in situ hybridization. Mod Pathol. 14:1246–1251. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Morerio C, Rapella A, Rosanda C, Tassano
E, Conte M, Gambini C and Panarello C: Differential diagnosis of
congenital fibrosarcoma. Cancer Genet Cytogenet. 152:167–168. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Mariño-Enríquez A, Li P, Samuelson J,
Rossi MR and Reyes-Múgica M: Congenital fibrosarcoma with a novel
complex 3-way translocation t(12;15;19) and unusual histologic
features. Hum Pathol. 39:1844–1848. 2008.PubMed/NCBI
|
|
38
|
Knezevich SR, Garnett MJ, Pysher TJ,
Beckwith JB, Grundy PE and Sorensen PH: ETV6-NTRK3 gene fusions and
trisomy 11 establish a histogenetic link between mesoblastic
nephroma and congenital fibrosarcoma. Cancer Res. 58:5046–5048.
1998.PubMed/NCBI
|
|
39
|
Sheng WQ, Hisaoka M, Okamoto S, Tanaka A,
Meis-Kindblom JM, Kindblom LG, Ishida T, Nojima T and Hashimoto H:
Congenital-infantile fibrosarcoma. A clinicopathologic study of 10
cases and molecular detection of the ETV6-NTRK3 fusion transcripts
using paraffin-embedded tissues. Am J Clin Pathol. 115:348–355.
2001. View Article : Google Scholar
|
|
40
|
Rubin BP, Chen CJ, Morgan TW, Xiao S,
Grier HE, Kozakewich HP, Perez-Atayde AR and Fletcher JA:
Congenital mesoblastic nephroma t(12;15) is associated with
ETV6-NTRK3 gene fusion: cytogenetic and molecular relationship to
congenital (infantile) fibrosarcoma. Am J Pathol. 153:1451–1458.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ramphal R, Manson D, Viero S, Zielenska M,
Gerstle T and Pappo A: Retroperitoneal infantile fibrosarcoma:
clinical, molecular, and therapeutic aspects of an unusual tumor.
Pediatr Hematol Oncol. 20:635–642. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Rizkalla H, Wildgrove H, Quinn F, Capra M
and O’Sullivan MJ: Congenital fibrosarcoma of the ileum: case
report with molecular confirmation and literature review. Fetal
Pediatr Pathol. 30:156–160. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Simon MP, Pedeutour F, Sirvent N,
Grosgeorge J, Minoletti F, Coindre JM, Terrier-Lacombe MJ, Mandahl
N, Craver RD, Blin N, Sozzi G, Turc-Carel C, O’Brien KP, Kedra D,
Fransson I, Guilbaud C and Dumanski JP: Deregulation of the
platelet-derived growth factor B-chain gene via fusion with
collagen gene COL1A1 in dermatofibrosarcoma protuberans and
giant-cell fibroblastoma. Nat Genet. 15:95–98. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Navarro M, Simon MP, Migeon C, Turc-Carel
C and Pedeutour F: COL1A1-PDGFB fusion in a ring chromosome 4 found
in a dermatofibrosarcoma protuberans. Genes Chromosomes Cancer.
23:263–266. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Salgado R, Llombart B, M Pujol R,
Fernández-Serra A, Sanmartín O, Toll A, Rubio L, Segura S, Barranco
C, Serra-Guillén C, Yébenes M, Salido M, Traves V, Monteagudo C,
Sáez E, Hernández T, de Álava E, Llombart-Bosch A, Solé F, Guillén
C, Espinet B and López-Guerrero JA: Molecular diagnosis of
dermatofibrosarcoma protuberans: a comparison between reverse
transcriptase-polymerase chain reaction and fluorescence in situ
hybridization methodologies. Genes Chromosomes Cancer. 50:510–517.
2011. View Article : Google Scholar
|
|
46
|
Segura S, Salgado R, Toll A,
Martín-Ezquerra G, Yébenes M, Sáez A, Solé F, Barranco C, Umbert P,
Espinet B and Pujol RM: Identification of t(17;22)(q22;q13)
(COL1A1/PDGFB) in dermatofibrosarcoma protuberans by fluorescence
in situ hybridization in paraffin-embedded tissue microarrays. Hum
Pathol. 42:176–184. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Walluks K, Chen Y, Woelfel C, Yang L, Cui
T, Seliger C, Geier C, Knösel T, Hauke S and Petersen I: Molecular
and clinicopathological analysis of dermatofibrosarcoma
protuberans. Pathol Res Pract. 209:30–35. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
O’Brien KP, Seroussi E, Dal Cin P, Sciot
R, Mandahl N, Fletcher JA, Turc-Carel C and Dumanski JP: Various
regions within the alpha-helical domain of the COL1A1 gene are
fused to the second exon of the PDGFB gene in dermatofibrosarcomas
and giant-cell fibroblastomas. Genes Chromosomes Cancer.
23:187–193. 1998.PubMed/NCBI
|
|
49
|
Macarenco RS, Zamolyi R, Franco MF,
Nascimento AG, Abott JJ, Wang X, Erickson-Johnson MR and Oliveira
AM: Genomic gains of COL1A1-PDFGB occur in the histologic evolution
of giant cell fibroblastoma into dermatofibrosarcoma protuberans.
Genes Chromosomes Cancer. 47:260–265. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Nishio J, Iwasaki H, Ohjimi Y, Ishiguro M,
Isayama T, Naito M, Kaneko Y and Kikuchi M: Supernumerary ring
chromosomes in dermatofibrosarcoma protuberans may contain
sequences from 8q11.2-qter and 17q21-qter: a combined cytogenetic
and comparative genomic hybridization study. Cancer Genet
Cytogenet. 129:102–106. 2001. View Article : Google Scholar
|
|
51
|
Kaur S, Vauhkonen H, Böhling T, Mertens F,
Mandahl N and Knuutila S: Gene copy number changes in
dermatofibrosarcoma protuberans - a fine-resolution study using
array comparative genomic hybridization. Cytogenet Genome Res.
115:283–288. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Wang J, Hisaoka M, Shimajiri S, Morimitsu
Y and Hashimoto H: Detection of COL1A1-PDGFB fusion transcripts in
dermatofibrosarcoma protuberans by reverse transcription-polymerase
chain reaction using archival formalin-fixed, paraffin-embedded
tissues. Diagn Mol Pathol. 8:113–119. 1999. View Article : Google Scholar
|
|
53
|
Szollosi Z, Scholtz B, Egervari K and
Nemes Z: Transformed dermatofibrosarcoma protuberans: real time
polymerase chain reaction detection of COL1A1-PDGFB fusion
transcripts in sarcomatous areas. J Clin Pathol. 60:190–194. 2007.
View Article : Google Scholar
|
|
54
|
Craver R, Dewenter T, Ebran N and
Pedeutour F: COL1A1-PDGFB fusion in a pediatric Bednar tumor with 2
copies of a der(22)t(17;22). Cancer Genet Cytogenet. 168:155–157.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Patel KU, Szabo SS, Hernandez VS, Prieto
VG, Abruzzo LV, Lazar AJ and López-Terrada D: Dermatofibrosarcoma
protuberans COL1A1-PDGFB fusion is identified in virtually all
dermatofibrosarcoma protuberans cases when investigated by newly
developed multiplex reverse transcription polymerase chain reaction
and fluorescence in situ hybridization assays. Hum Pathol.
39:184–193. 2008.
|
|
56
|
Gibson S, Sebire NJ and Anderson J:
Platelet-derived growth factor receptors and ligands are
up-regulated in paediatric fibromatoses. Histopathology.
51:752–757. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Takahira T, Oda Y, Tamiya S, Higaki K,
Yamamoto H, Kobayashi C, Izumi T, Tateishi N, Iwamoto Y and
Tsuneyoshi M: Detection of COL1A1-PDGFB fusion transcripts and
PDGFB/PDGFRB mRNA expression in dermatofibrosarcoma protuberans.
Mod Pathol. 20:668–675. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Muchemwa FC, Wakasugi S, Honda Y and Ihn
H: PDGFB quantification is a useful tool in the diagnosis of
dermatofibrosarcoma protuberans: a study of 10 cases. Clin Exp
Dermatol. 35:295–299. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Engel R, Ritterbach J, Schwabe D and
Lampert F: Chromosome translocation (2;13)(q37;q14) in a
disseminated alveolar rhabdomyosarcoma. Eur J Pediatr. 148:69–71.
1988. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Mehra S, de la Roza G, Tull J, Shrimpton
A, Valente A and Zhang S: Detection of FOXO1 (FKHR) gene
break-apart by fluorescence in situ hybridization in
formalin-fixed, paraffin-embedded alveolar rhabdomyosarcomas and
its clinicopathologic correlation. Diagn Mol Pathol. 17:14–20.
2008.
|
|
61
|
Liu J, Guzman MA, Pezanowski D, Patel D,
Hauptman J, Keisling M, Hou SJ, Papenhausen PR, Pascasio JM,
Punnett HH, Halligan GE and de Chadarévian JP: FOXO1-FGFR1 fusion
and amplification in a solid variant of alveolar rhabdomyosarcoma.
Mod Pathol. 24:1327–1335. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Biegel JA, Nycum LM, Valentine V, Barr FG
and Shapiro DN: Detection of the t(2;13)(q35;q14) and PAX3-FKHR
fusion in alveolar rhabdomyosarcoma by fluorescence in situ
hybridization. Genes Chromosomes Cancer. 12:186–192. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
McManus AP, O’Reilly MA, Jones KP,
Gusterson BA, Mitchell CD, Pinkerton CR and Shipley JM: Interphase
fluorescence in situ hybridization detection of t(2;13)(q35;q14) in
alveolar rhabdomyosarcoma - a diagnostic tool in minimally invasive
biopsies. J Pathol. 178:410–414. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Matsumura T, Yamaguchi T, Seki K, Shimoda
T, Wada T, Yamashita T and Hasegawa T: Advantage of FISH analysis
using FKHR probes for an adjunct to diagnosis of rhabdomyosarcomas.
Virchows Arch. 452:251–258. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Miura Y, Keira Y, Ogino J, Nakanishi K,
Noguchi H, Inoue T and Hasegawa T: Detection of specific genetic
abnormalities by fluorescence in situ hybridization in soft tissue
tumors. Pathol Int. 62:16–27. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Barr FG, Chatten J, D’Cruz CM, Wilson AE,
Nauta LE, Nycum LM, Biegel JA and Womer RB: Molecular assays for
chromosomal translocations in the diagnosis of pediatric soft
tissue sarcomas. JAMA. 273:553–557. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Arden KC, Anderson MJ, Finckenstein FG,
Czekay S and Cavenee WK: Detection of the t(2;13) chromosomal
translocation in alveolar rhabdomyosarcoma using the reverse
transcriptase-polymerase chain reaction. Genes Chromosomes Cancer.
16:254–260. 1996. View Article : Google Scholar
|
|
68
|
Kelly KM, Womer RB and Barr FG: Minimal
disease detection in patients with alveolar rhabdomyosarcoma using
a reverse transcriptase-polymerase chain reaction method. Cancer.
78:1320–1327. 1996. View Article : Google Scholar
|
|
69
|
Sorensen PH, Lynch JC, Qualman SJ,
Tirabosco R, Lim JF, Maurer HM, Bridge JA, Crist WM, Triche TJ and
Barr FG: PAX3-FKHR and PAX7-FKHR gene fusions are prognostic
indicators in alveolar rhabdomyosarcoma: a report from the
children’s oncology group. J Clin Oncol. 20:2672–2679.
2002.PubMed/NCBI
|
|
70
|
Thway K, Rockcliffe S, Gonzalez D,
Swansbury J, Min T, Thompson L and Fisher C: Utility of
sarcoma-specific fusion gene analysis in paraffin-embedded material
for routine diagnosis at a specialist centre. J Clin Pathol.
63:508–512. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Eguía-Aguilar P, Ponce-Castañeda V,
Nájera-García N, Nieto-Martínez K, Kofman-Alfaro S, Sadowinski-Pine
S, Valencia-Mayoral P, Arenas-Huertero F and Perezpeña-Diazconti M:
Detection of fusion genes in formalin-fixed paraffin-embedded
tissue sections of rhabdomyosarcoma by RT-PCR and fluorescence in
situ hybridization in Mexican patients. Arch Med Res. 41:119–124.
2010.PubMed/NCBI
|
|
72
|
Downing JR, Khandekar A, Shurtleff SA,
Head DR, Parham DM, Webber BL, Pappo AS, Hulshof MG, Conn WP and
Shapiro DN: Multiplex RT-PCR assay for the differential diagnosis
of alveolar rhabdomyosarcoma and Ewing’s sarcoma. Am J Pathol.
146:626–634. 1995.
|
|
73
|
Anderson J, Renshaw J, McManus A, Carter
R, Mitchell C, Adams S and Pritchard-Jones K: Amplification of the
t(2;13) and t(1;13) translocations of alveolar rhabdomyosarcoma in
small formalin-fixed biopsies using a modified reverse
transcriptase polymerase chain reaction. Am J Pathol. 150:477–482.
1997.
|
|
74
|
Athale UH, Shurtleff SA, Jenkins JJ,
Poquette CA, Tan M, Downing JR and Pappo AS: Use of reverse
transcriptase polymerase chain reaction for diagnosis and staging
of alveolar rhabdomyosarcoma, Ewing sarcoma family of tumors, and
desmoplastic small round cell tumor. J Pediatr Hematol Oncol.
23:99–104. 2001. View Article : Google Scholar
|
|
75
|
Edwards RH, Chatten J, Xiong QB and Barr
FG: Detection of gene fusions in rhabdomyosarcoma by reverse
transcriptase-polymerase chain reaction assay of archival samples.
Diagn Mol Pathol. 6:91–97. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Chen BF, Chen ML, Liang DC, Huang YW, Liu
HC and Chen SH: Detection of PAX3-FKHR and PAX7-FKHR fusion
transcripts in rhabdomyosarcoma by reverse transcriptase-polymerase
chain reaction using paraffin-embedded tissue. Zhonghua Yi Xue Za
Zhi (Taipei). 62:86–91. 1999.PubMed/NCBI
|
|
77
|
Jin L, Majerus J, Oliveira A, Inwards CY,
Nascimento AG, Burgart LJ and Lloyd RV: Detection of fusion gene
transcripts in fresh-frozen and formalin-fixed paraffin-embedded
tissue sections of soft-tissue sarcomas after laser capture
microdissection and rt-PCR. Diagn Mol Pathol. 12:224–230. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Fritsch MK, Bridge JA, Schuster AE,
Perlman EJ and Argani P: Performance characteristics of a reverse
transcriptase-polymerase chain reaction assay for the detection of
tumor-specific fusion transcripts from archival tissue. Pediatr Dev
Pathol. 6:43–53. 2003. View Article : Google Scholar
|
|
79
|
Peter M, Gilbert E and Delattre O: A
multiplex real-time pcr assay for the detection of gene fusions
observed in solid tumors. Lab Invest. 81:905–912. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Krsková L, Mrhalová M, Hilská I, Sumerauer
D, Drahokoupilová E, Múdry P and Kodet R: Detection and clinical
significance of bone marrow involvement in patients with
rhabdomyosarcoma. Virchows Arch. 456:463–472. 2010.PubMed/NCBI
|
|
81
|
Hostein I, Andraud-Fregeville M, Guillou
L, Terrier-Lacombe MJ, Deminière C, Ranchère D, Lussan C,
Longavenne E, Bui NB, Delattre O and Coindre JM: Rhabdomyosarcoma:
value of myogenin expression analysis and molecular testing in
diagnosing the alveolar subtype: an analysis of 109
paraffin-embedded specimens. Cancer. 101:2817–2824. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Turc-Carel C, Dal Cin P, Limon J, Rao U,
Li FP, Corson JM, Zimmerman R, Parry DM, Cowan JM and Sandberg AA:
Involvement of chromosome X in primary cytogenetic change in human
neoplasia: nonrandom translocation in synovial sarcoma. Proc Natl
Acad Sci USA. 84:1981–1985. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Clark J, Rocques PJ, Crew AJ, Gill S,
Shipley J, Chan AM, Gusterson BA and Cooper CS: Identification of
novel genes, SYT and SSX, involved in the t(X;18)(p11.2;q11.2)
translocation found in human synovial sarcoma. Nat Genet.
7:502–508. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
de Leeuw B, Balemans M, Olde Weghuis D and
Geurts van Kessel A: Identification of two alternative fusion
genes, SYT-SSX1 and SYT-SSX2, in t(X;18)(p11.2;q11.2)-positive
synovial sarcomas. Hum Mol Genet. 4:1097–1099. 1995.PubMed/NCBI
|
|
85
|
Lee W, Han K, Harris CP, Shim S, Kim S and
Meisner LF: Use of FISH to detect chromosomal translocations and
deletions. Analysis of chromosome rearrangement in synovial sarcoma
cells from paraffin-embedded specimens. Am J Pathol. 143:15–19.
1993.
|
|
86
|
Knight JC, Reeves BR, Kearney L, Monaco
AP, Lehrach H and Cooper CS: Localization of the synovial sarcoma
t(X;18)(p11.2;q11.2) breakpoint by fluorescence in situ
hybridization. Hum Mol Genet. 1:633–637. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
de Leeuw B, Suijkerbuijk RF, Balemans M,
Sinke RJ, de Jong B, Molenaar WM, Meloni AM, Sandberg AA, Geraghty
M and Hofker M: Sublocalization of the synovial sarcoma-associated
t(X;18) chromosomal breakpoint in Xp11.2 using cosmid cloning and
fluorescence in situ hybridization. Oncogene. 8:1457–1463.
1993.
|
|
88
|
Motoi T, Kumagai A, Tsuji K, Imamura T and
Fukusato T: Diagnostic utility of dual-color break-apart
chromogenic in situ hybridization for the detection of rearranged
SS18 in formalin-fixed, paraffin-embedded synovial sarcoma. Hum
Pathol. 41:1397–1404. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Terry J, Barry TS, Horsman DE, Hsu FD,
Gown AM, Huntsman DG and Nielsen TO: Fluorescence in situ
hybridization for the detection of t(X;18)(p11.2;q11.2) in a
synovial sarcoma tissue microarray using a breakapart-style probe.
Diagn Mol Pathol. 14:77–82. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Geiersbach K, Rector LS, Sederberg M,
Hooker A, Randall RL, Schiffman JD and South ST: Unknown partner
for USP6 and unusual SS18 rearrangement detected by fluorescence in
situ hybridization in a solid aneurysmal bone cyst. Cancer Genet.
204:195–202. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Kanemitsu S, Hisaoka M, Shimajiri S,
Matsuyama A and Hashimoto H: Molecular detection of SS18-SSX fusion
gene transcripts by cRNA in situ hybridization in synovial sarcoma
using formalin-fixed, paraffin-embedded tumor tissue specimens.
Diagn Mol Pathol. 16:9–17. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Argani P, Zakowski MF, Klimstra DS, Rosai
J and Ladanyi M: Detection of the SYT-SSX chimeric RNA of synovial
sarcoma in paraffin-embedded tissue and its application in
problematic cases. Mod Pathol. 11:65–71. 1998.PubMed/NCBI
|
|
93
|
Guillou L, Coindre J, Gallagher G, Terrier
P, Gebhard S, de Saint Aubain Somerhausen N, Michels J, Jundt G,
Vince DR, Collin F, Trassard M, Le Doussal V and Benhattar J:
Detection of the synovial sarcoma translocation t(X;18) (SYT;SSX)
in paraffin-embedded tissues using reverse transcriptase-polymerase
chain reaction: a reliable and powerful diagnostic tool for
pathologists. A molecular analysis of 221 mesenchymal tumors fixed
in different fixatives. Hum Pathol. 32:105–112. 2001.
|
|
94
|
Fligman I, Lonardo F, Jhanwar SC, Gerald
WL, Woodruff J and Ladanyi M: Molecular diagnosis of synovial
sarcoma and characterization of a variant SYT-SSX2 fusion
transcript. Am J Pathol. 147:1592–1599. 1995.PubMed/NCBI
|
|
95
|
Safar A, Wickert R, Nelson M, Neff JR and
Bridge JA: Characterization of a variant SYT-SSX1 synovial sarcoma
fusion transcript. Diagn Mol Pathol. 7:283–287. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Tsuji S, Hisaoka M, Morimitsu Y, Hashimoto
H, Shimajiri S, Komiya S, Ushijima M and Nakamura T: Detection of
SYT-SSX fusion transcripts in synovial sarcoma by reverse
transcription-polymerase chain reaction using archival
paraffin-embedded tissues. Am J Pathol. 153:1807–1812. 1998.
View Article : Google Scholar
|
|
97
|
Sanders ME, van de Rijn M and Barr FG:
Detection of a variant SYT-SSX1 fusion in a case of predominantly
epithelioid synovial sarcoma. Mol Diagn. 4:65–70. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Katenkamp K, Richter P, Slatosch T,
Katenkamp D and Berndt A: Simultaneous analysis of t(X;18) by FISH-
und SYT/SSX-RT-PCR in synovial sarcoma. Pathologe. 26:111–116.
2005.(In German).
|
|
99
|
Cummings TJ, Brown NM and Stenzel TT:
TaqMan junction probes and the reverse transcriptase polymerase
chain reaction: detection of alveolar rhabdomyosarcoma, synovial
sarcoma, and desmoplastic small round cell tumor. Ann Clin Lab Sci.
32:219–224. 2002.
|
|
100
|
Coindre JM, Hostein I, Benhattar J, Lussan
C, Rivel J and Guillou L: Malignant peripheral nerve sheath tumors
are t(X;18)-negative sarcomas. Molecular analysis of 25 cases
occurring in neurofibromatosis type 1 patients, using two different
RT-PCR-based methods of detection. Mod Pathol. 15:589–592. 2002.
View Article : Google Scholar
|
|
101
|
Hostein I, Menard A, Bui BN, Lussan C,
Wafflart J, Delattre O, Peter M, Benhattar J, Guillou L and Coindre
JM: Molecular detection of the synovial sarcoma translocation
t(X;18) by real-time polymerase chain reaction in paraffin-embedded
material. Diagn Mol Pathol. 11:16–21. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Bijwaard KE, Fetsch JF, Przygodzki R,
Taubenberger JK and Lichy JH: Detection of SYT-SSX fusion
transcripts in archival synovial sarcomas by real-time reverse
transcriptase-polymerase chain reaction. J Mol Diagn. 4:59–64.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Wang WL, Mayordomo E, Zhang W, Hernandez
VS, Tuvin D, Garcia L, Lev DC, Lazar AJ and López-Terrada D:
Detection and characterization of EWSR1/ATF1 and EWSR1/CREB1
chimeric transcripts in clear cell sarcoma (melanoma of soft
parts). Mod Pathol. 22:1201–1209. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Hiraga H, Nojima T, Abe S, Yamashiro K,
Yamawaki S, Kaneda K and Nagashima K: Establishment of a new
continuous clear cell sarcoma cell line. Morphological and
cytogenetic characterization and detection of chimaeric EWS/ATF-1
transcripts. Virchows Arch. 431:45–51. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Yamaguchi U, Hasegawa T, Morimoto Y,
Tateishi U, Endo M, Nakatani F, Kawai A, Chuman H, Beppu Y, Endo M,
Kurotaki H and Furuta K: A practical approach to the clinical
diagnosis of Ewing’s sarcoma/primitive neuroectodermal tumour and
other small round cell tumours sharing EWS rearrangement using new
fluorescence in situ hybridisation probes for EWSR1 on formalin
fixed, paraffin wax embedded tissue. J Clin Pathol. 58:1051–1056.
2005.
|
|
106
|
Song JS, Choi J, Kim JH, Jang SJ and Cho
KJ: Diagnostic utility of EWS break-apart fluorescence in situ
hybridization in distinguishing between non-cutaneous melanoma and
clear cell sarcoma. Pathol Int. 60:608–613. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Patel RM, Downs-Kelly E, Weiss SW, Folpe
AL, Tubbs RR, Tuthill RJ, Goldblum JR and Skacel M: Dual-color,
break-apart fluorescence in situ hybridization for EWS gene
rearrangement distinguishes clear cell sarcoma of soft tissue from
malignant melanoma. Mod Pathol. 18:1585–1590. 2005.PubMed/NCBI
|
|
108
|
Speleman F, Delattre O, Peter M, Hauben E,
Van Roy N and Van Marck E: Malignant melanoma of the soft parts
(clear-cell sarcoma): confirmation of EWS and ATF-1 gene fusion
caused by a t(12;22) translocation. Mod Pathol. 10:496–499.
1997.PubMed/NCBI
|
|
109
|
Panagopoulos I, Mertens F, Dêbiec-Rychter
M, Isaksson M, Limon J, Kardas I, Domanski HA, Sciot R, Perek D,
Crnalic S, Larsson O and Mandahl N: Molecular genetic
characterization of the EWS/ATF1 fusion gene in clear cell sarcoma
of tendons and aponeuroses. Int J Cancer. 99:560–567. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Antonescu CR, Tschernyavsky SJ, Woodruff
JM, Jungbluth AA, Brennan MF and Ladanyi M: Molecular diagnosis of
clear cell sarcoma: detection of EWS-ATF1 and MITF-M transcripts
and histopathological and ultrastructural analysis of 12 cases. J
Mol Diagn. 4:44–52. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Coindre JM, Hostein I, Terrier P,
Bouvier-Labit C, Collin F, Michels JJ, Trassard M, Marques B,
Ranchere D and Guillou L: Diagnosis of clear cell sarcoma by
real-time reverse transcriptase-polymerase chain reaction analysis
of paraffin embedded tissues: clinicopathologic and molecular
analysis of 44 patients from the French sarcoma group. Cancer.
107:1055–1064. 2006. View Article : Google Scholar
|
|
112
|
Ladanyi M and Gerald W: Fusion of the EWS
and WT1 genes in the desmoplastic small round cell tumor. Cancer
Res. 54:2837–2840. 1994.PubMed/NCBI
|
|
113
|
Karnieli E, Werner H, Rauscher FJ III,
Benjamin LE and LeRoith D: The IGF-I receptor gene promoter is a
molecular target for the Ewing’s sarcoma-Wilms’ tumor 1 fusion
protein. J Biol Chem. 271:19304–19309. 1996.
|
|
114
|
Benjamin LE, Fredericks WJ, Barr FG and
Rauscher FJ III: Fusion of the EWS1 and WT1 genes as a result of
the t(11;22)(p13;q12) translocation in desmoplastic small round
cell tumors. Med Pediatr Oncol. 27:434–439. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Barnoud R, Delattre O, Péoc’h M, Pasquier
D, Plantaz D, Leroux D and Pasquier B: Desmoplastic small round
cell tumor: RT-PCR analysis and immunohistochemical detection of
the Wilm’s tumor gene WT1. Pathol Res Pract. 194:693–700.
1998.PubMed/NCBI
|
|
116
|
Gerald WL, Rosai J and Ladanyi M:
Characterization of the genomic breakpoint and chimeric transcripts
in the EWS-WT1 gene fusion of desmoplastic small round cell tumor.
Proc Natl Acad Sci USA. 92:1028–1032. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Rauscher FJ III, Benjamin LE, Fredericks
WJ and Morris JF: Novel oncogenic mutations in the WT1 Wilms’ tumor
suppressor gene: a t(11;22) fuses the Ewing’s sarcoma gene, EWS1,
to WT1 in desmoplastic small round cell tumor. Cold Spring Harb
Symp Quant Biol. 59:137–146. 1994.
|
|
118
|
Antonescu CR, Gerald WL, Magid MS and
Ladanyi M: Molecular variants of the EWS-WT1 gene fusion in
desmoplastic small round cell tumor. Diagn Mol Pathol. 7:24–28.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Hill DA, Pfeifer JD, Marley EF, Dehner LP,
Humphrey PA, Zhu X and Swanson PE: WT1 staining reliably
differentiates desmoplastic small round cell tumor from Ewing
sarcoma/primitive neuroectodermal tumor. An immunohistochemical and
molecular diagnostic study. Am J Clin Pathol. 114:345–353.
2000.
|
|
120
|
Brodie SG, Stocker SJ, Wardlaw JC, Duncan
MH, McConnell TS, Feddersen RM and Williams TM: EWS and WT-1 gene
fusion in desmoplastic small round cell tumor of the abdomen. Hum
Pathol. 26:1370–1374. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Liu J, Nau MM, Yeh JC, Allegra CJ, Chu E
and Wright JJ: Molecular heterogeneity and function of EWS-WT1
fusion transcripts in desmoplastic small round cell tumors. Clin
Cancer Res. 6:3522–3529. 2000.PubMed/NCBI
|
|
122
|
Su LD, Atayde-Perez A, Sheldon S, Fletcher
JA and Weiss SW: Inflammatory myofibroblastic tumor: cytogenetic
evidence supporting clonal origin. Mod Pathol. 11:364–368.
1998.PubMed/NCBI
|
|
123
|
Souid AK, Ziemba MC, Dubansky AS, Mazur M,
Oliphant M, Thomas FD, Ratner M and Sadowitz PD: Inflammatory
myofibroblastic tumor in children. Cancer. 72:2042–2048. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Griffin CA, Hawkins AL, Dvorak C, Henkle
C, Ellingham T and Perlman EJ: Recurrent involvement of 2p23 in
inflammatory myofibroblastic tumors. Cancer Res. 59:2776–2780.
1999.PubMed/NCBI
|
|
125
|
Tan LH, Do E, Tan SY, Chong SM and Koay
ES: Multi-lineage interrogation of the performance characteristics
of a split-signal fluorescence in situ hybridization probe for
anaplastic lymphoma kinase gene rearrangements: a study of 101
cases characterized by immunohistomorphology on fixed archival
tissue. Mol Diagn. 8:213–229. 2004.
|
|
126
|
Sirvent N, Hawkins AL, Moeglin D, Coindre
JM, Kurzenne JY, Michiels JF, Barcelo G, Turc-Carel C, Griffin CA
and Pedeutour F: ALK probe rearrangement in a
t(2;11;2)(p23;p15;q31) translocation found in a prenatal
myofibroblastic fibrous lesion: toward a molecular definition of an
inflammatory myofibroblastic tumor family? Genes Chromosomes
Cancer. 31:85–90. 2001. View Article : Google Scholar
|
|
127
|
Stoll LM and Li QK: Cytology of
fine-needle aspiration of inflammatory myofibroblastic tumor. Diagn
Cytopathol. 39:663–672. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Borak S, Siegal GP, Reddy V, Jhala N and
Jhala D: Metastatic inflammatory myofibroblastic tumor identified
by EUS-FNA in mediastinal lymph nodes with ancillary FISH studies
for ALK rearrangement. Diagn Cytopathol. 40(Suppl 2): S118–S125.
2012. View Article : Google Scholar
|
|
129
|
Li XQ, Hisaoka M, Shi DR, Zhu XZ and
Hashimoto H: Expression of anaplastic lymphoma kinase in soft
tissue tumors: an immunohistochemical and molecular study of 249
cases. Hum Pathol. 35:711–721. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Alaggio R, Barisani D, Ninfo V, Rosolen A
and Coffin CM: Morphologic overlap between infantile
myofibromatosis and infantile fibrosarcoma: A pitfall in diagnosis.
Pediatr Dev Pathol. 11:355–362. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Lawrence B, Perez-Atayde A, Hibbard MK,
Rubin BP, Dal Cin P, Pinkus JL, Pinkus GS, Xiao S, Yi ES, Fletcher
CD and Fletcher JA: TPM3-ALK and TPM4-ALK oncogenes in inflammatory
myofibroblastic tumors. Am J Pathol. 157:377–384. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Cools J, Wlodarska I, Somers R, Mentens N,
Pedeutour F, Maes B, De Wolf-Peeters C, Pauwels P, Hagemeijer A and
Marynen P: Identification of novel fusion partners of ALK, the
anaplastic lymphoma kinase, in anaplastic large-cell lymphoma and
inflammatory myofibroblastic tumor. Genes Chromosomes Cancer.
34:354–362. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Lamant L, Dastugue N, Pulford K, Delsol G
and Mariamé B: A new fusion gene TPM3-ALK in anaplastic large cell
lymphoma created by a (1;2)(q25;p23) translocation. Blood.
93:3088–3095. 1999.PubMed/NCBI
|
|
134
|
Drexler HG, Gignac SM, von Wasielewski R,
Werner M and Dirks WG: Pathobiology of NPM-ALK and variant fusion
genes in anaplastic large cell lymphoma and other lymphomas.
Leukemia. 14:1533–1559. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Bridge JA, Kanamori M, Ma Z, Pickering D,
Hill DA, Lydiatt W, Lui MY, Colleoni GW, Antonescu CR, Ladanyi M
and Morris SW: Fusion of the ALK gene to the clathrin heavy chain
gene, CLTC, in inflammatory myofibroblastic tumor. Am J Pathol.
159:411–415. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Wang X, Krishnan C, Nguyen EP, Meyer KJ,
Oliveira JL, Yang P, Yi ES, Erickson-Johnson MR, Yaszemski MJ,
Maran A and Oliveira AM: Fusion of dynactin 1 to anaplastic
lymphoma kinase in inflammatory myofibroblastic tumor. Hum Pathol.
43:2047–2052. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Panagopoulos I, Nilsson T, Domanski HA,
Isaksson M, Lindblom P, Mertens F and Mandahl N: Fusion of the
SEC31L1 and ALK genes in an inflammatory myofibroblastic tumor. Int
J Cancer. 118:1181–1186. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Butrynski JE, D’Adamo DR, Hornick JL, Dal
Cin P, Antonescu CR, Jhanwar SC, Ladanyi M, Capelletti M, Rodig SJ,
Ramaiya N, Kwak EL, Clark JW, Wilner KD, Christensen JG, Jänne PA,
Maki RG, Demetri GD and Shapiro GI: Crizotinib in ALK-rearranged
inflammatory myofibroblastic tumor. N Engl J Med. 363:1727–1733.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Tothova Z and Wagner AJ: Anaplastic
lymphoma kinase-directed therapy in inflammatory myofibroblastic
tumors. Curr Opin Oncol. 24:409–413. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Lieberman PH, Brennan MF, Kimmel M,
Erlandson RA, Garin-Chesa P and Flehinger BY: Alveolar soft-part
sarcoma. A clinico-pathologic study of half a century. Cancer.
63:1–13. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Ordonez NG: Alveolar soft part sarcoma: a
review and update. Adv Anat Pathol. 6:125–139. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Joyama S, Ueda T, Shimizu K, Kudawara I,
Mano M, Funai H, Takemura K and Yoshikawa H: Chromosome
rearrangement at 17q25 and xp11.2 in alveolar soft-part sarcoma: A
case report and review of the literature. Cancer. 86:1246–1250.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Uppal S, Aviv H, Patterson F, Cohen S,
Benevenia J, Aisner S and Hameed M: Alveolar soft part sarcoma -
reciprocal translocation between chromosome 17q25 and Xp11. Report
of a case with metastases at presentation and review of the
literature. Acta Orthop Belg. 69:182–187. 2003.PubMed/NCBI
|
|
144
|
Amin MB, Patel RM, Oliveira P, Cabrera R,
Carneiro V, Preto M, Balzer B and Folpe AL: Alveolar soft-part
sarcoma of the urinary bladder with urethral recurrence: a unique
case with emphasis on differential diagnoses and diagnostic utility
of an immunohistochemical panel including TFE3. Am J Surg Pathol.
30:1322–1325. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Heimann P, Devalck C, Debusscher C,
Sariban E and Vamos E: Alveolar soft-part sarcoma: further evidence
by FISH for the involvement of chromosome band 17q25. Genes
Chromosomes Cancer. 23:194–197. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Ladanyi M, Lui MY, Antonescu CR,
Krause-Boehm A, Meindl A, Argani P, Healey JH, Ueda T, Yoshikawa H,
Meloni-Ehrig A, Sorensen PH, Mertens F, Mandahl N, van den Berghe
H, Sciot R, Dal Cin P and Bridge J: The der(17)t(X;17)(p11;q25) of
human alveolar soft part sarcoma fuses the TFE3 transcription
factor gene to ASPL, a novel gene at 17q25. Oncogene. 20:48–57.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Argani P, Antonescu CR, Illei PB, Lui MY,
Timmons CF, Newbury R, Reuter VE, Garvin AJ, Perez-Atayde AR,
Fletcher JA, Beckwith JB, Bridge JA and Ladanyi M: Primary renal
neoplasms with the ASPL-TFE3 gene fusion of alveolar soft part
sarcoma: a distinctive tumor entity previously included among renal
cell carcinomas of children and adolescents. Am J Pathol.
159:179–192. 2001. View Article : Google Scholar
|
|
148
|
Aulmann S, Longerich T, Schirmacher P,
Mechtersheimer G and Penzel R: Detection of the ASPSCR1-TFE3 gene
fusion in paraffin-embedded alveolar soft part sarcomas.
Histopathology. 50:881–886. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Hoshino M, Ogose A, Kawashima H, Izumi T,
Hotta T, Hatano H, Morita T, Otsuka H, Umezu H, Yanoma S, Tsukuda M
and Endo N: Molecular analyses of cell origin and detection of
circulating tumor cells in the peripheral blood in alveolar soft
part sarcoma. Cancer Genet Cytogenet. 190:75–80. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Pink D, Bertz-Lepel J, Busemann C, Bitz U
and Reichardt P: Efficacy of trabectedin in patients with advanced
or metastatic alveolar soft-part sarcoma. Onkologie. 35:249–252.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Tsuneyoshi M, Enjoji M, Iwasaki H and
Shinohara N: Extraskeletal myxoid chondrosarcoma - a
clinicopathologic and electron microscopic study. Acta Pathol Jpn.
31:439–447. 1981.PubMed/NCBI
|
|
152
|
Saleh G, Evans HL, Ro JY and Ayala AG:
Extraskeletal myxoid chondrosarcoma. A clinicopathologic study of
ten patients with long-term follow-up. Cancer. 70:2827–2830. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Gebhardt MC, Parekh SG, Rosenberg AE and
Rosenthal DI: Extraskeletal myxoid chondrosarcoma of the knee.
Skeletal Radiol. 28:354–358. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Meis-Kindblom JM, Bergh P, Gunterberg B
and Kindblom LG: Extraskeletal myxoid chondrosarcoma: a reappraisal
of its morphologic spectrum and prognostic factors based on 117
cases. Am J Surg Pathol. 23:636–650. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Sciot R, Dal Cin P, Fletcher C, Samson I,
Smith M, De Vos R, Van Damme B and Van den Berghe H:
t(9;22)(q22-31;q11-12) is a consistent marker of extraskeletal
myxoid chondrosarcoma: evaluation of three cases. Mod Pathol.
8:765–768. 1995.PubMed/NCBI
|
|
156
|
Rao UN, Surti U, Hoffner L, Howard T,
Leger W, Contis L and Yaw K: Extraskeletal and skeletal myxoid
chondrosarcoma: A multiparameter analysis of three cases including
cytogenetic analysis and fluorescence in situ hybridization. Mol
Diagn. 1:99–107. 1996. View Article : Google Scholar
|
|
157
|
Harris M, Coyne J, Tariq M, Eyden BP,
Atkinson M, Freemont AJ, Varley J, Attwooll C and Telford N:
Extraskeletal myxoid chondrosarcoma with neuroendocrine
differentiation: a pathologic, cytogenetic, and molecular study of
a case with a novel translocation t(9;17)(q22;q11.2). Am J Surg
Pathol. 24:1020–1026. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Sjögren H, Wedell B, Meis-Kindblom JM,
Kindblom LG and Stenman G: Fusion of the NH2-terminal domain of the
basic helix-loop-helix protein TCF12 to TEC in extraskeletal myxoid
chondrosarcoma with translocation t(9;15)(q22;q21). Cancer Res.
60:6832–6835. 2000.PubMed/NCBI
|
|
159
|
Gan TI, Rowen L, Nesbitt R, Roe BA, Wu H,
Hu P, Yao Z, Kim UJ, O’Sickey T and Bina M: Genomic organization of
human TCF12 gene and spliced mRNA variants producing isoforms of
transcription factor HTF4. Cytogenet Genome Res. 98:245–248. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Attwooll C, Tariq M, Harris M, Coyne JD,
Telford N and Varley JM: Identification of a novel fusion gene
involving hTAFII68 and CHN from a t(9;17)(q22;q11.2) translocation
in an extraskeletal myxoid chondrosarcoma. Oncogene. 18:7599–7601.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Lim B, Jun HJ, Kim AY, Kim S, Choi J, Kim
J, et al: The TFG-TEC fusion gene created by the t(3;9)
translocation in human extraskeletal myxoid chondrosarcomas encodes
a more potent transcriptional activator than TEC.
Carcinogenesis. 33:1450–1458. 2012.
|
|
162
|
Brody RI, Ueda T, Hamelin A, Jhanwar SC,
Bridge JA, Healey JH, Huvos AG, Gerald WL and Ladanyi M: Molecular
analysis of the fusion of EWS to an orphan nuclear receptor gene in
extraskeletal myxoid chondrosarcoma. Am J Pathol. 150:1049–1058.
1997.PubMed/NCBI
|
|
163
|
Panagopoulos I, Mertens F, Isaksson M,
Domanski HA, Brosjö O, Heim S, Bjerkehagen B, Sciot R, Dal Cin P,
Fletcher JA, Fletcher CD and Mandahl N: Molecular genetic
characterization of the EWS/CHN and RBP56/CHN fusion genes in
extraskeletal myxoid chondrosarcoma. Genes Chromosomes Cancer.
35:340–352. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Jakowski JD and Wakely PE Jr:
Cytopathology of extraskeletal myxoid chondrosarcoma: report of 8
cases. Cancer. 111:298–305. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Wang WL, Mayordomo E, Czerniak BA, Abruzzo
LV, Dal Cin P, Araujo DM, Lev DC, López-Terrada D and Lazar AJ:
Fluorescence in situ hybridization is a useful ancillary diagnostic
tool for extraskeletal myxoid chondrosarcoma. Mod Pathol.
21:1303–1310. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Noguchi H, Mitsuhashi T, Seki K, Tochigi
N, Tsuji M, Shimoda T and Hasegawa T: Fluorescence in situ
hybridization analysis of extraskeletal myxoid chondrosarcomas
using EWSR1 and NR4A3 probes. Hum Pathol. 41:336–342. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Sjögren H, Meis-Kindblom JM, Orndal C,
Bergh P, Ptaszynski K, Aman P, Kindblom LG and Stenman G: Studies
on the molecular pathogenesis of extraskeletal myxoid
chondrosarcoma-cytogenetic, molecular genetic, and cDNA microarray
analyses. Am J Pathol. 162:781–792. 2003.PubMed/NCBI
|
|
168
|
Okamoto S, Hisaoka M, Ishida T, Imamura T,
Kanda H, Shimajiri S and Hashimoto H: Extraskeletal myxoid
chondrosarcoma: a clinicopathologic, immunohistochemical, and
molecular analysis of 18 cases. Hum Pathol. 32:1116–1124. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
169
|
Matsukuma S, Hisaoka M, Obara K, Kono T,
Takeo H, Sato K and Hata Y: Primary pulmonary myxoid sarcoma with
EWSR1-CREB1 fusion, resembling extraskeletal myxoid chondrosarcoma:
Case report with a review of Literature. Pathol Int. 62:817–822.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Aurias A, Rimbaut C, Buffe D, Zucker JM
and Mazabraud A: Translocation involving chromosome 22 in Ewing’s
sarcoma. A cytogenetic study of four fresh tumors. Cancer Genet
Cytogenet. 12:21–25. 1984.
|
|
171
|
Whang-Peng J, Triche TJ, Knutsen T, Miser
J, Kao-Shan S, Tsai S and Israel MA: Cytogenetic characterization
of selected small round cell tumors of childhood. Cancer Genet
Cytogenet. 21:185–208. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Burchill SA: Ewing’s sarcoma: diagnostic,
prognostic, and therapeutic implications of molecular
abnormalities. J Clin Pathol. 56:96–102. 2003.
|
|
173
|
Gamberi G, Cocchi S, Benini S, Magagnoli
G, Morandi L, Kreshak J, Gambarotti M, Picci P, Zanella L and
Alberghini M: Molecular diagnosis in Ewing family tumors: the
Rizzoli experience-222 consecutive cases in four years. J Mol
Diagn. 13:313–324. 2011.PubMed/NCBI
|
|
174
|
Kojima T, Asami S, Chin M, Yoshida Y,
Mugishima H and Suzuki T: Detection of chimeric genes in Ewing’s
sarcoma and its clinical applications. Biol Pharm Bull. 25:991–994.
2002.
|
|
175
|
Davison JM, Morgan TW, Hsi BL, Xiao S and
Fletcher JA: Subtracted, unique-sequence, in situ hybridization:
experimental and diagnostic applications. Am J Pathol.
153:1401–1409. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
176
|
Cantile M, Marra L, Franco R, Ascierto P,
Liguori G, De Chiara A and Botti G: Molecular detection and
targeting of EWSR1 fusion transcripts in soft tissue tumors. Med
Oncol. 30:4122013. View Article : Google Scholar : PubMed/NCBI
|
|
177
|
Kumar S, Pack S, Kumar D, Walker R,
Quezado M, Zhuang Z, Meltzer P and Tsokos M: Detection of EWS-FLI-1
fusion in Ewing’s sarcoma/peripheral primitive neuroectodermal
tumor by fluorescence in situ hybridization using formalin-fixed
paraffin-embedded tissue. Hum Pathol. 30:324–330. 1999.
|
|
178
|
Hattinger CM, Rumpler S, Kovar H and
Ambros PF: Fine-mapping of cytogenetically undetectable EWS/ERG
fusions on DNA fibers of Ewing tumors. Cytogenet Cell Genet.
93:29–35. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
179
|
Newby R, Rowe D, Paterson L, Farquharson
MA, MacDuff E, Coupe A, Hale J, Dildey P and Bown N: Cryptic
EWSR1-FLI1 fusions in Ewing sarcoma: potential pitfalls in the
diagnostic use of fluorescence in situ hybridization probes. Cancer
Genet Cytogenet. 200:60–64. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
180
|
Mangham DC, Williams A, McMullan DJ,
McClure J, Sumathi VP, Grimer RJ and Davies AM: Ewing’s sarcoma of
bone: the detection of specific transcripts in a large, consecutive
series of formalin-fixed, decalcified, paraffin-embedded tissue
samples using the reverse transcriptase-polymerase chain reaction.
Histopathology. 48:363–376. 2006.
|
|
181
|
Park YK, Chi SG, Park HR, Yang MH and Unni
KK: Detection of t(11;22)(q24;q12) translocation of Ewing’s sarcoma
in paraffin embedded tissue by nested reverse
transcription-polymerase chain reaction. J Korean Med Sci.
13:395–399. 1998.PubMed/NCBI
|
|
182
|
Stegmaier S, Leuschner I, Aakcha-Rudel E,
Münch P, Kazanowska B, Bekassy A, Treuner J and Koscielniak E:
Identification of various exon combinations of the ews/fli1
translocation: an optimized RT-PCR method for paraffin embedded
tissue - a report by the CWS-study group. Klin Padiatr.
216:315–322. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
183
|
Hisaoka M, Tsuji S, Morimitsu Y, Hashimoto
H, Shimajiri S, Komiya S and Ushijima M: Molecular detection of
EWS-FLI1 chimeric transcripts in Ewing family tumors by nested
reverse transcription-polymerase chain reaction: application to
archival paraffin-embedded tumor tissues. APMIS. 107:577–584. 1999.
View Article : Google Scholar
|
|
184
|
Montanaro L, Pession A, Trerè D, Vici M,
Prete A, Paolucci G and Derenzini M: Detection of EWS chimeric
transcripts by nested RT-PCR to allow reinfusion of uncontaminated
peripheral blood stem cells in high-risk Ewing’s tumor in
childhood. Haematologica. 84:1012–1015. 1999.PubMed/NCBI
|
|
185
|
Yang Y, Zhang L, Wei Y, Wang H, Xiong W,
Chen Z, Hes O and Zheng J: Detection of EWSR1 translocation with
nuclear extraction-based fluorescence in situ hybridization for
diagnosis of Ewing’s sarcoma/primitive neuroectodermal tumor. Anal
Quant Cytol Histol. 29:221–230. 2007.PubMed/NCBI
|
|
186
|
Meier VS, Kühne T, Jundt G and Gudat F:
Molecular diagnosis of Ewing tumors: improved detection of
EWS-FLI-1 and EWS-ERG chimeric transcripts and rapid determination
of exon combinations. Diagn Mol Pathol. 7:29–35. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
187
|
Wang M, Nilsson G, Carlberg M, Dricu A,
Wejde J, Kreicbergs A and Larsson O: Specific and sensitive
detection of the EWS/FLI1 fusion protein in Ewing’s sarcoma by
western blotting. Virchows Arch. 432:131–134. 1998.
|
|
188
|
Silva DS, Sawitzki FR, De Toni EC, Graebin
P, Picanco JB, Abujamra AL, de Farias CB, Roesler R, Brunetto AL
and Alho CS: Ewing’s sarcoma: analysis of single nucleotide
polymorphism in the EWS gene. Gene. 509:263–266. 2012.
|
|
189
|
Lewis TB, Coffin CM and Bernard PS:
Differentiating Ewing’s sarcoma from other round blue cell tumors
using a RT-PCR translocation panel on formalin-fixed
paraffin-embedded tissues. Mod Pathol. 20:397–404. 2007.
|
|
190
|
Angervall L and Kindblom LG: Principles
for pathologic-anatomic diagnosis and classification of soft-tissue
sarcomas. Clin Orthop Relat Res. 289:9–18. 1993.PubMed/NCBI
|
|
191
|
Sreekantaiah C, Ladanyi M, Rodriguez E and
Chaganti RS: Chromosomal aberrations in soft tissue tumors.
Relevance to diagnosis, classification, and molecular mechanisms.
Am J Pathol. 144:1121–1134. 1994.PubMed/NCBI
|
|
192
|
Ladanyi M and Bridge JA: Contribution of
molecular genetic data to the classification of sarcomas. Hum
Pathol. 31:532–538. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
193
|
Bridge JA and Sandberg AA: Cytogenetic and
molecular genetic techniques as adjunctive approaches in the
diagnosis of bone and soft tissue tumors. Skeletal Radiol.
29:249–258. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
194
|
Singer S: New diagnostic modalities in
soft tissue sarcoma. Semin Surg Oncol. 17:11–22. 1999. View Article : Google Scholar : PubMed/NCBI
|