Introduction
The endoplasmic reticulum (ER) is an organelle in
eukaryotic cells that is involved in the synthesis, maturation,
quality control and trafficking of a wide range of proteins
(1, 2). Disruptions in ER function resulting
from increased protein synthesis, misfolded protein overload,
changes in calcium concentration or hypoxia induce ER stress and
activate the ER stress response [also known as the unfolded protein
response (UPR)] to restore ER homeostasis and protect the cell.
Mechanisms activated during the ER stress response include
translational attenuation, activation of ER chaperones, such as
immunoglobulin heavy chain-binding protein (BiP, also known as
GRP78) and the degradation of unfolded proteins (1,3).
Chronic or unresolved ER stress, however, can lead to apoptosis.
Apoptosis resulting from ER stress is mediated by factors including
C/EBP homologous protein (CHOP), IRE1α and caspase-12 (4–6).
Accumulating evidence indicates that ER stress-induced apoptosis
contributes to the pathogenesis of various diseases, such as type 2
diabetes mellitus (T2D), atherosclerosis, liver disease,
neurodegenerative disorders and cancer (2,7–12).
It has also been shown that many cellular regulatory
processes depend on the post-translational functions of ubiquitin
and ubiquitin-like proteins (Ubls), including transcription, DNA
repair, signal transduction, autophagy, cell proliferation,
differentiation, apoptosis, ER regulation, inflammation, antigen
processing and stress responses (13–15). Ubiquitin-fold modifier 1 (Ufm1)
has recently been identified as a novel Ubl with a molecular mass
of 9.1 kDa, and it appears to have a similar tertiary structure to
ubiquitin, despite having little amino acid sequence identity
(16). Similar to the process of
protein ubiquitination, Ufm1 is first synthesized in a pro-form and
cleaved at the C terminus by the specific cysteine proteases, UfSP1
and UfSP2, to expose the conserved glycine residue that is
essential for its subsequent conjugating reactions (17). The mature form of Ufm1 is
successively activated by a specific E1-activating enzyme (Uba5),
an E2-conjugating enzyme (Ufc1) and an E3-ligating enzyme (Ufl1),
and then it is covalently attached to a cellular target protein,
such as C20orf116 (also known as UFBP1), CDK5RAP3 and mitochondrial
trifunctional protein (MTP) (18–20). Finally, Ufm1 is cleaved by UfSPs
from the conjugation (16,18).
Previous studies have suggested that there is an
association between Ufm1 expression and the ER stress response in
mice suffering from ischemic heart injury and in the pancreatic
islets of an animal model of T2D (21,22). Recently, it has been shown that
Ufm1 expression is induced by ER stress and protects against ER
stress-induced apoptosis in the mouse pancreatic β-cell line,
INS-1E (19). Moreover, ER stress
upregulates the Ufm1 system in multiple cancer cell lines, and the
knockdown of the Ufm1 system results in the activation of the UPR
(23). These results indicate
that Ufm1 is linked to ER stress.
Macrophages are found in all tissues and have been
shown to play important roles in development, metabolic
homeostasis, tissue repair and immunity (24). These findings also indicated that
macrophages play important roles in T2D and atherosclerosis, and a
number of studies have focused on the role of macrophage ER
stress-induced apoptosis in diabetic macrovascular diseases, such
as atherosclerosis (25–28). The expression pattern and
potential biological function of Ufm1 in macrophages under ER
stress, however, remains unclear. Therefore, in the present study,
we evaluated Ufm1 expression in diabetic mouse resident peritoneal
macrophages (RPMs), as well as the effects of Ufm1 on the ER stress
response in the cultured mouse macrophage cell line, Raw264.7.
Furthermore, the lentiviral short hairpin RNA (shRNA)-mediated
knockdown of Ufm1 increased the apoptosis of Raw264.7 cells exposed
to ER stress inducers and these cells had higher expression levels
of BiP and CHOP, which are markers of the ER stress response. The
overexpression of Ufm1 by the lentiviral infection of Raw264.7
cells treated with ER stress inducers resulted in the opposite
effect. Thus, our results demonstrate that Ufm1 suppresses
macrophage apoptosis by inhibiting the ER stress response in
vitro.
Materials and methods
Materials
Dulbecco’s modified Eagle’s medium (DMEM), fetal
bovine serum (FBS), penicillin, streptomycin, phosphate-buffered
saline (PBS) and puromycin were purchased from Gibco BRL (Grant
Island, NY, USA). Lipofectamine 2000, Opti-MEM and TRIzol reagent
were obtained from Invitrogen (Carlsbad, CA, USA). The GoScript™
Reverse Transcription kit and the GoTaq® qPCR Master mix
kit were purchased from Promega Corp. (Madison, WI, USA).
Ethylenediaminetetraacetic acid (EDTA), trypsin, thapsigargin (TG)
and tunicamycin (TM) were obtained from Sigma-Aldrich (St. Louis,
MO, USA).
Animals
For our experiments, 8- to 12-week-old male db/db
(C57BL/6J-Leprdb/Leprdb) mice and littermate
control db/m (C57BL/6J background) mice were purchased from the
Model Animal Research Center of Nanjing University (Nanjing, China)
and fed standard chow (LabDiet 5001; LabDiet, St. Louis, MO, USA).
All the mice were kept in a specific pathogen-free facility and
allowed free access to food and water. The experiments were
performed in strict accordance with the Institutional Guidelines
for the Care and Use of Laboratory Animals of Shanghai Jiao Tong
University (Shanghai, China).
Isolation and culture of mouse RPMs
RPMs from db/db mice and db/m mice were isolated 3
days following an intraperitoneal injection of 4% thioglycollate
solution, as previously described (29). Briefly, approximately 5 ml of cold
serum-free DMEM were injected intraperitoneally after the mice
under were euthanized ether anesthesia. Following a gentle massage,
the peritoneal fluid containing the cells was harvested and
centrifuged at 700 × g for 5 min at 4°C. The pellet was resuspended
in 5 ml of DMEM containing 10% FBS, 100 U/ml penicillin and 100
μg/ml streptomycin and then cultured at 37°C in a 60-mm dish
(Corning Life Sciences, Oneonta, NY, USA) for 1 h. Only the
attached macrophages were used for the following experiments.
Cell culture
The human embryonic kidney 293T (HEK293T) cell line
and mouse macrophage cell line, Raw264.7, were obtained from the
American Type Culture Collection (ATCC; Rockville, MD, USA). The
cells were cultured in DMEM supplemented with 10% FBS, 100 U/ml
penicillin and 100 μg/ml streptomycin in a humidified atmosphere of
5% CO2 at 37°C. When the cells reached approximately
70–80% confluency, they were dissociated with trypsinization and
subcultured.
RNA isolation and quantitative reverse
transcription-polymerase chain reaction (qRT-PCR)
The cells were collected and washed with ice-cold
PBS. Total RNA was isolated from the cells using TRIzol reagent.
cDNA was synthesized using the GoScript™ Reverse Transcription
System according to the manufacturer’s instructions and then used
as the template for qPCR. The GoTaq® qPCR Master mix kit
was used for the qPCR assays. The thermal cycling conditions
consisted of 2 min at 95°C, followed by 40 cycles of 15 sec at 95°C
and 1 min at 60°C. Each sample was run in duplicate on an ABI ViiA
7 Real-Time PCR system and analyzed by the relevant software. The
2−ΔΔCT method was used to analyze the relative changes
in gene expression. Either GAPDH or β-actin was used as an internal
standard. The following primers for each target gene were used:
Ufm1 forward, 5′-TTCCTG CAGCTACAAGTGCG-3′ and reverse,
5′-TCCAACTCGGT CTCTAGGAATGAT-3′; β-actin forward, 5′-TGTGACGTT
GACATCCGTAAAGAC-3′ and reverse, 5′-TCCACACAGAG TACTTGCGCTC-3′; BiP
forward, 5′-GAGTTCTTCAA TGGCAAGGAGC-3′ and reverse,
5′-GGACAAACATCAA GCAGTACCAGAT-3′; CHOP forward, 5′-CTCATCCCCA
GGAAACGAAGAG-3′ and reverse, 5′-TTGGGATGTGCG TGTGACC-3′; and GAPDH
forward, 5′-TGGTGAAGGT CGGTGTGAAC-3′ and reverse, 5′-GCTCCTGGAAGAT
GGTGATGG-3′;
Western blot analysis
The cells were harvested in RIPA lysis buffer
supplemented with phenylmethanesulfonyl fluoride (PMSF) and
protease inhibitor cocktail. Proteins were separated by 10–15%
SDS-PAGE, transferred onto PVDF membranes, and stained with the
following antibodies: anti-Ufm1 (1:10,000; Abcam, Cambridge, MA,
USA), anti-BiP (1:1,000), anti-CHOP (1:1,000), anti-cleaved
caspase-3 (1:1,000) (all from Cell Signaling Technology, Inc.,
Danvers, MA, USA), anti-GAPDH (1:5,000; Santa Cruz Biotechnology,
Inc., Santa Cruz, CA, USA) and anti-β-actin (1:10,000; Cell
Signaling Technology). Secondary antibodies conjugated to
horseradish peroxidase and ECL western blot analysis reagents
(Millipore Corp., Billerica, MA, USA) were used for detection.
Immunoreactive bands were quantified using Gel-Pro 32 software
(Media Cybernetics, Inc., Rockville, MD, USA).
Construction of recombinant lentivirus
and cell infection
Three candidate small interfering RNA (siRNA)
sequences for mouse Ufm1 were designed following the procedure from
the Dharmacon siDESIGN Center: siRNA-1 (5′-CGGCTCAGAA
CTGAGAATCAT-3′), siRNA-2 (5′-CGCCGTTCACAGCAGT GCTAA-3′), and
siRNA-3 (5′-AGTTGGAAGCTGCTAAT ATAT-3′). Scrambled siRNA
(5′-TTCTCCGAACGTGTC ACGT-3′) was used as the negative control.
shRNAs that corresponded to the siRNA sequences for Ufm1 and the
negative control were then generated. These shRNAs were inserted
into the lentiviral vector, pFU-GW-RNAi (Genechem Co., Ltd.,
Shanghai, China). The vectors containing the Ufm1-shRNA sequences
were designated as pFU-GW-RNAi-Ufm1 and the vector containing the
negative control was designated as pFU-GW-RNAi-NC. Lentiviruses
were generated by the triple transfection of 80% confluent HEK293T
cells with the modified pFU-GW-RNAi plasmid and pHelper 1.0 and
pHelper 2.0 helper plasmids (Genechem Co.) using Lipofectamine 2000
according to the manufacturer’s instructions. Lentiviruses were
harvested 72 h after transfection. The lentiviruses were purified
using ultracentrifugation and the titers of the lentiviruses were
determined. The Raw264.7 cells were infected with either the
Ufm1-shRNA lentivirus (Lv-shUfm1) or the negative control
lentivirus (Lv-shNC).
The sequence of the mouse Ufm1 gene was obtained
from GenBank (NM_026435) and oligonucleotides were designed and
synthesized based on this sequence. A restriction enzyme site
(NotI and NsiI) was added to each end of the
oligonucleotides. PCR was performed to connect the synthesized
oligonucleotides into a complete sequence. Following a
NotI/NsiI (Fermentas, Glen Burnie, MD, USA)
digestion, the produced fragment was ligated into the pLV5 vector
(GenePharma Co.), producing the pLV5-UFM1 construct. The pLV5
vector was used as a negative control. The lentiviral vector
(pLV5-UFM1 or pLV5) was added to Opti-MEM, and then packing
plasmids (pGag/Pol, pRev, pVSV-G; GenePharma Co.) and Lipofectamine
2000 were added sequentially. The resulting mixture was
co-transfected into the HEK293T cells, which were incubated for 72
h. The supernatant was collected, centrifuged and filtered, and the
titers of the lentiviruses were determined. The Raw264.7 cells were
then infected with either the Ufm1 lentivirus (Lv-Ufm1) or the
negative control lentivirus (Lv-NC).
Detection of apoptosis by flow
cytometry
Apoptosis was analyzed by double staining with
Annexin V-PE and 7-AAD (BD Biosciences, Franklin Lakes, NJ, USA),
which was detected by a FACSCalibur flow cytometer (BD
Biosciences). Briefly, the cells were stained with 100 μl binding
buffer containing 5 μl Annexin V-PE and 5 μl 7-AAD at room
temperature in the dark for 10–15 min. The data were analyzed using
CellQuest software (BD Biosciences).
Statistical analysis
All data are expressed as the means ± standard
deviation (SD) of 2 or 3 independent experiments. The Student’s
t-test was used to evaluate the differences in each group using
SPSS 17.0 software. A value of p<0.05 was considered to indicate
a statistically significant difference.
Results
Increase in Ufm1 expression in the RPMs
of db/db mice
To investigate the functional role of Ufm1 in T2D
macrophages, we determined the Ufm1 mRNA and protein expression
levels in the RPMs from the db/db mice and the littermate controls,
db/m mice. We first identified the RPMs using the previously
reported surface antigen F4/80+ detection technique
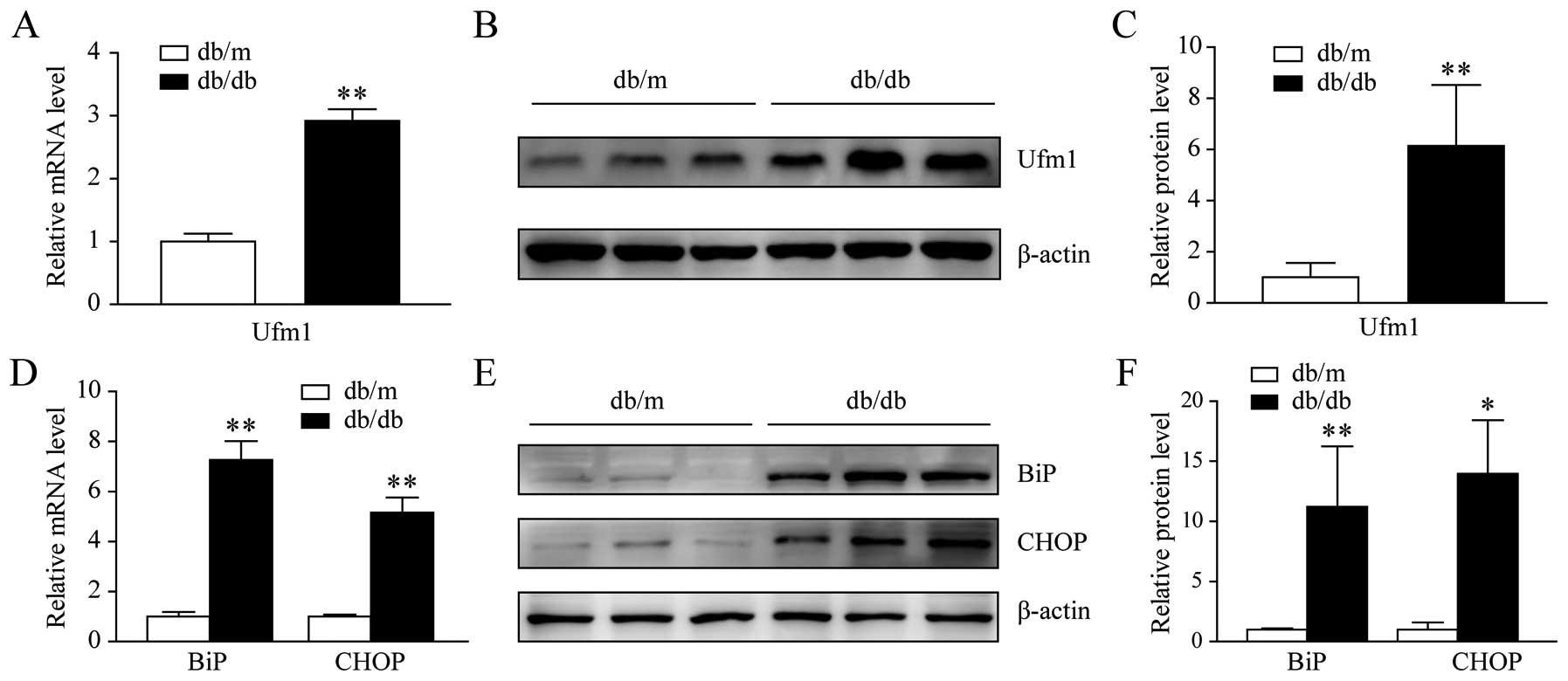
(29). Total RNA and protein were
extracted from the RPMs of the db/db and db/m mice. The results
from qRT-PCR and western blot analysis showed that there was a
significant increase in the Ufm1 mRNA and protein expression levels
(Fig. 1A–C). We also determined
the expression levels of BiP and CHOP, which are markers of the ER
stress response. As shown in Fig.
1D–F, their expression levels in the RPMs from the db/db mice
were higher than those from the db/m mice. These results indicated
that the Ufm1 mRNA and protein levels were higher in the diabetic
mice and that the ER stress response was activated in the RPMs from
the db/db mice.
Ufm1 expression is upregulated by ER
stress inducers in Raw264.7 cells
It has been reported that the Ufm1 expression level
is higher in animal models and cultured cells under ER stress
(19,21–23); however, the effects of ER stress
on Ufm1 expression in macrophages remain unknown. In this study, to
investigate the association between Ufm1 expression and macrophage
ER stress, we examined whether Ufm1 expression is affected by ER
stress in Raw264.7 cells. We used TG, an inhibitor of ER
Ca2+ ATPase, and TM, an inhibitor of glycosylation, to
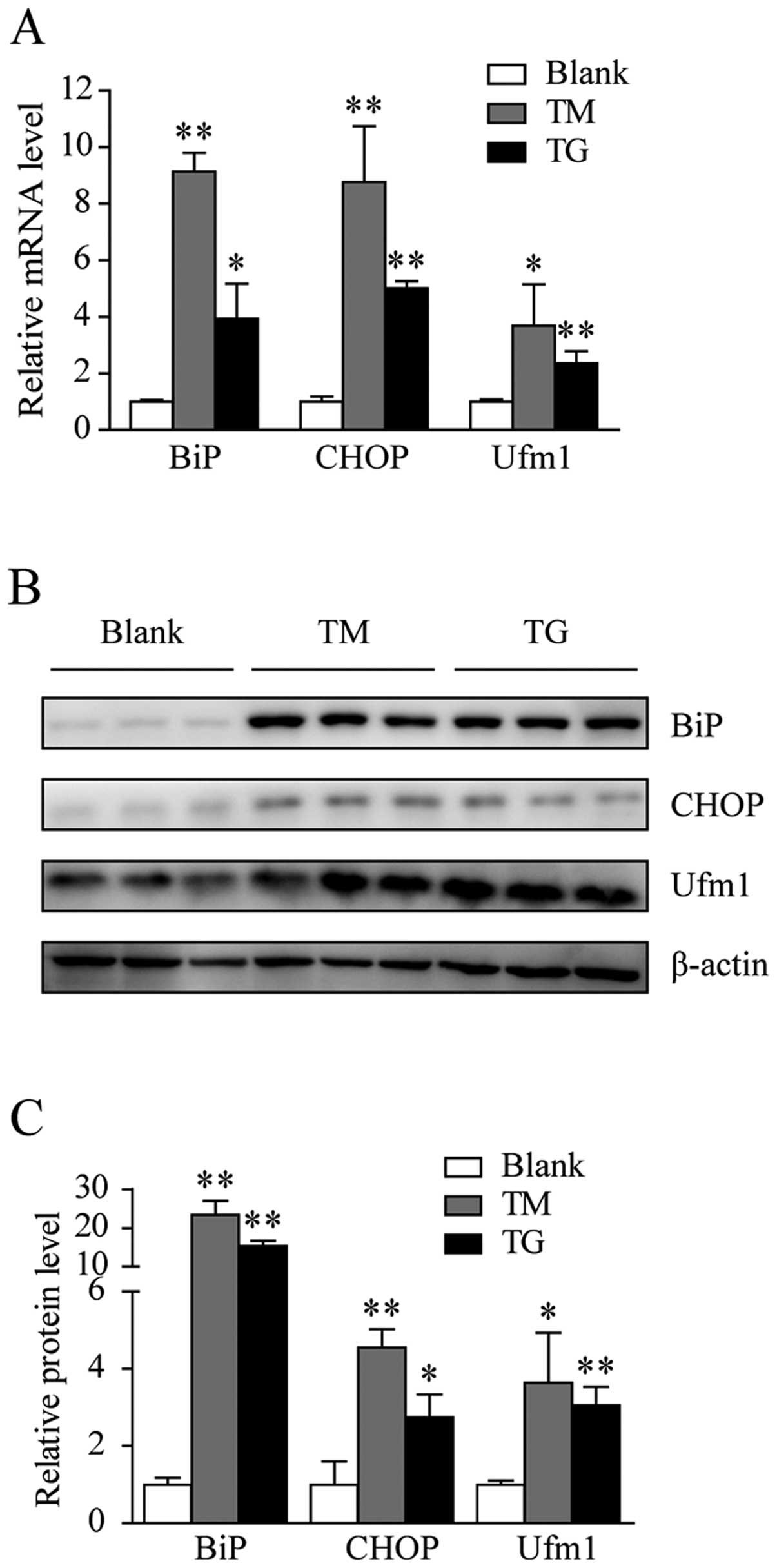
induce the ER stress response in Raw264.7 cells. As shown in
Fig. 2A, Ufm1 mRNA expression
increased by >2-fold in the Raw264.7 cells treated with 0.5 μM
TG for 12 h and by approximately 4-fold in those treated with 8
μg/ml TM for 12 h. In addition, the Ufm1 protein level increased
(Fig. 2B and C). To confirm that
the Raw264.7 cells treated with either TG or TM were under ER
stress, we also examined the BiP and CHOP expression levels. As
expected, their expression levels had markedly increased (Fig. 2A–C), and this result is consistent
with the results previously discussed in this study. Taken
together, these data suggest that an upregulation in Ufm1
expression is associated with the ER stress response.
ER stress-induced apoptosis increases
following the knockdown of Ufm1 expression in Raw264.7 cells
Apoptosis is triggered when the ER stress response
fails to restore ER homeostasis. To further analyze the
contributions of Ufm1 to the regulation of macrophage ER stress, as
well as the effects of a reduced Ufm1 expression level on
macrophage function, we examined the ER stress response and ER
stress-induced apoptosis in Raw264.7 cells in which Ufm1 expression
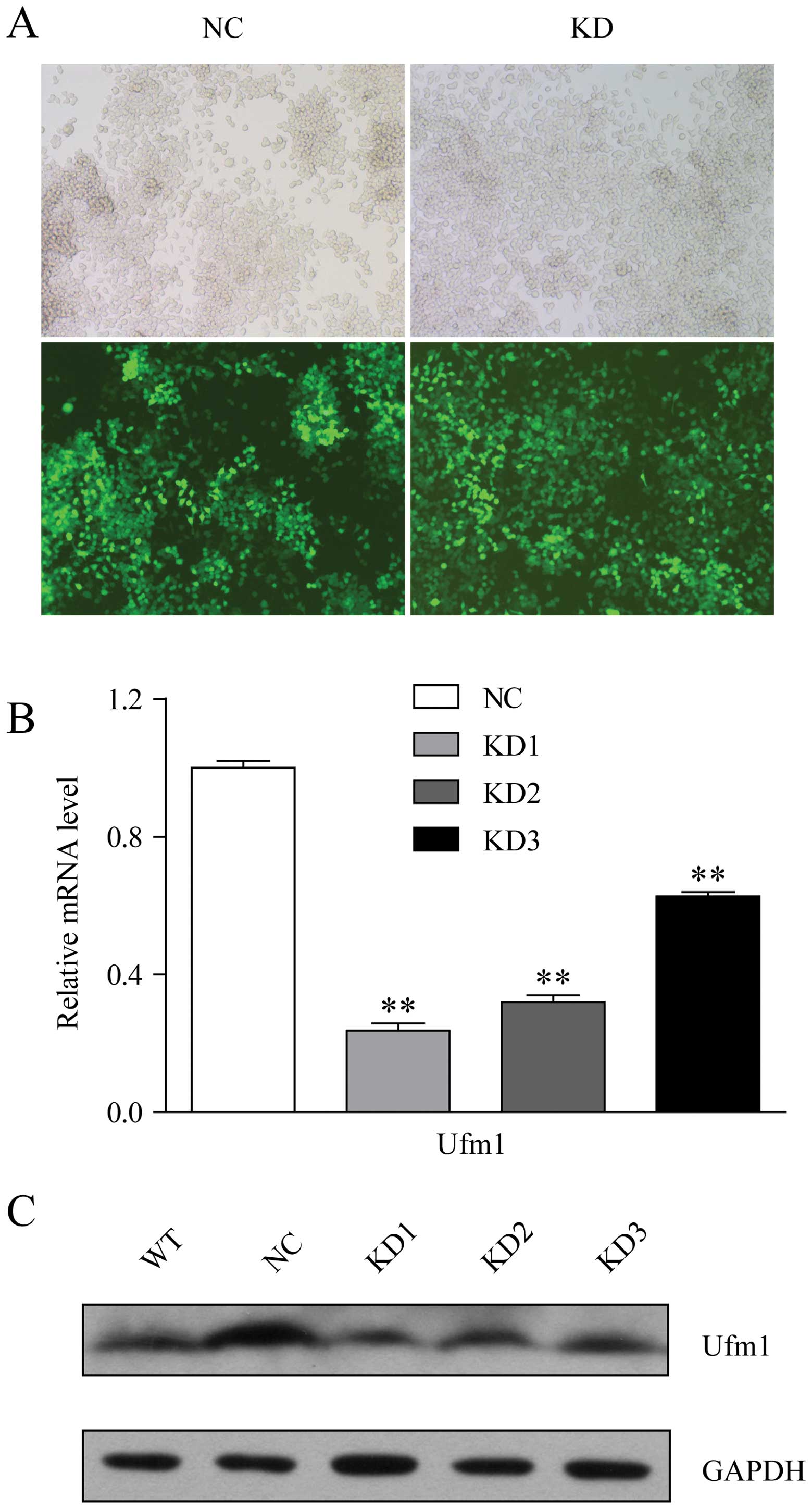
had been knocked down by lentivirus-mediated shRNA. Following viral
infection, >90% of the cells were eGFP-positive, which indicated
a high efficiency of infection (Fig.
3A). Among the 3 shRNAs examined, shUfm1–1 was the most
effective in reducing the Ufm1 expression level (Fig. 3B and C). Therefore, this shRNA
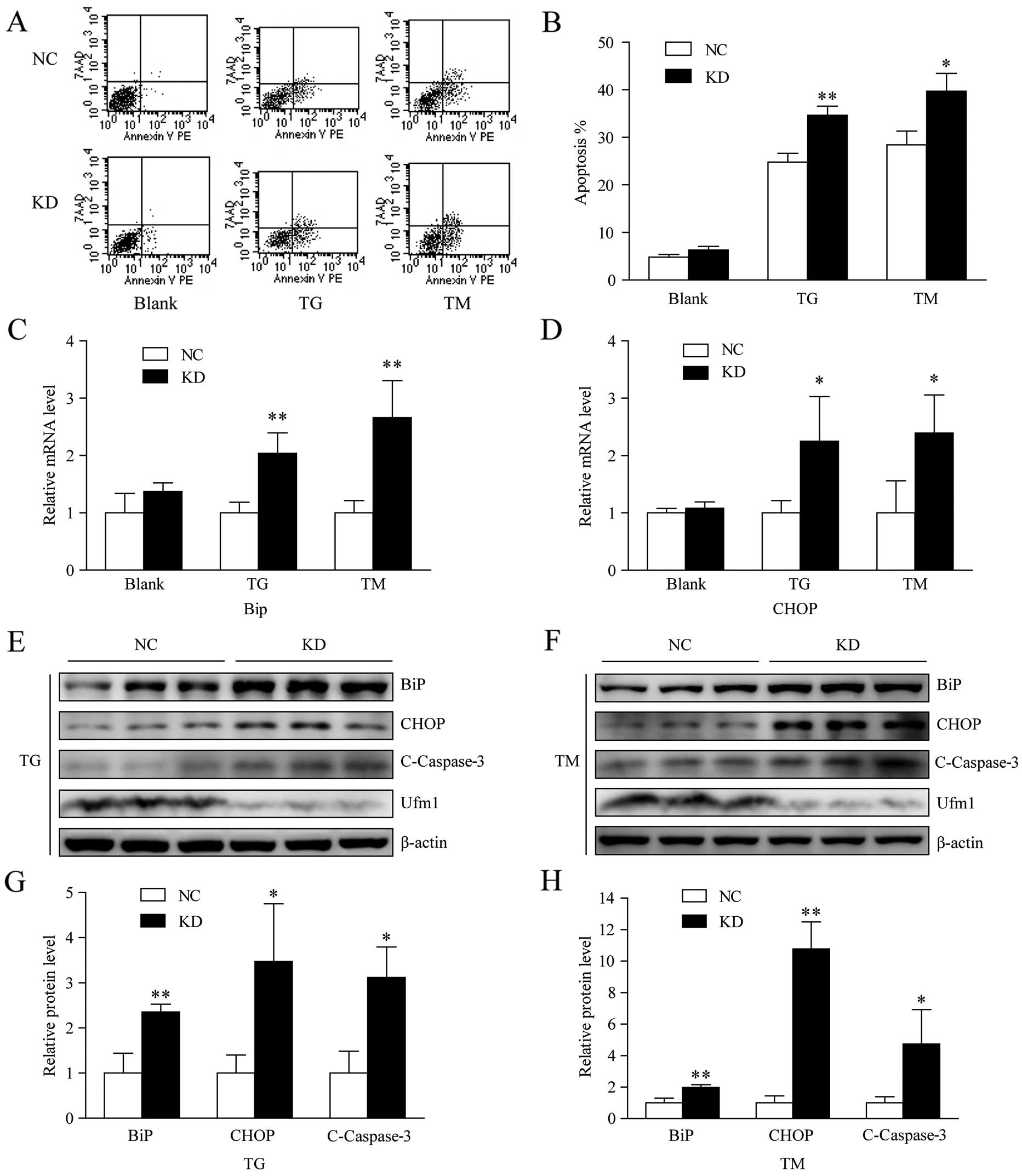
sequence was selected for further study. According to the results
from flow cytometry, decreasing the Ufm1 expression level markedly
enhanced TG- and TM-induced apoptosis by 10 and 12%, respectively,
but it had no significant effect on the basal apoptosis when
compared with the negative control (Fig. 4A and B). The sensitization of
macrophages to ER stress-induced apoptosis was also confirmed by
the level of cleaved caspase-3 detected by western blot analysis
(Fig. 4E–H). Furthermore, when
the Ufm1 expression was knocked down, there was no effect on the
expression levels of BiP and CHOP under no treatment conditions
(Fig. 4C and D), but after the
cells were treated with TG or TM, there was a higher expression
level of BiP and CHOP compared with the negative control (Fig. 4C–H). These results indicate that
Ufm1 knockdown sensitizes macrophages to ER stress. Taken together,
these data demonstrate that Ufm1 is at least partially involved in
regulating ER stress-induced apoptosis.
Overexpression of Ufm1 prevents ER
stress-induced apoptosis in Raw264.7 cells
To further substantiate the effects of Ufm1 on ER
stress-induced macrophage apoptosis, we constructed a lentiviral
vector system that expresses Ufm1 cDNA and GFP and is puromycin
resistant. The Raw264.7 cells were transfected with the Lv-Ufm1 or
Lv-NC vectors, and after 4 days of transfection, >80% of the
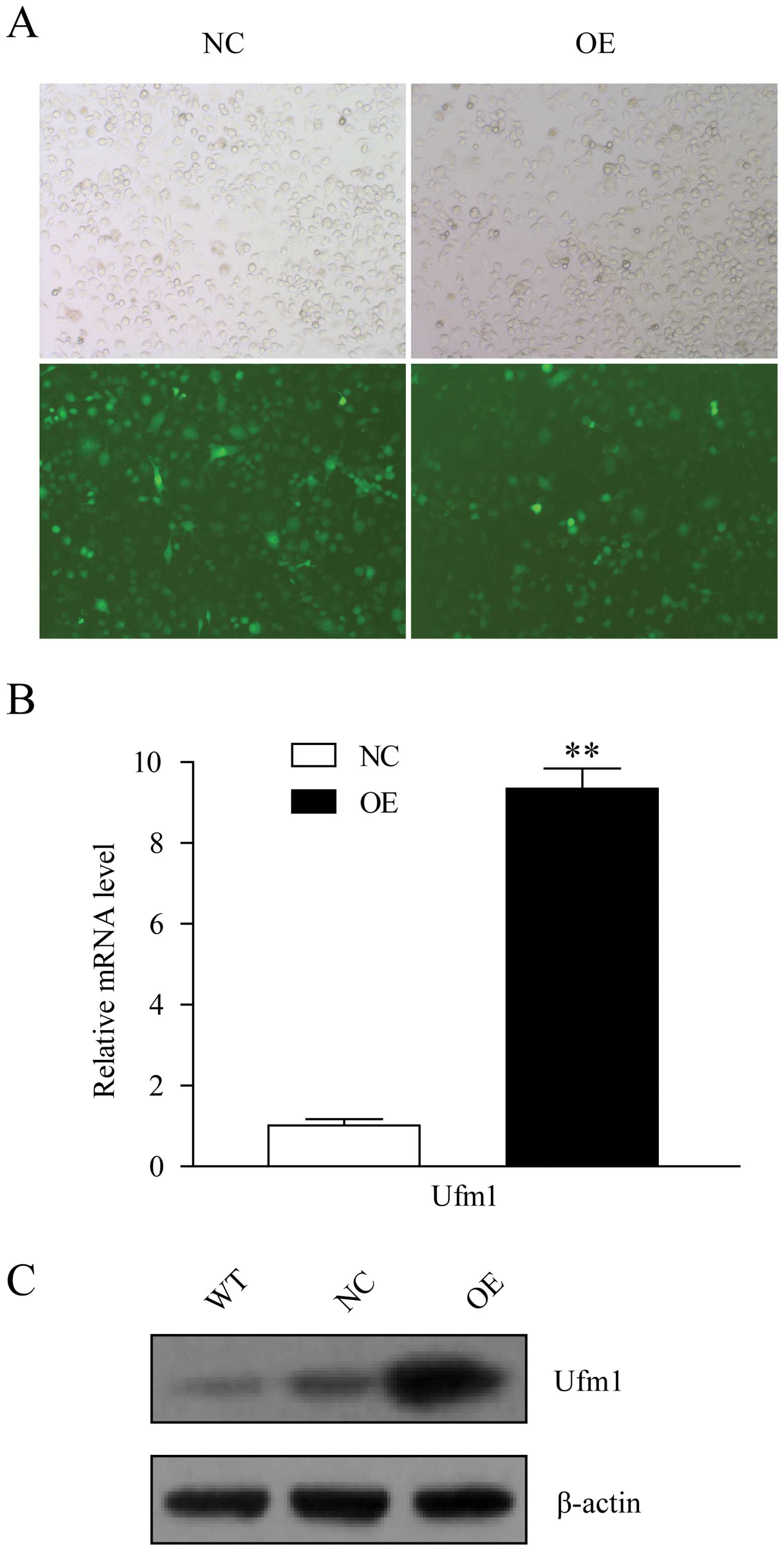
cells expressed GFP (Fig. 5A),
which indicated that the lentiviral infections were effective.
After another 5 days of 4 μg/ml puromycin selection, the Ufm1 mRNA
and protein expression levels in the Lv-Ufm1-infected cells were
markedly increased, when compared with those in the Lv-NC-infected
cells (Fig. 5B and C). To
determine whether the overexpression of Ufm1 affects ER
stress-induced macrophage apoptosis, the Lv-Ufm1 and Lv-NC cells
were exposed to 0.5 μM TG or 8 μg/ml TM for 12 h, and the
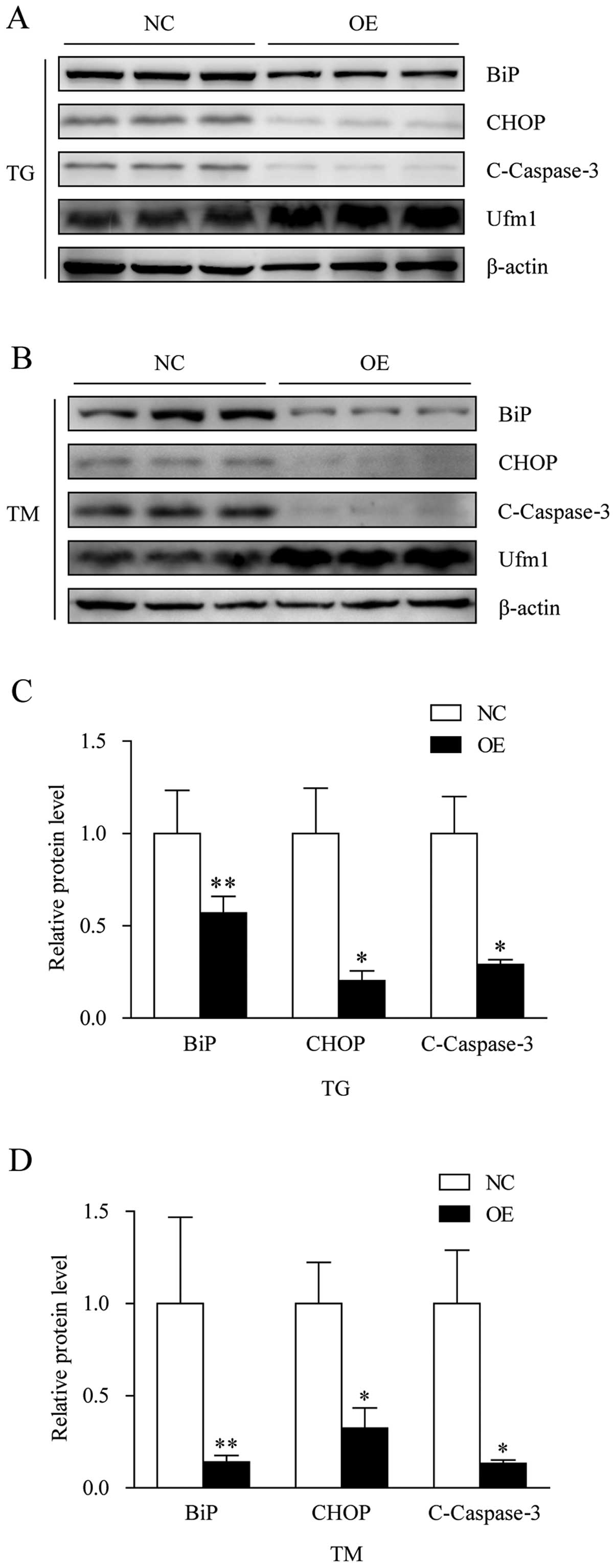
expression levels of BiP, CHOP and cleaved caspase-3 were
determined. As predicted, in comparison with the negative control,
the overexpression of Ufm1 attenuated macrophage ER stress and
prevented the apoptosis induced by exposure to TG or TM (Fig. 6A–D).
Discussion
In the present study, we investigated the expression
pattern and potential biological function of Ufm1 in macrophages.
We found that Ufm1 expression was markedly upregulated in diabetic
RPMs, and its expression increased in response to ER stress
inducers in cultured macrophages. Further investigation revealed
that ER stress-induced macrophage apoptosis increased after the
knockdown of Ufm1 and that the overexpression of Ufm1 attenuated ER
stress-induced macrophage apoptosis.
Ufm1 is a new member of the Ubl family, whose
biological functions are poorly understood, particularly in
macrophages. It has been reported that Ufm1 is expressed in many
tissues and cell lines (16,19) and that an upregulation in Ufm1
expression is linked with the activation of the ER stress response
in ischemic heart injury and T2D (21,22). Consistent with these previous
studies, our results demonstrate that Ufm1 is expressed in RPMs
from normal mice and that its expression is upregulated in RPMs
from diabetic mice. Additionally, the expression levels of BiP and
CHOP were upregulated in the RPMs from diabetic mice compared with
those from the control mice. Elevated BiP and CHOP expression
levels may potentially result from insulin resistance or glucose
and lipid metabolic abnormalities, which are known ER stress
inducers and may contribute to impeding macrophage ER function.
These findings are also consistent with those of our previous
studies using cDNA microarray expression profiling in RPMs from
diabetic mice (unpublished data). Chemicals such as TG and TM are
commonly used to induce the ER stress response in cultured cells
and animals for experimental purposes (30). In our study, we used TG and TM to
induce the ER stress response in the Raw264.7 macrophage cell line.
Following exposure to TG or TM, the Ufm1 mRNA and protein
expression levels increased; these results are consistent with
those of previous studies (19,23) and the results presented above. The
BiP and CHOP expression levels were also higher in the TG- or
TM-treated groups, which indicated that the ER stress response was
activated in these cells. Taken together, these findings suggest
that Ufm1 is associated with macrophage ER stress.
It has been reported that prolonged ER stress can
trigger apoptosis. CHOP is a member of the C/EBP family of bZIP
transcription factors (5).
Mounting evidence suggests that CHOP plays an essential role in
regulating macrophage ER stress-induced apoptosis, including the
release of ER calcium, the mitochondrial release of apoptogens and
the activation of the death receptor, Fas (31–35). Caspase-3 is a well-known apoptosis
effector, and it has been reported to be involved in ER
stress-induced apoptosis (36,37). Based on our findings that Ufm1
expression was upregulated by ER stress inducers and that the Ufm1
expression pattern was similar to that of several UPR genes and the
known functions of activated CHOP, we initially hypothesized that
Ufm1 may be a negative regulator in macrophage ER stress-induced
apoptosis and that the knockdown of Ufm1 may reduce the amount of
ER stress and apoptosis observed following treatment with TG or TM.
To our surprise, however, our flow cytometry and western blot
analysis results revealed that the knockdown of Ufm1 expression
exacerbated ER stress-induced apoptosis in the cultured
macrophages. To confirm this finding, we established an Ufm1
overexpression model. The overexpressing of Ufm1 significantly
reduced the BiP and CHOP expression levels, as well as the
cleaved-caspase-3 expression level under ER stress conditions and
reduced ER stress-induced apoptosis. These results are slightly
different from those of a previous study in which there was no
change in the UPR when the Ufm1 expression was knocked down in
INS-1E cells, even though Ufm1 knockdown sensitized β-cells to
apoptosis (19). Our results,
however, are similar to the findings of a recent study by Zhang
et al (23). The authors
found that knocking down the expression of Ufm1 components in U2OS
cells triggered the activation of the UPR and amplified the ER
network (23). This discrepancy
between studies may be attributed to differences in the protocols
used in each study, such as the cell lines, chemical inducers, RNA
interference and target gene knockdown efficiency. Additionally,
Ufm1 is expressed in many tissues and cells under physiological
conditions (16,19), and its Uba5 plays a critical role
in cell differentiation (38),
which provides indirect evidence that Ufm1 may play a key role in
normal cellular function and that Ufm1 knockdown may affect
macrophage function. Taken together, these findings establish a
link between Ufm1 and ER stress and suggest that Ufm1 is a novel
protective gene whose expression is upregulated in response to ER
stress-induced macrophage apoptosis. Further studies are required
to elucidate the precise mechanisms involved.
In conlcusion, our study demonstrates that Ufm1
expression is involved in macrophage ER stress and that Ufm1
inhibits apoptosis by suppressing the ER stress response in
macrophages. These results demonstrate the significance of Ufm1 in
regulating macrophage function during ER stress, indicating that an
increase in Ufm1 expression protects a compensatory response in
diabetic mice and suggests that Ufm1 be considered as a potential
therapeutic target against diabetes.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (grant no. 81270908). The authors would
like to acknowledge the expert assistance provided by Professor
Jinke Cheng (Department of Biochemistry and Molecular Cell Biology,
School of Medicine, Shanghai Jiao Tong University, Shanghai,
China).
References
|
1
|
Schröder M and Kaufman RJ: The mammalian
unfolded protein response. Annu Rev Biochem. 74:739–789. 2005.
|
|
2
|
Hotamisligil GS: Endoplasmic reticulum
stress and the inflammatory basis of metabolic disease. Cell.
140:900–917. 2010. View Article : Google Scholar
|
|
3
|
Ron D and Walter P: Signal integration in
the endoplasmic reticulum unfolded protein response. Nat Rev Mol
Cell Biol. 8:519–529. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Nakagawa T, Zhu H, Morishima N, et al:
Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and
cytotoxicity by amyloid-beta. Nature. 403:98–103. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Oyadomari S and Mori M: Roles of
CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ.
11:381–389. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tabas I and Ron D: Integrating the
mechanisms of apoptosis induced by endoplasmic reticulum stress.
Nat Cell Biol. 13:184–190. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ozcan L and Tabas I: Role of endoplasmic
reticulum stress in metabolic disease and other disorders. Annu Rev
Med. 63:317–328. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hotamisligil GS: Endoplasmic reticulum
stress and atherosclerosis. Nat Med. 16:396–399. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Back SH and Kaufman RJ: Endoplasmic
reticulum stress and type 2 diabetes. Annu Rev Biochem. 81:767–793.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kammoun HL, Chabanon H, Hainault I, et al:
GRP78 expression inhibits insulin and ER stress-induced SREBP-1c
activation and reduces hepatic steatosis in mice. J Clin Invest.
119:1201–1215. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Imai Y, Soda M and Takahashi R: Parkin
suppresses unfolded protein stress-induced cell death through its
E3 ubiquitin-protein ligase activity. J Biol Chem. 275:35661–35664.
2000. View Article : Google Scholar
|
|
12
|
Lee AS: GRP78 induction in cancer:
therapeutic and prognostic implications. Cancer Res. 67:3496–3499.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Weissman AM: Themes and variations on
ubiquitylation. Nat Rev Mol Cell Biol. 2:169–178. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kerscher O, Felberbaum R and Hochstrasser
M: Modification of proteins by ubiquitin and ubiquitin-like
proteins. Annu Rev Cell Dev Biol. 22:159–180. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hochstrasser M: Origin and function of
ubiquitin-like proteins. Nature. 458:422–429. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Komatsu M, Chiba T, Tatsumi K, et al: A
novel protein-conjugating system for Ufm1, a ubiquitin-fold
modifier. EMBO J. 23:1977–1986. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kang SH, Kim GR, Seong M, et al: Two novel
ubiquitin-fold modifier 1 (Ufm1)-specific proteases, UfSP1 and
UfSP2. J Biol Chem. 282:5256–5262. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tatsumi K, Sou YS, Tada N, et al: A novel
type of E3 ligase for the Ufm1 conjugation system. J Biol Chem.
285:5417–5427. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lemaire K, Moura RF, Granvik M, et al:
Ubiquitin fold modifier 1 (UFM1) and its target UFBP1 protect
pancreatic beta cells from ER stress-induced apoptosis. PLoS One.
6:e185172011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gannavaram S, Connelly PS, Daniels MP,
Duncan R, Salotra P and Nakhasi HL: Deletion of mitochondrial
associated ubiquitin fold modifier protein Ufm1 in Leishmania
donovani results in loss of beta-oxidation of fatty acids and
blocks cell division in the amastigote stage. Mol Microbiol.
86:187–198. 2012. View Article : Google Scholar
|
|
21
|
Azfer A, Niu J, Rogers LM, Adamski FM and
Kolattukudy PE: Activation of endoplasmic reticulum stress response
during the development of ischemic heart disease. Am J Physiol
Heart Circ Physiol. 291:H1411–H1420. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lu H, Yang Y, Allister EM, Wijesekara N
and Wheeler MB: The identification of potential factors associated
with the development of type 2 diabetes: a quantitative proteomics
approach. Mol Cell Proteomics. 7:1434–1451. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang Y, Zhang M, Wu J, Lei G and Li H:
Transcriptional regulation of the Ufm1 conjugation system in
response to disturbance of the endoplasmic reticulum homeostasis
and inhibition of vesicle trafficking. PLoS One. 7:e485872012.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wynn TA, Chawla A and Pollard JW:
Macrophage biology in development, homeostasis and disease. Nature.
496:445–455. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Senokuchi T, Liang CP, Seimon TA, et al:
Forkhead transcription factors (FoxOs) promote apoptosis of
insulin-resistant macrophages during cholesterol-induced
endoplasmic reticulum stress. Diabetes. 57:2967–2976. 2008.
View Article : Google Scholar
|
|
26
|
Liang CP, Han S, Li G, Tabas I and Tall
AR: Impaired MEK signaling and SERCA expression promote ER stress
and apoptosis in insulin-resistant macrophages and are reversed by
exenatide treatment. Diabetes. 61:2609–2620. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Seimon TA, Nadolski MJ, Liao X, et al:
Atherogenic lipids and lipoproteins trigger CD36-TLR2-dependent
apoptosis in macrophages undergoing endoplasmic reticulum stress.
Cell Metab. 12:467–482. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tabas I: The role of endoplasmic reticulum
stress in the progression of atherosclerosis. Circ Res.
107:839–850. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Liu HF, Zhang HJ, Hu QX, et al: Altered
polarization, morphology, and impaired innate immunity germane to
resident peritoneal macrophages in mice with long-term type 2
diabetes. J Biomed Biotechnol. 2012:8670232012.PubMed/NCBI
|
|
30
|
Yoshida H: ER stress and diseases. FEBS J.
274:630–658. 2007. View Article : Google Scholar
|
|
31
|
Ozcan L and Tabas I: Pivotal role of
calcium/calmodulin-dependent protein kinase II in ER stress-induced
apoptosis. Cell Cycle. 9:223–224. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Timmins JM, Ozcan L, Seimon TA, et al:
Calcium/calmodulin-dependent protein kinase II links ER stress with
Fas and mitochondrial apoptosis pathways. J Clin Invest.
119:2925–2941. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
33
|
Li G, Mongillo M, Chin KT, et al: Role of
ERO1-alpha-mediated stimulation of inositol 1,4,5-triphosphate
receptor activity in endoplasmic reticulum stress-induced
apoptosis. J Cell Biol. 186:783–792. 2009. View Article : Google Scholar
|
|
34
|
Thorp E, Li G, Seimon TA, Kuriakose G, Ron
D and Tabas I: Reduced apoptosis and plaque necrosis in advanced
atherosclerotic lesions of Apoe−/− and Ldlr−/− mice lacking CHOP.
Cell Metab. 9:474–481. 2009.
|
|
35
|
Tsukano H, Gotoh T, Endo M, et al: The
endoplasmic reticulum stress-C/EBP homologous protein
pathway-mediated apoptosis in macrophages contributes to the
instability of atherosclerotic plaques. Arterioscler Thromb Vasc
Biol. 30:1925–1932. 2010. View Article : Google Scholar
|
|
36
|
Song L, De Sarno P and Jope RS: Central
role of glycogen synthase kinase-3beta in endoplasmic reticulum
stress-induced caspase-3 activation. J Biol Chem. 277:44701–44708.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Morishima N, Nakanishi K, Takenouchi H,
Shibata T and Yasuhiko Y: An endoplasmic reticulum stress-specific
caspase cascade in apoptosis. Cytochrome c-independent activation
of caspase-9 by caspase-12. J Biol Chem. 277:34287–34294. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Tatsumi K, Yamamoto-Mukai H, Shimizu R, et
al: The Ufm1-activating enzyme Uba5 is indispensable for erythroid
differentiation in mice. Nat Commun. 2:1812011. View Article : Google Scholar : PubMed/NCBI
|