Introduction
Orofacial clefts (OFCs), primarily cleft lip and
cleft palate, are among the most common congenital anomalies in
individuals in China and worldwide (1). The etiology of clefts in humans is a
complex interaction between genetic and environmental factors
(2). Nonetheless, our knowledge
of their etiology and pathogenesis remains scarce, limiting our
efforts at prevention (3). In
many cases, the underlying molecular and cellular mechanisms that
result in these debilitating anomalies remain largely unknown
(4). OFCs lead to many
complications including speech, hearing and feeding problems that
require a team of maxillofacial and oral surgeons, head and neck
surgeons, speech therapists and phoniatricians for treatment
(2). Although surgical treatment
has improved over the centuries, facial scarring remains a problem.
Facial scars lead to cosmetic problems as well as to psychological
effects such as poor self-esteem, emotional debilitation,
embarrassment, and social isolation (5). Therefore, identifying ways to reduce
scar formation is of utmost significance.
Scars are formed via the transformation of
fibroblasts into myofibroblasts, accompanied by the proliferation
of fibroblasts and excessive deposition of extracellular matrix
(ECM) (6).Fibroblasts derived
from surgical scar tissue produce high levels of α-smooth muscle
actin (α-SMA) and transforming growth factor (TGF)-β1 (7). TGF-β1 is one of the key cytokines in
scar formation, and can act at different levels to increase
collagen deposition. TGF-β1 promotes fibroblast transformation into
myofibroblasts, induces the synthesis of glycoproteins and matrix
proteins, and inhibits collagen degradation by the reduction of
metalloproteases and induction of protease inhibitors (8). Various anti-TGF-β approaches have
been successfully used to prevent fibrotic disorders of the skin
and to reduce scar formation.
The CCN protein family comprises the members:
CCN1/CYR61, CCN2/CTGF, CCN3/NOV, CCN4/WISP1, CCN5/WISP2 and
CCN6/WISP3. These proteins are induced by growth factors and
cytokines such as TGF-β and endothelin-1, and affect connective
tissues, resulting in fibrosis and scarring (9). CCN2 is a member of the CCN family,
which plays an important role in a variety of cell processes
including angiogenesis, chondrogenesis and wound healing (10,11). Findings of previous studies
provided strong evidence that CCN2 plays a role in scars,
particularly as a co-factor of TGF-β1 (12). In addition, CCN2 is likely to
mediate the ability of TGF-β1 to stimulate ECM synthesis and is a
moderate inducer of collagen synthesis (11,13). CCN2 and CCN3 are opposing factors
in regulating collagen promoter activity and the secretion of this
ECM protein. Riser et al demonstrated that CCN3, an
endogenous regulator of pro-fibrotic changes, markedly
downregulates collagen production and deposition (14). Collectively, these data suggest
that CCN3 is a potentially important regulatory molecule in the
TGF-β1/Smad signaling pathway and a target for treatment in scar
formation.
Materials and methods
Antibodies and reagents
Mouse anti-collagen I monoclonal antibody, mouse
anti-collagen III monoclonal antibody, mouse anti-α-SMA monoclonal
antibody, mouse anti-Smad1 monoclonal antibody, mouse anti-Bcl-2
monoclonal antibody, mouse anti-Bax monoclonal antibody, mouse
anti-CCN3 monoclonal antibody, mouse anti-β-actin monoclonal
antibody and HRP-conjugated goat anti-mouse IgG were obtained from
Abcam (Cambridge, UK). Dimethylsulfoxide (DMSO), propidium iodide
(PI) and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium
bromide (MTT) assay were purchased from Bio-Rad (Hercules, CA,
USA). The enhanced chemiluminescence western blot analysis
detection reagents were obtained from Pierce Biotechnology, Inc.
(Rockford, IL, USA). The fluorescein isothiocyanate (FITC)-Annexin
V/PI apoptosis assay kit was purchased from Abcam.
Cell culture
Palatal fibroblasts were obtained from the explants
of the oral palatal mucosa of 8-week-old male Sprague-Dawley rats
(Shanghai Laboratory Animal Center, the Chinese Academy of
Sciences), as reported previously (15), with minor modifications. Cells
were cultured at 37°C under a humidified atmosphere of 5%
CO2 and 95% air and were grown in Dulbecco’s modified
Eagle’s medium (DMEM; Gibco-BRL, Gaithersburg, MD, USA)
supplemented with 10% heat-inactivated fetal bovine serum (CSL
Ltd., Melbourne, Australia), 2 mM L-glutamine, 50 mg/ml
streptomycin and 50 U/ml penicillin (Gibco-BRL). The medium was
changed every 2–3 days, and the cells were passaged with
trypsin-EDTA (Gibco-BRL) when they became confluent. Experiments
were performed using early passage cells (passage 5–10) derived
from at least three independent animals without freezing.
Construction of CCN3 adenoviral
vectors
Recombinant adenovirus was constructed as previously
described (16).Briefly, the full
length of CCN3 cDNA was amplified and subcloned into pAdTrack-CMV,
an adenoviral shuttle plasmid. A recombinant adenovirus expressing
green fluorescent protein (GFP) was also used to assess infection
efficiency. Then, the recombinant shuttle plasmids pAdTrack-CMV and
pAdEasy-1 were homologously recombined in the Escherichia
coli strain BJ5183. After sequencing, recombinant adenoviruses
were packaged and produced in 293A cells.
Determination of cell proliferation
Cell proliferation was determined via the MTT assay
and cell counting, as described previously with minor modifications
(17). Briefly, 2×103
cells/well (3-wells per condition) were seeded in 96-well plates,
and the cells were transfected with CCN3 adenoviral or blank
vector, using Lipofectamine 2000 according to the manufacturer’s
instructions (Invitrogen Life Technologies, Grand Island, NY, USA).
Culture medium was replaced every 2 days to avoid mitogen
deprivation. At the indicated times, 10 μl of an MTT solution (5
mg/ml in phosphate-buffered solution) was added to the wells and
cells were incubated for 4 h at 37°C, 5% CO2. Following
incubation, the culture medium in each well was replaced with 150
μl of DMSO, and the plates were agitated to dissolve the dark blue
crystals (formazan). Absorbance was measured at 570 nm using an
enzyme-linked immunosorbent assay plate reader (Roche Diagnostics
GmbH, Penzberg,Germany). Cell proliferation was expressed as the
percentage relative to the control. Each concentration was analyzed
in triplicate and the experiment was repeated three times. All the
experiments were performed at least three times and the results
were averaged.
Analysis of cell apoptosis
The rate of apoptosis was measured via Annexin
V-FITC and PI staining, followed by flow cytometry (Beckman
Coulter, Inc., Brea, CA, USA). Briefly, the cells were seeded in a
6-well plate at a density of 2×104 cells per well. After
the cells reached 70% confluency, they were transfected with CCN3
adenoviral or blank vector. The cells were then trypsinized and
suspended in 500 μl of binding buffer containing 5 μl Annexin
V-FITC and 5 μl PI (Abcam, Cambridge, MA, USA). After incubation in
the dark for 1 h, the cells were subjected to flow cytometry and
the rate of cell apoptosis was determined.
RT-PCR
qRT-PCR was performed as previously described
(18). Total-RNA was isolated
using the RNeasy Plus Micro kit (Qiagen, Hilden, Germany) according
to the manufacturer’s instructions. Reverse transcription was
performed using the High Capacity cDNA kit (Pierce Biotechnology,
Inc.) according to the manufacturer’s instructions. Real-time PCR
was performed using the 7900HT Fast Real-Time PCR System (Bio-Rad)
and was carried out using the SYBR Premix Ex Taq kit [Takara
Biotechnology Co., Ltd., Dalian, China] at a final volume of 20 μl.
Real-time PCRs were performed in triplicate. The expression levels
of the relative genes were calculated using the 2−ΔΔCT
method and the housekeeping gene β-actin was utilized as a control.
The primer sets used were: collagen I forward, 5′-TCCCCAG
CCACAAAGAGTCTACA-3′; and reverse, 5′-GTGATTGGGTGGGATGTCTTCGTC-3′;
collagen III forward, 5′-CTGCCATCCTGAACTCAA GAGTGG-3′; and reverse,
5′-CCATCCTCCAGAACTGTG TAGG-3′; TGF-β1 forward, 5′-CAACAATTCCTGGCG
ATACCTCA-3′; and reverse, 5′-GGTAGTGAACCCGTTGATGTCCA-3′ (19); α-SMA forward, 5′-GACAATGGCTCT
GGGCTCTGTAA-3′; and reverse, 5′-TGTGCTTCGTCACCCACGTA-3′ (19); Smad1 forward, 5′-ATGGACACGAACATG
ACGAA-3′; and reverse, 5′-GCACCAGTGTTTTGGTT CCT-3′ (20).
Western blot analysis
Total cellular proteins were extracted by incubating
cells in lysis buffer according to the operating protocols. The
protein concentrations in the cell lysates were determined using
abicinchoninic acid assay protein assay kit (Hitachi, Tokyo,
Japan). The same amount of protein from each sample was separated
by sodium SDS-PAGE on a 12% agarose gel and electrophoretically
transferred to a nitrocellulose membrane (Olympus, Tokyo, Japan).
Mouse monoclonal anti-collagen I, anti-collagen III, anti-TGF-β1,
anti-α-SMA, anti-CCN3, anti-Bcl-2, anti-Bax and anti-Smad1 were
used as the primary antibody and anti-mouse IgG monoclonal antibody
conjugated with horseradish peroxidase was used as the secondary
antibody. Protein bands were detected using the West Femto system
(Pierce Biotechnology, Inc.). Bands were visualized using ECL and
semi-quantitative analysis was performed using β-actin as a protein
loading control (21).
Statistical analysis
Experiments were carried out at least in triplicate
and the results were expressed as mean ± SD.Statistical analysis
was performed using the SPSS 16.0 (SPSS, Inc., Chicago, IL, USA).
Pairs of samples were compared using the Student’s t-test.
P<0.05 was considered to indicate statistical significance.
Results
Determination of transfection
effects
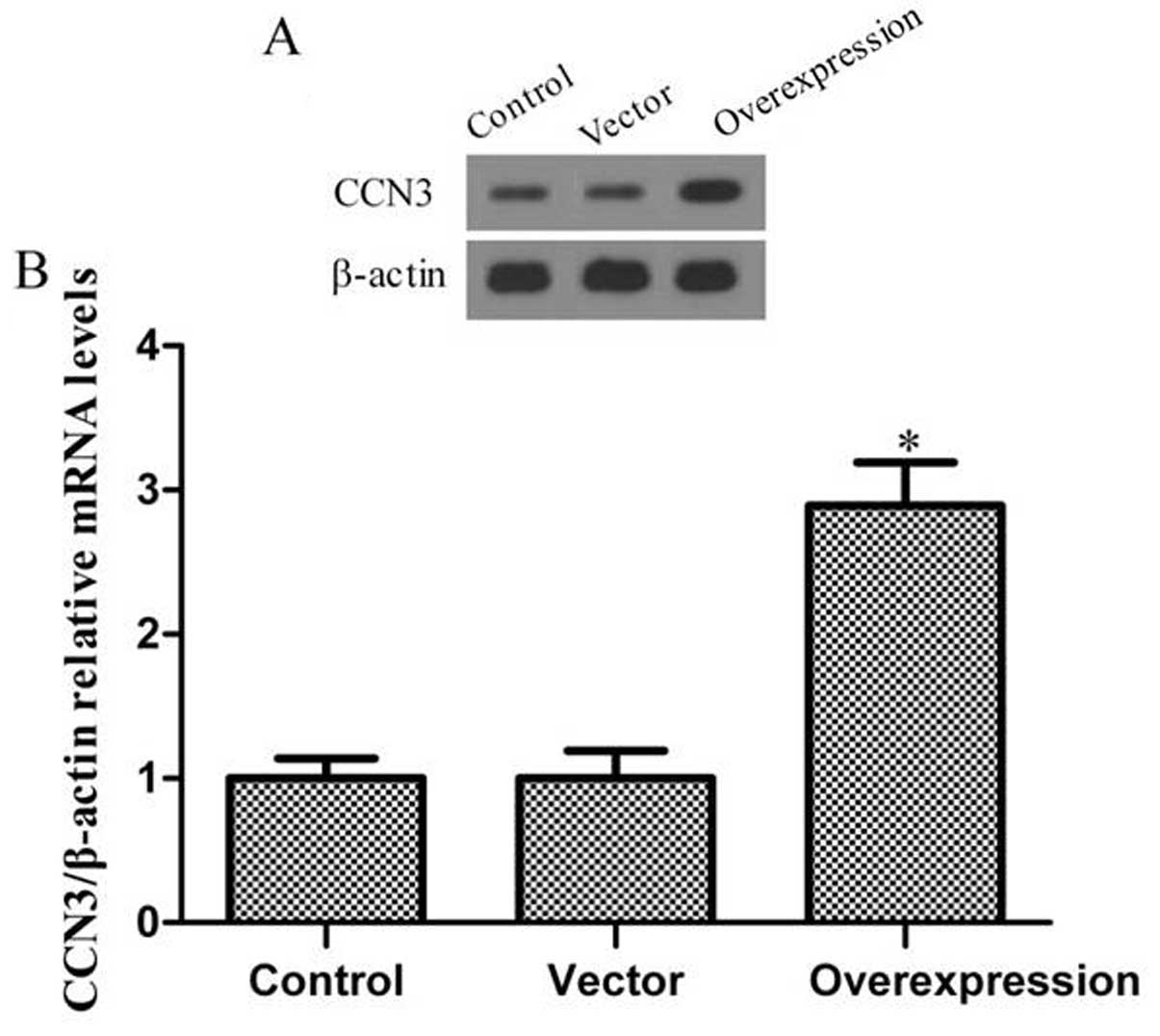
To test the efficiency of CCN3 transfection, western
blot analysis and RT-PCR were employed to determine the expression
level of the protein and mRNA. Fig.
1 shows that the fibroblast expressed only low levels of CCN3
in the control group. Moreover, transfection of the cells with
adenoviral vector without CCN3 barely increased CCN3 expression. Of
note, CCN3 expression significantly increased following
transfection of adenoviral vector with CCN3 into fibroblasts.
Collectively, these results indicate that transfection with
CCN3-adenoviral vector effectively increased CCN3 expression in
fibroblasts.
Effect of CCN3 on fibroblast
proliferation
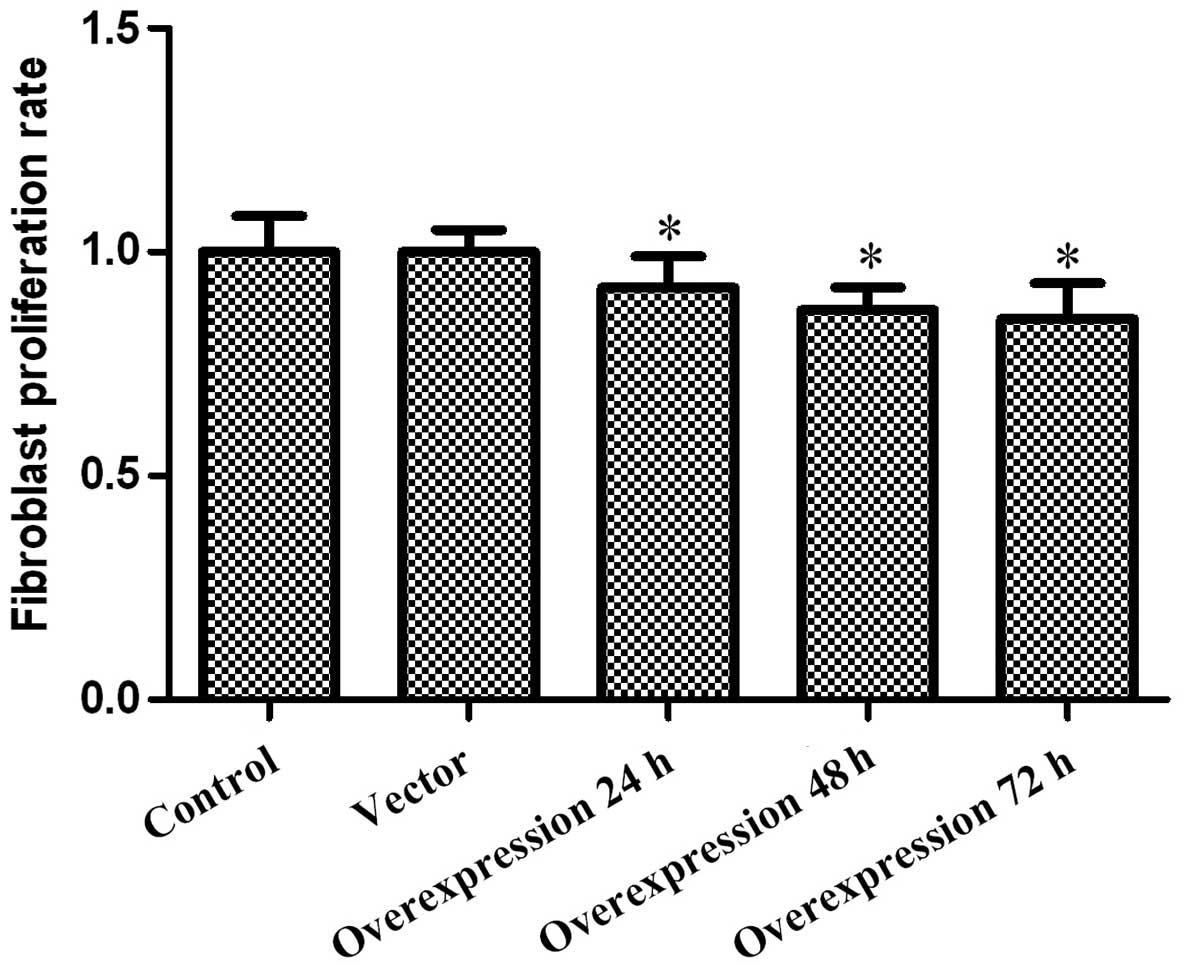
The impact of CCN3 on fibroblast proliferation was
determined via MTT assay every 24 h after transfection, for up to
72 h. The obtained results revealed that cell viability was, to a
certain extent, inhibited by CCN3 in a time-dependent manner.As
shown in Fig. 2, the
CCN3-transfected group grew more slowly than the control groups.
These results demonstrated that the overexpression of CCN3
inhibited fibroblast proliferation.
Effect of CCN3 on fibroblast
apoptosis
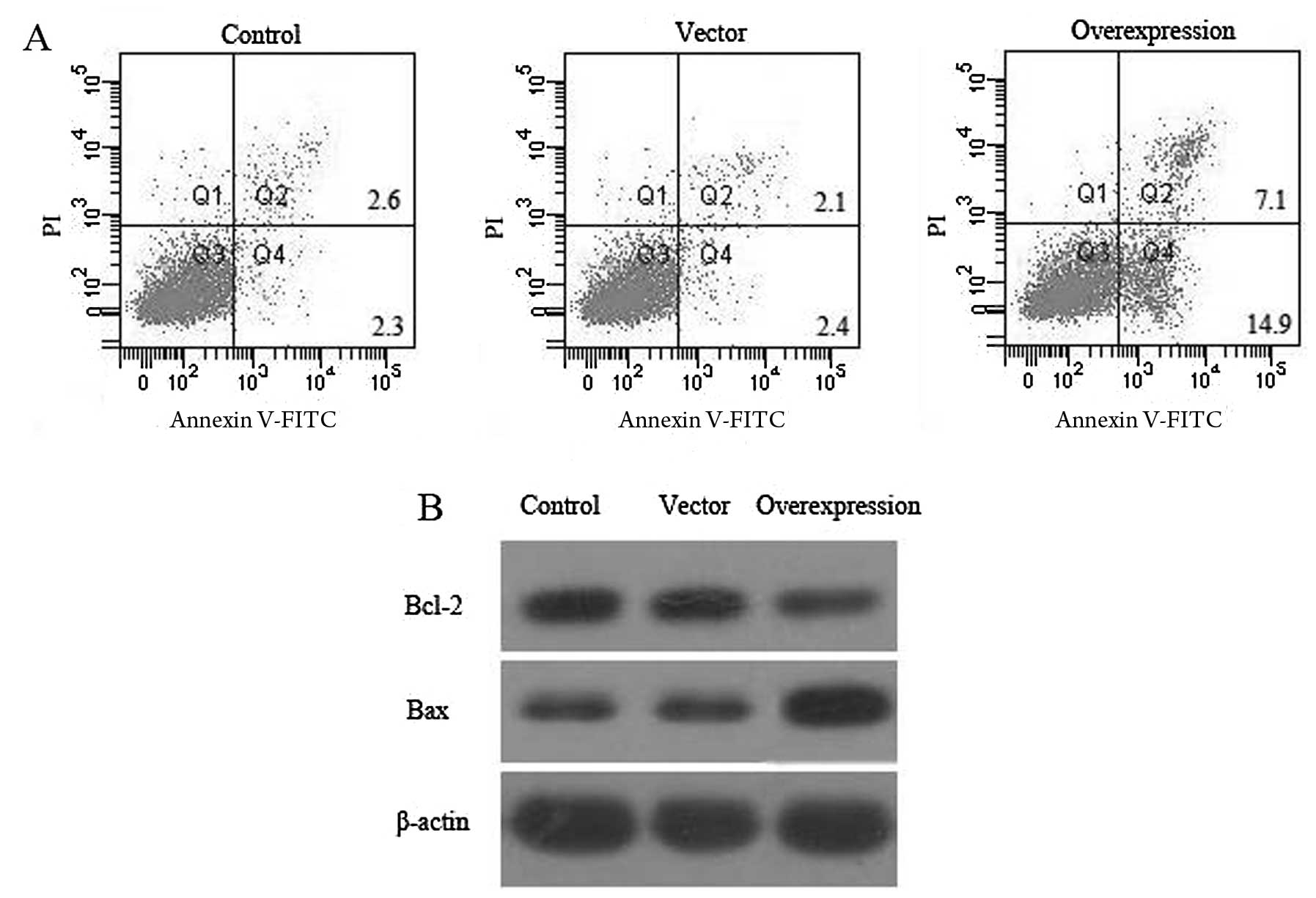
Fibroblast apoptosis was detected via PI staining
and the Annexin V method after 48 h of CCN3 transfection, followed
by flow cytometry.As shown in Fig.
3A, there was a low level (4.9 and 4.5%) of fibroblast
apoptosis in the control and vector groups, although the percentage
of apoptosis significantly increased to 22% (P<0.05) after 48 h
of CCN3 transfection.
To investigate whether CCN3 induces apoptosis in
fibroblasts, the possible molecular mechanisms of CCN3 associated
with apoptosis were assessed. Thus, we measured the expression of
Bcl-2 and Bax proteins in cells 48 h post-transfection (Fig. 3B). The results showed that the
expression of Bcl-2 decreased and the expression of Bax was
simultaneously upregulated in the CCN3-transfected group compared
with the control and vector groups (P<0.05). The apoptotic rate
was significantly higher because of the upregulation of
pro-apoptotic genes. These data revealed that CCN3 plays a critical
role in promoting fibroblast apoptosis.
Effect of CCN3 on collagen I and collagen
III
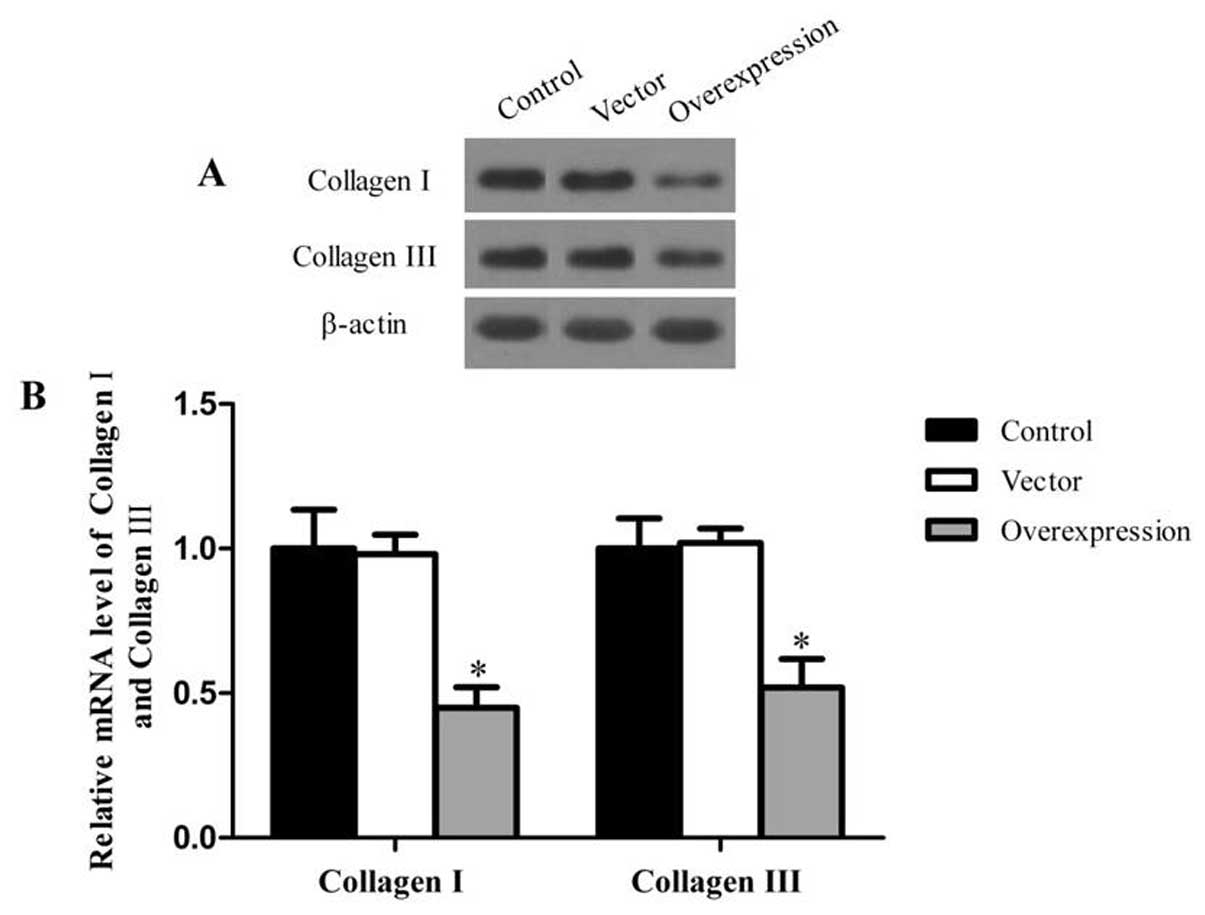
In order to evaluate the effects of CCN3 on matrix
production, we measured the protein and mRNA expression of collagen
I and III, which constitute the bulk of the scar ECM. In RT-PCR and
western blot analysis, the protein and mRNA levels of collagen I
and III were significantly lower in the CCN3-transfected group
compared with the control and vector groups (P<0.05) (Fig. 4).
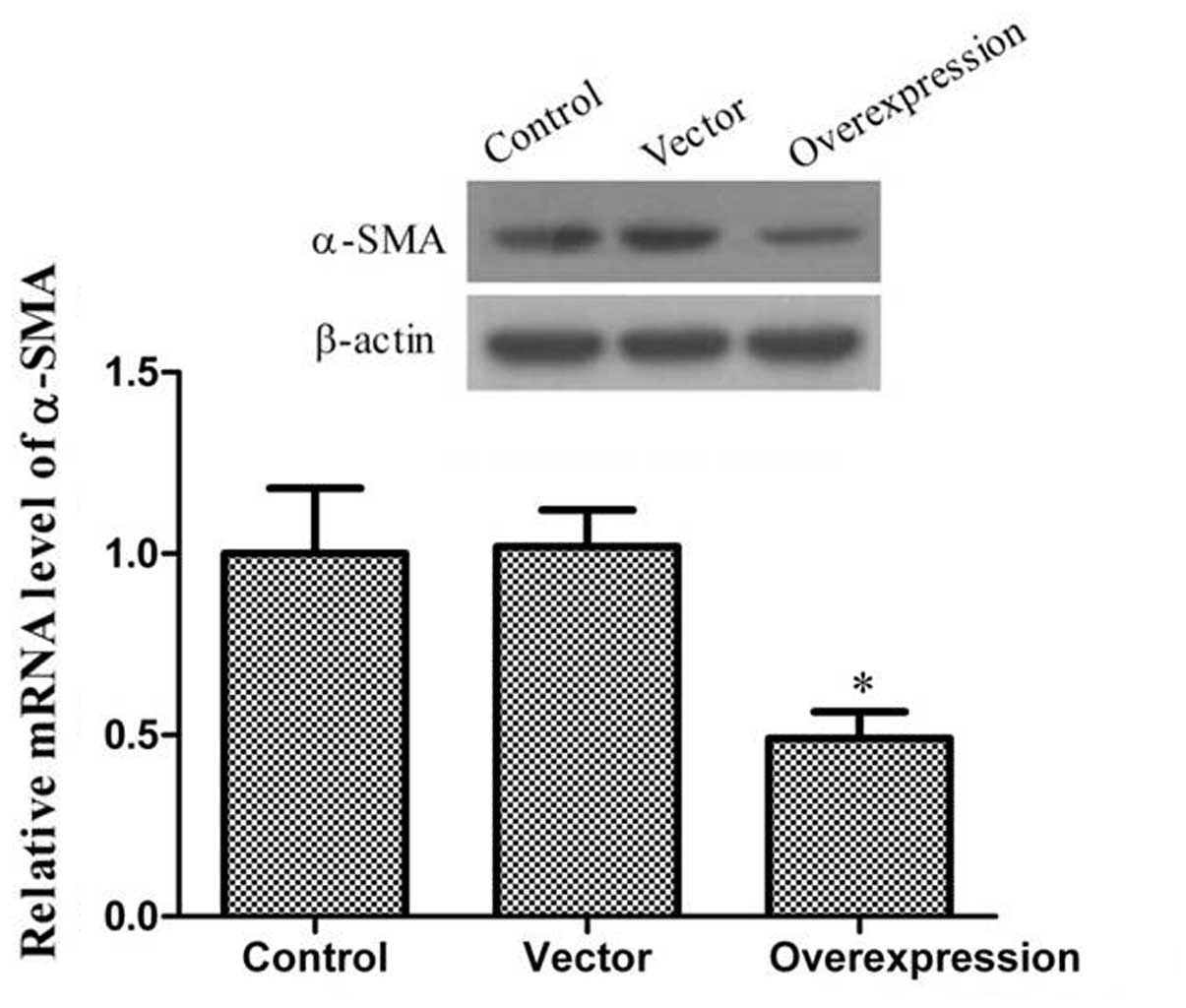
Effect of CCN3 on α-SMA
α-SMA, which plays a key role in the pathogenesis of
scars, is the most widely used marker for myofibroblasts. The
persistent presence of myofibroblasts is a distinctive feature of
scars that contributes to excessive matrix production. As shown in
Fig. 5, these results showed that
the levels of α-SMA expression were lower in the CCN3-transfected
group as compared to the control and vector groups.
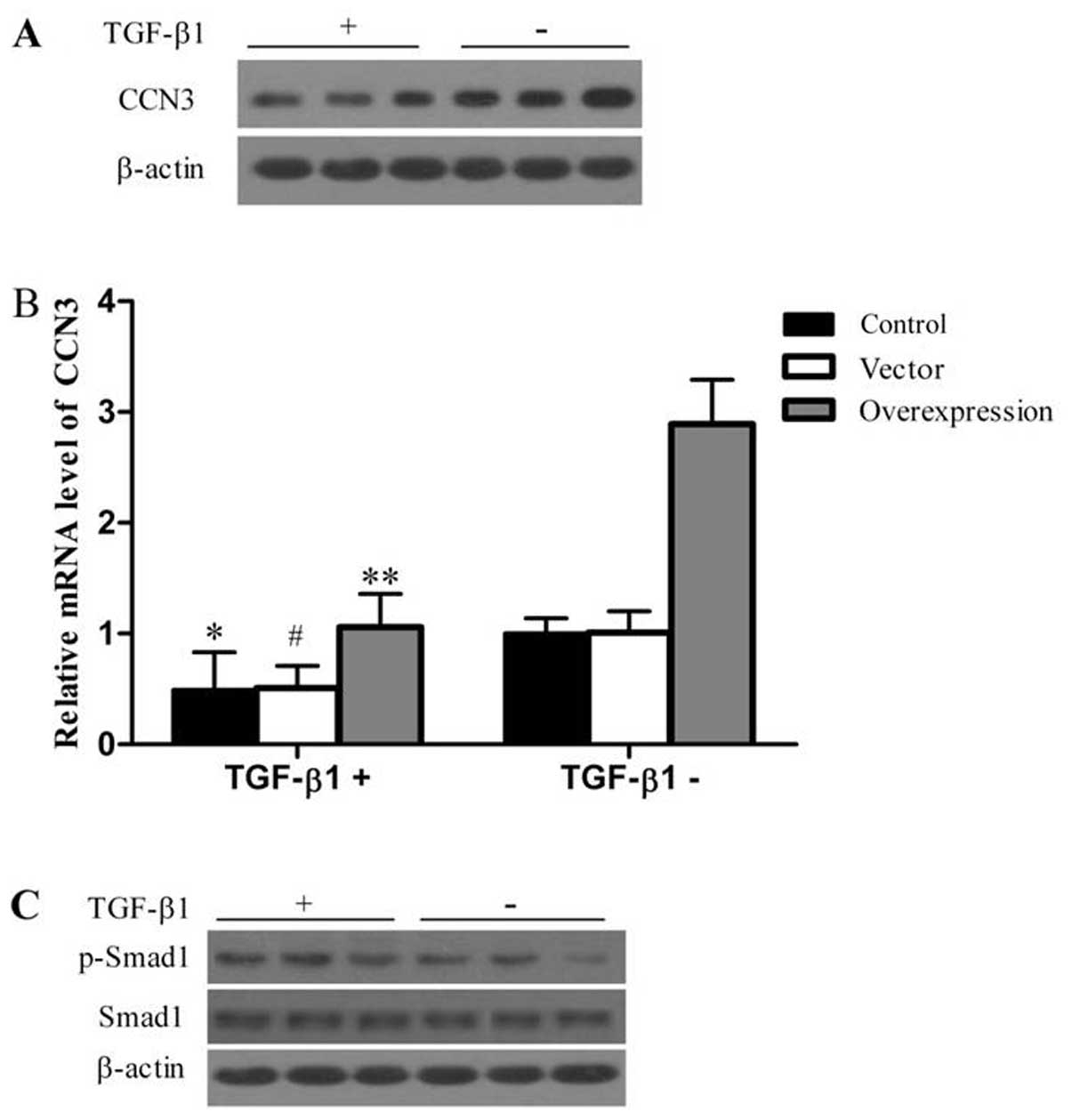
CCN3 induces cell apoptosis and
downregulates collagen I, collagen III and a-SMA via TGF-β1/Smad
signaling pathway
TGF-β1 promotes fibroblast transformation into
myofibroblasts and the synthesis of collagen (8). Therefore, we investigated the role
of CCN3 in mediating the TGF-β1-induced formation of collagen and
α-SMA. Under exogenous TGF-β1 stimulation, we determined the
protein and mRNA levels of CCN3.TGF-β1 stimulation efficiently
suppressed the expression of CCN3 at the mRNA and protein levels
(Fig. 6A and B). Previously it
was shown that the classical Smad1 pathway plays a role in the
TGF-β1 regulation of collagen and α-SMA expression (11). We then determined the activation
of Smad1 pathways in response to TGF-β1 stimulation. As shown in
Fig. 6C, the phosphorylation of
Smad1 was significantly lower in the CCN3-transfected group
compared with the control and vector groups. Furthermore, the
phosphorylation of Smad1 in response to TGF-β1 was higher than that
without TGF-β1 stimulation. However, there was no apparent change
in the protein level of Smad1 following TGF-β1 stimulation.
Discussion
To the best of our knowledge, this study has shown
for the first time that CCN3 induces cell apoptosis and
downregulates collagen I, collagen III and α-SMA via the
TGF-β1/Smad signaling pathway.
Treatment of scars following cleft lip and palate
operations is problematic, with no single modality having uniformly
satisfactory results. The techniques currently used to correct
cleft lip cannot successfully solve the problem of scarring.
Several effective therapeutic measures can be offered for the
clinical treatment of exuberant scars; however, these therapies are
far from satisfactory. Therefore, there is a need to find
approaches that are suitable for the treatment and better control
of scars.
Studies on the mechanism of scar formation have
shown that TGF-β1 is one of the most important elements in the
process of scar formation (22).
Previous studies have shown that CCN2 plays a crucial role in
concert with TGF-β1 during the formation and development of
fibrotic disorders or scarring (18). CCN2 is known to mediate the
ability of TGF-β to stimulate ECM synthesis, leading to keloid
formation (13). However, CCN3
may perform opposing functions that modulate the effects of CCN2.
Thus, we hypothesize that CCN3 exerts an anti-scarring effect.
However, the exact mechanism of involvement of CCN3 in scarring is
poorly understood. This study aimed to address this issue.
In the present study, we evaluated the effects of
the overexpression of CCN3 on the inhibition of proliferation and
promotion of apoptosis in palatal fibroblasts, along with its
mechanism of action. The results showed that the overexpression of
CCN3 inhibited palatal fibroblast proliferation and induced cell
apoptosis. Overexpression of CCN3 can dowregulate Bcl-2 expression
and upregulate Bax expression. It is likely that CCN3 induces
palatal fibroblast apoptosis by inhibiting Bcl-2 expression and
activating Bax.
Type I and III collagens are the major proteins
comprising the ECM (23).
However, excess deposition of ECM components, particularly
collagen, can result in scarring (24). Therefore, we assessed the protein
and mRNA expression of collagen I and III, and found that CCN3
downregulates the expression of the two proteins.
α-SMA is a hallmark of myofibroblast
differentiation, which plays a key role in the pathogenesis of
scars (25). The persistent
presence of myofibroblasts is a distinctive feature of scars that
contributes to excessive matrix production. In our study, the
downregulation of α-SMA by CCN3 was considered one of the signals
inhibiting fibroblast differentiation. These results suggest that
CCN3 is crucial in scarring.
Moreover, results of this study suggest that CCN3
induces cell apoptosis and downregulates collagen I, collagen III
and α-SMA via the TGF-β1/Smad signaling pathway. Under exogenous
TGF-β1 stimulation, CCN3 expression was suppressed and Smad1
phosphorylation increased. Thus, CCN3 induces cell apoptosis and
downregulates collagen I, collagen III and α-SMA via the
TGF-β1/Smad signaling pathway.
In summary, this study has demonstrated that CCN3 is
involved in the proliferation and apoptosis of fibroblasts and the
synthesis of ECM proteins. Therefore, CCN3 may play an important
role in the development of scars, and may represent a novel
therapeutic target for reducing scar formation.
References
|
1
|
Marazita ML: The evolution of human
genetic studies of cleft lip and cleft palate. Annu Rev Genomics
Hum Genet. 13:263–283. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Reiter R, Haase S and Brosch S: [Orofacial
clefts]. Laryngorhinootologie. 91:84–95. 2012. View Article : Google Scholar
|
|
3
|
Cox TC, Luquetti DV and Cunningham ML:
Perspectives and challenges in advancing research into craniofacial
anomalies. Am J Med Genet C Semin Med Genet. 163:213–217. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Metzis V, Courtney AD, Kerr MC, Ferguson
C, Rondón Galeano GM, Parton RG, Wainwright BJ and Wicking C:
Patched1 is required in neural crest cells for the prevention of
orofacial clefts. Hum Mol Genet. 22:5026–5035. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Díaz Casado GH and Díaz Grávalos GJ:
[Orofacial closure defects: cleft lip and palate. A literature
review]. Semergen. 39:267–271. 2013.
|
|
6
|
Hoogewerf CJ, van Baar ME, Middelkoop E
and van Loey NE: Patient reported facial scar assessment:
directions for the professional. Burns. 40:347–353. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Goldberg MT, Han YP, Yan C, Shaw MC and
Garner WL: TNF-alpha suppresses alpha-smooth muscle actin
expression in human dermal fibroblasts: an implication for abnormal
wound healing. J Invest Dermatol. 127:2645–2655. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Omori S, Kitagawa H, Koike J, Fujita H,
Hida M, Pringle KC and Awazu M: Activated extracellular
signal-regulated kinase correlates with cyst formation and
transforming growth factor-beta expression in fetal obstructive
uropathy. Kidney Int. 73:1031–1037. 2008. View Article : Google Scholar
|
|
9
|
Leask A and Abraham DJ: All in the CCN
family: essential matricellular signaling modulators emerge from
the bunker. J Cell Sci. 119:4803–4810. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Moussad EE and Brigstock DR: Connective
tissue growth factor: what’s in a name? Mol Genet Metab.
71:276–292. 2000.
|
|
11
|
Nakerakanti SS, Bujor AM and Trojanowska
M: CCN2 is required for the TGF-β induced activation of
Smad1-Erk1/2 signaling network. PLoS One. 6:e219112011.
|
|
12
|
Hahn A, Heusinger-Ribeiro J, Lanz T,
Zenkel S and Goppelt-Struebe M: Induction of connective tissue
growth factor by activation of heptahelical receptors. Modulation
by Rho proteins and the actin cytoskeleton. J Biol Chem.
275:37429–37435. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sisco M, Kryger ZB, O’Shaughnessy KD, Kim
PS, Schultz GS, Ding XZ, Roy NK, Dean NM and Mustoe TA: Antisense
inhibition of connective tissue growth factor (CTGF/CCN2) mRNA
limits hypertrophic scarring without affecting wound healing in
vivo. Wound Repair Regen. 16:661–673. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Riser BL, Bhagavathula N, Perone P,
Garchow K, Xu Y, Fisher GJ, Najmabadi F, Attili D and Varani J:
Gadolinium-induced fibrosis is counter-regulated by CCN3 in human
dermal fibroblasts: a model for potential treatment of nephrogenic
systemic fibrosis. J Cell Commun Signal. 6:97–105. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kanda T, Funato N, Baba Y and Kuroda T:
Evidence for fibroblast growth factor receptors in myofibroblasts
during palatal mucoperiosteal repair. Arch Oral Biol. 48:213–221.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liu L, Zhang G, Liang Z, Liu X, Li T, Fan
J, Bai J and Wang Y: MicroRNA-15b enhances
hypoxia/reoxygenation-induced apoptosis of cardiomyocytes via a
mitochondrial apoptotic pathway. Apoptosis. 19:19–29. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Crowe J, Aubareda A, McNamee K, Przybycien
PM, Lu X, Williams RO, Bou-Gharios G, Saklatvala J and Dean JL:
Heat shock protein B1-deficient mice display impaired wound
healing. PLoS One. 8:e773832013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hu X, Wang H, Liu J, Fang X, Tao K, Wang
Y, Li N, Shi J, Wang Y, Ji P, Cai W, Bai X, Zhu X, Han J and Hu D:
The role of ERK and JNK signaling in connective tissue growth
factor induced extracellular matrix protein production and scar
formation. Arch Dermatol Res. 305:433–445. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yang G, Crawford RC and Wang JH:
Proliferation and collagen production of human patellar tendon
fibroblasts in response to cyclic uniaxial stretching in serum-free
conditions. J Biomech. 37:1543–1550. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Fujiwara N, Doi T, Gosemann JH, Kutasy B,
Friedmacher F and Puri P: Smad1 and WIF1 genes are downregulated
during saccular stage of lung development in the nitrofen rat
model. Pediatr Surg Int. 28:189–193. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yuan J, Huang G, Xiao Z, Lin L and Han T:
Overexpression of β-NGF promotes differentiation of bone marrow
mesenchymal stem cells into neurons through regulation of AKT and
MAPK pathway. Mol Cell Biochem. 383:201–211. 2013.
|
|
22
|
Cannata A, Petrella D, Russo CF, Bruschi
G, Fratto P, Gambacorta M and Martinelli L: Postsurgical
intrapericardial adhesions: mechanisms of formation and prevention.
Ann Thorac Surg. 95:1818–1826. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Pickering JG: Regulation of vascular cell
behavior by collagen: form is function. Circ Res. 88:458–459. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sato M: Upregulation of the
Wnt/beta-catenin pathway induced by transforming growth factor-beta
in hypertrophic scars and keloids. Acta Derm Venereol. 86:300–307.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Abdalla M, Goc A, Segar L and Somanath PR:
Akt1 mediates α-smooth muscle actin expression and myofibroblast
differentiation via myocardin and serum response factor. J Biol
Chem. 288:33483–33493. 2013.
|