Introduction
Ischemic stroke is a major cause of death and
disability worldwide, and the clinical prognosis of acute cerebral
ischemia is poor (1,2). The early reperfusion after cerebral
ischemia is essential for the viability and functional recovery of
the brain; however, the arrival of blood oxygen to the ischemic
tissue causes ischemia/reperfusion injury (IRI), which induces
further damage (3). Since 1986,
when Murray et al (4)
first described a classic phenomenon they termed ischemic
preconditioning (IPC), it has been repeatedly confirmed in animal
models that IPC and ischemic postconditioning (IPTC) are powerful
endogenous protective strategies against IRI of multiple organs,
including the heart, brain and kidneys (8). However, the clinical implementation
of IPC and IPTC as a means of protection against IRI is not
desirable or feasible in most clinical circumstances due to the
unpredictability of organ ischemia and the possibility that added
ischemia may result in dangerous complications (9). The application of brief episodes of
ischemia to a tissue remote from the primary ischemic organ (e.g.,
heart or brain) using the techniques of remote ischemic
preconditioning (RIP) or remote ischemic postconditioning (RIPoC)
may confer cerebral/cardiac protection, and RIPoC can overcome the
aforementioned issues previously associated with IPC and IPTC
(10–17). Both RIP and RIPoC have been
recognized as applicable strategies for the protection against
cerebral and myocardial IRI (11,13,18–22). A further refinement of RIPoC,
non-invasive remote ischemic postconditioning (NRIPoC), can be
applied in a wider range of clinical settings for cerebral and
myocardial IRI due to its practicality as a non-invasive technique.
NRIPoC is a relatively recent innovation (11,23–25), and its fundamental biology is not
yet well understood.
Apoptosis and the inflammatory response both play
fundamental pathogenic roles in cerebral IRI (26–32). Tumor necrosis factor-α (TNF-α) is
the most important pro-inflammatory cytokine involved in cerebral
IRI, and its effects are observed throughout the development of
inflammation (33–35). Nuclear factor-κB (NF-κB) regulates
the gene expression of inflammatory factors involved in the
ischemia/reperfusion (I/R) of the cerebral tissue (36,37). The inflammatory response can
induce apoptosis by regulating apoptotic signals during cerebral
IRI (38–41). Evidence indicates that the Janus
kinase (JAK)/signal transducer and activator of transcription 3
(STAT3) signaling pathway transduces stress-activating
extracellular chemical signals into cellular responses for a number
of pathophysiological processes, such as immunity, inflammation and
apoptosis, and is involved in cerebral IRI (42–45). We hypothesized that the
NRIPoC-induced neuroprotective effects may be associated with the
activation of the STAT3 signaling pathway, the inhibition of the
inflammatory response and the regulation of apoptosis. To explore
this hypothesis, we performed experiments to evaluate the roles of
apoptosis, the inflammatory response and STAT3 signaling in
cerebral protection conferred by NRIPoC during focal cerebral IRI
in an in vivo rat model.
Materials and methods
Animals
Male Sprague-Dawley rats weighting 250–300 g were
purchased from the Center of Laboratory Animals of Central South
University, Changsha, China. The rats were placed in a room with a
controlled environment with a 12-/12-h light/dark cycle and allowed
access ad libitum to standard rodent chow and tap water.
This study was approved by the Institutional Animals Ethics
Committee of Central South University and was conducted in
accordance with the Guidelines for the Care and Use of Laboratory
Animals provided by the National Institutes of Health (NIH
publication no. 80–82).
Model of focal cerebral ischemia
Middle cerebral artery (MCA) occlusion (MCAO) was
carried out as previously described (46). The animals were anesthetized by an
intraperitoneal injection of 300 mg/kg chloral hydrate. Heating
lamps were used to maintain rectal temperature at 37–37.5°C. The
right common carotid artery (CCA), external carotid artery (ECA)
and internal carotid artery (ICA) were exposed through a midline
neck incision, and the ECA was ligated close to its origin with a
3-0 silk suture. A 0.26-mm monofilament nylon suture with a blunt
tip (Beijing Shandong Industrial Corp., Beijing, China) was
inserted into the ICA, and advanced 18–20 mm until mild resistance
was felt, effectively occluding the MCA. After 90 min of MCAO, the
monofilament nylon suture was removed and ICA perfusion was
restored.
Experimental protocol
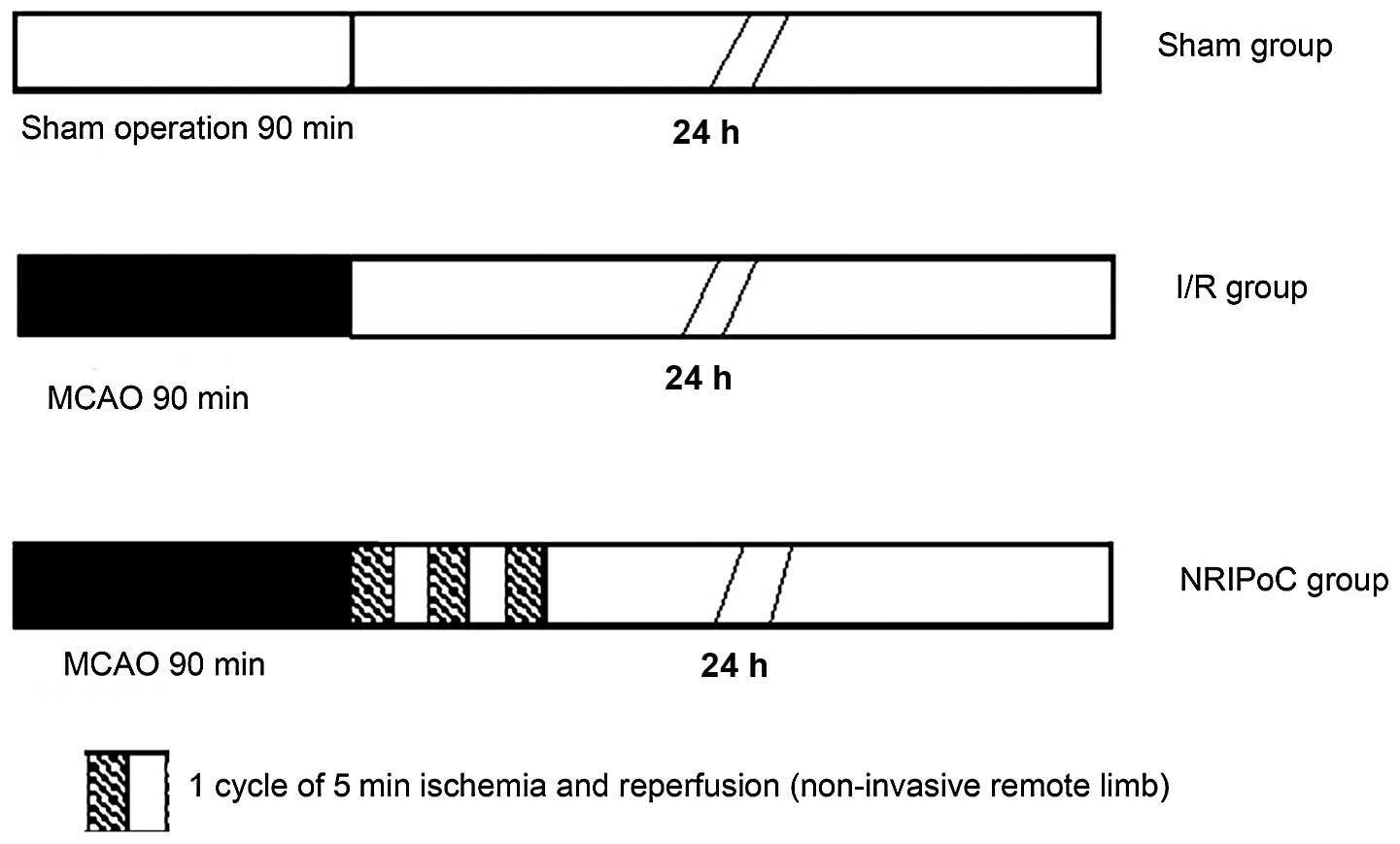
In total, 45 rats were randomly assigned to 3 groups
(n=15 in each group): the sham-operated, I/R and NRIPoC groups
(Fig. 1). The I/R group underwent
MCAO by occlusion of the right MCA for 90 min, followed by 24 h of
cerebral reperfusion. The sham-operated group underwent the same
procedure as the I/R rats, but without occlusion of the right MCA.
The NRIPoC group underwent the same procedure as the I/R group, but
was also subjected to sequential I/R, which involved 3 applications
of ischemia for 5 min and reperfusion for 5 min to the right hind
limb during MCA recovery reperfusion.
To carry out this procedure, a modified non-invasive
blood pressure cuff was strapped to the base of the right hind limb
of the rat, and pulse sensors were placed on the area of the
dorsalis pedis artery. NRIPoC was induced by increasing the
pressure of the cuff until blood flow in the limb was blocked. This
was maintained for 5 min and then released for 5 min, beginning the
onset of MCA reperfusion. This was carried out 3 times for 10-min
cycles. The blood flow of the right hind limb was completely
blocked by the cuff as confirmed by the disappearance of the pulse
and hypothermia and skin cyanosis in the limb.
Neurological evaluation
The animals were returned to their cages after the
procedures were finished and again allowed free access to food and
water. The neurological deficit score (NDS), as previously
described by Longa et al (46), was measured to assess neurological
evaluation at 24 h after reperfusion as follows: 0, no deficit; 1,
failure to extend left forepaw fully; 2, circling to the left; 3,
falling to the left; and 4, no spontaneous walking with a depressed
level of consciousness.
Measurement of infarct volume
After a reperfusion period of 24 h, the cerebral
infarct area was identified by 2,3,5-triphenyltetrazolium chloride
(TTC) staining as previously described (47,48). The infarct volume was calculated
according to the following formula: V = t × (A1 + A2... + An),
where ‘t’ is the brain slice thickness and ‘A’ is the infarct area.
The percentage cerebral infarct volume was calculated as follows:
(cerebral infarct total volume/whole brain volume) ×100.
Preparation of cerebral specimens
After a reperfusion period of 24 h, 5 rats in each
group were randomly selected and anesthetized with chloral hydrate.
Approximately 300–400 ml normal saline was infused via an aortic
root catheter until the liver appeared white, followed by 200 ml 4%
paraformaldehyde solution that had been cooled to 4°C. The brain
was removed, fixed in 4% paraformaldehyde solution for 24 h, and
4-mm-thick coronal sections were then cut from the front of the
brain through the optic chiasm. After gradient ethanol dehydration,
vitrification with isobutanol and n-butyl alcohol and embedding in
paraffin wax, the slices were cut into 5-μm-thick coronal sections
on a vibratome, and the sections were dried overnight in an
incubator at 37°C for later processing with hematoxylin and eosin
(H&E) and Nissl staining, terminal deoxynucleotidyl
transferase-mediated dUTP nick end-labelling (TUNEL).
Assessment of neuronal survival
The coronal brain sections were examined using a
standard hematoxylin and eosin (H&E) staining protocol to
examine cellular morphology by observing the number and shape of
the neurons. The brain sections were stained with toluidine blue
for Nissl bodies. Neuronal survival was assessed by observing the
Nissl bodies in the neurons. Images of the ischemic penumbras of
the 5 rats were captured, and quantitative analyses of the cells
were performed using Image-Pro Plus 6.0 software (Media
Cybernetics, Inc., Rockville, MD, USA).
Assessment of apoptosis
Apoptosis was measured by TUNEL assay using an
Apoptosis Detection kit (Roche Applied Science, Mannheim, Germany),
in accordance with the manufacturer’s instructions. TUNEL-positive
neurons were viewed under a light microscope (Leica DM 2000; Leica
Microsystems, Wetzlar, Germany) and were identified by
bluish-violet-stained nuclei. Five visual fields were randomly
selected from the ischemic penumbra of each slice at ×400
magnification. The apoptotic cells were counted in each field, and
the average value of the 5 fields was calculated.
Western blot analysis
After a reperfusion period of 24 h, the brains of
the 5 rats in each group selected randomly were rapidly removed
under deep anesthesia, and the right ischemic penumbra areas of the
parietal cortex were immediately isolated onto ice held at −20°C.
The pulverized brain samples weighing 0.25 g were homogenized with
500 μl protein lysis buffer. The homogenates were centrifuged at
12,000 rpm for 5 min at 4°C, and the supernatants were collected
for western blot analysis. The protein concentrations were
determined by bicinchoninic acid (BCA) protein assays (WellBiz
Brands, Inc., Highlands Ranch, CO, USA). Western blot analyses were
carried out using standard techniques (49). Briefly, a mixture of homogenate
sample containing 30 μg protein and 5× sodium dodecyl sulfate (SDS)
loading buffer was boiled at 100°C for 5 min, separated on 10%
SDS-polyacrylamide gels, transferred onto polyvinylidene fluoride
(PVDF) membranes (Pierce Biotechnology, Inc., Rockford, IL, USA)
and blocked for 1 h with 5% non-fat powdered milk in 100 mM
Tris-buffered saline containing 0.05% Tween-20 (TBST). The
membranes were incubated overnight at 4°C with antibodies against
Bcl-2 (Abcam plc., Cambridge, UK), Bax (Cell Signaling Technology,
Inc., Beverly, MA, USA), phosphorylated STAT3 (p-STAT3), or β-actin
(both Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA). After 3
washes in TBST, the membranes were incubated with secondary
antibody (HRP rabbit anti-goat IgG; Santa Cruz Biotechnology, Inc.)
for 1 h at room temperature, followed by 3 washes in TBST. Signals
were detected with an Enhanced Chemiluminescence kit (Pierce
Biotechnology, Inc.).
Statistical analysis
Data analyses were performed using SPSS software
13.0 (SPSS Inc., Chicago, IL, USA). NDS values are expressed as the
median (range) and were compared using Kruskal-Wallis tests. Other
data are presented as the means ± standard deviation (SD). One-way
analysis of variance (ANOVA) followed by Student’s t-tests were
applied to determine differences between groups. A value of
P<0.05 was considered to indicate a statistically significant
difference.
Results
Effects of NRIPoC on the IRI-induced
cerebral infarct volume
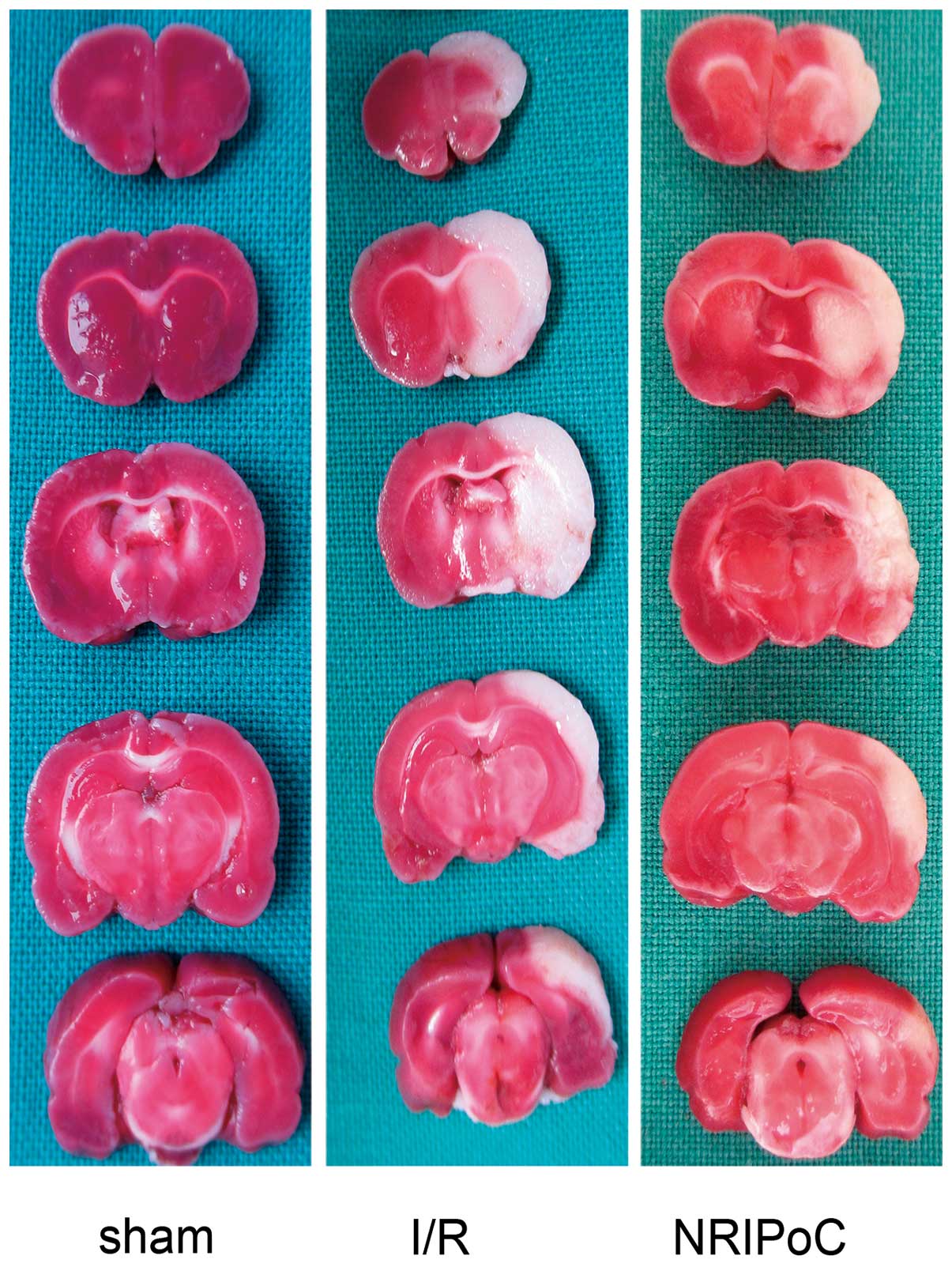
The cerebral infarct areas determined by TTC
staining are illustrated in Fig.
2. There were no conspicuous cerebral infarcts area in the
sham-operated group rats, while the cerebral infarct volumes of the
I/R and NRIPoC rats were 28.4±3.7 and 15.2±6.9, respectively. The
cerebral infarct volumes were significantly decreased in the NRIPoC
group compared with the I/R group (P<0.05).
Effects of NRIPoC on IRI-induced neuronal
loss
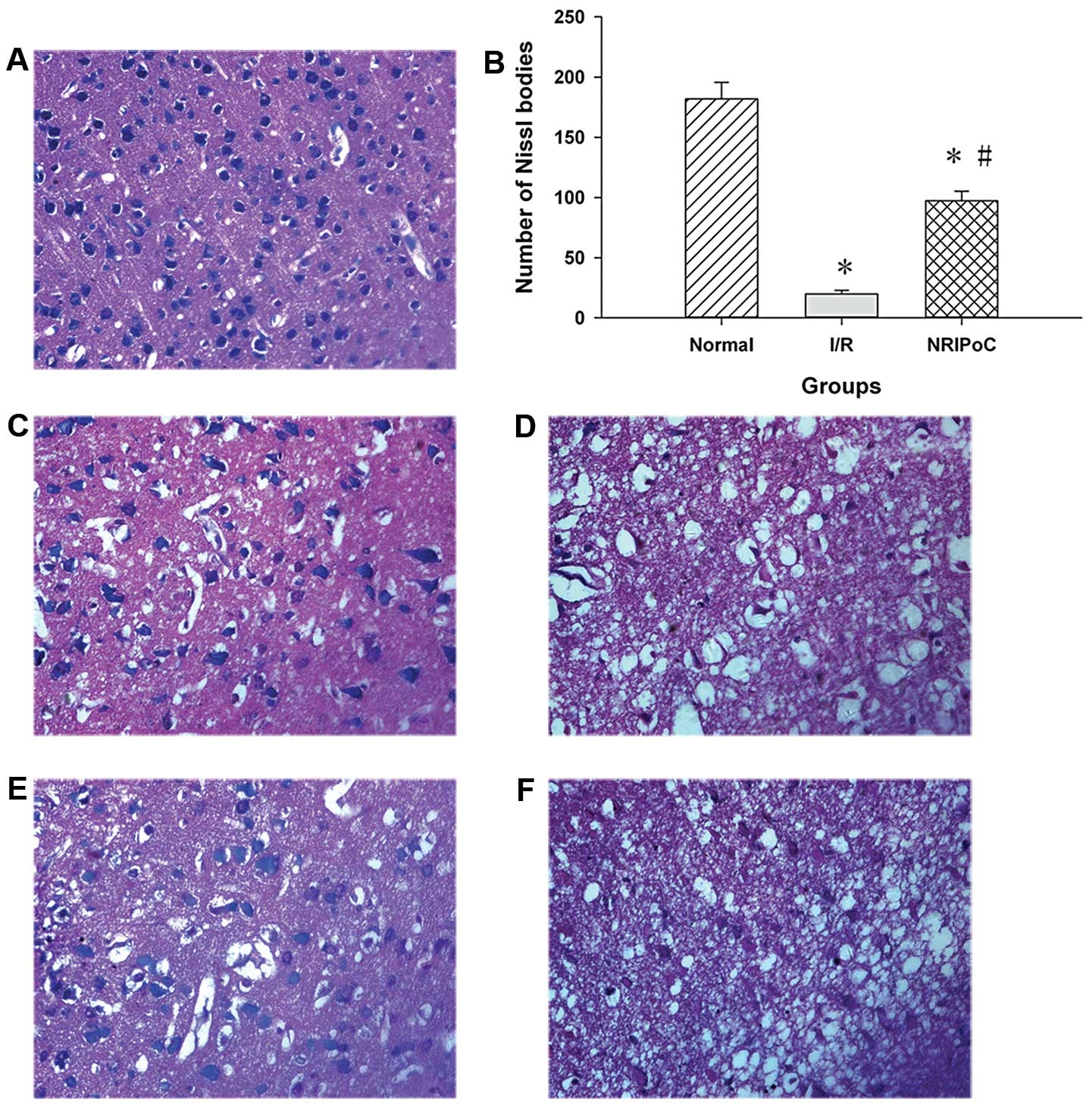
Nissl bodies were used as a morphological indicator
of neuronal survival. The number of Nissl-stained neurons in the
ischemic penumbra areas in the I/R and NRIPoC groups was
significantly reduced compared with the sham-operated group
(P<0.05); however, this decrease was less significant in the
NRIPoC group compared with the marked decrease observed in the I/R
group (Fig. 3A–C and E). The
Nissl bodies in the ischemic core areas in the I/R and NRIPoC
groups were wiped out in vast numbers, and there was no significant
difference between the 2 groups (Fig.
3D and F).
Effects of NRIPoC on IRI-induced neuronal
morphological changes
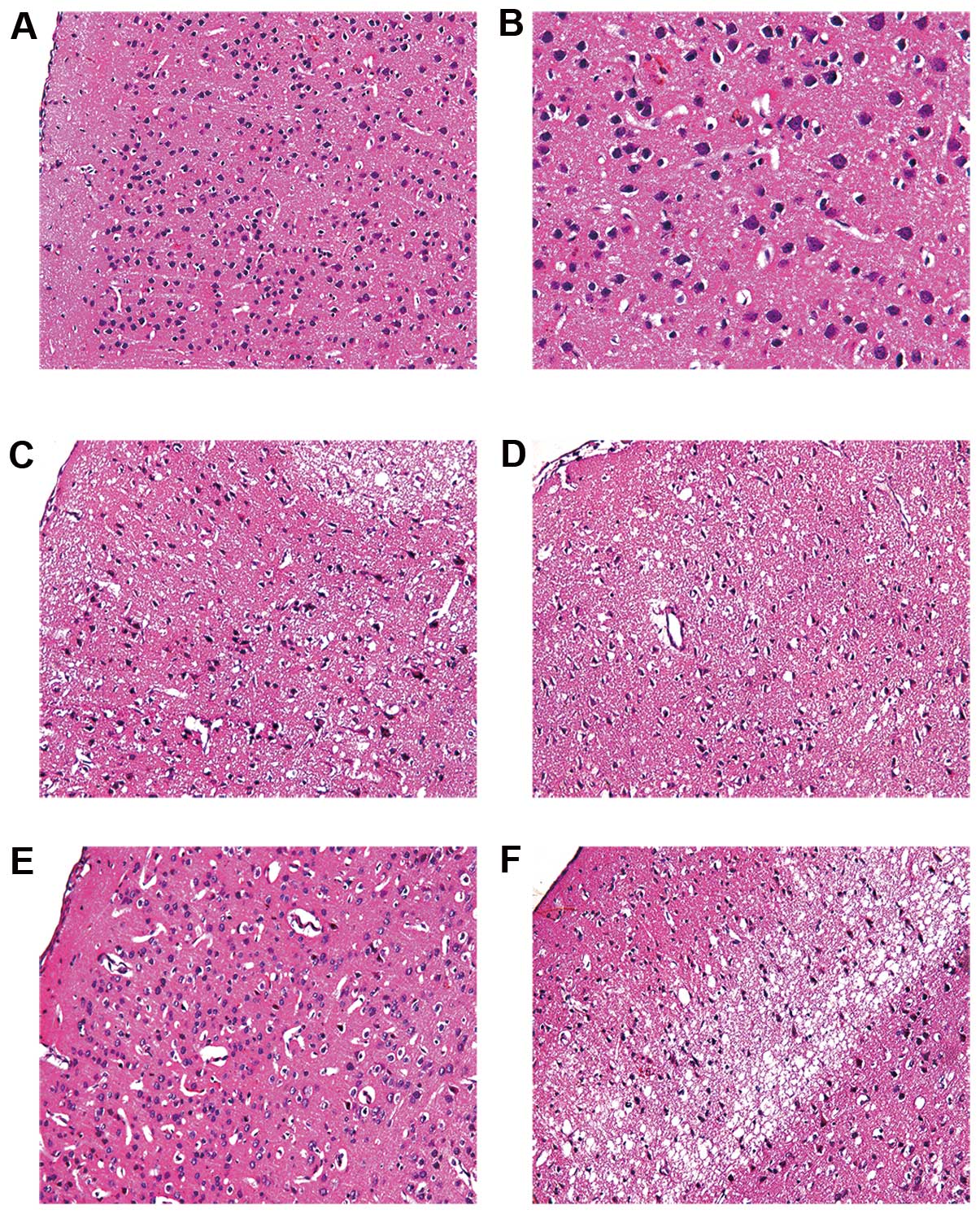
Neuronal morphology was assessed by H&E staining
(Fig. 4). The number of neurons
in the ischemic penumbra area of the I/R group was reduced. The
obvious characteristics of the neurons were: decreased cell size,
nuclear pyknosis, interstitial edema, cell disorder and chromatin
condensation. Compared with the I/R group, the NRIPoC group showed
a marked improvement in the cellular morphology of the ischemic
penumbra area, as only a few neurons were observed to have nuclear
pyknosis, hyperchromasis and extremely loose organization. There
was no obvious difference in cellular morphology in the ischemic
core area between the I/R and NRIPoC groups.
Effects of NRIPoC on IRI-induced neuronal
apoptosis
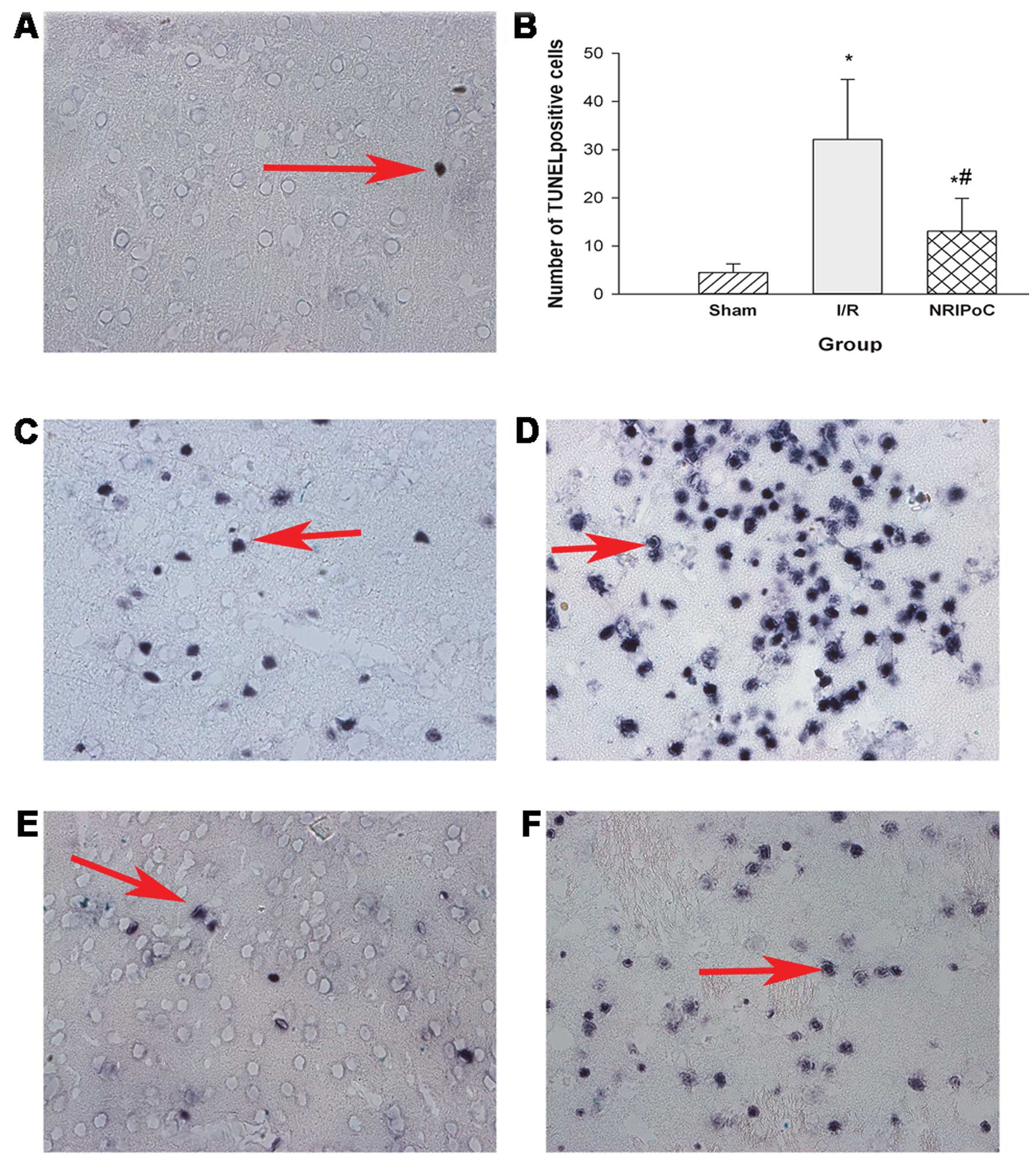
Apoptosis was determined by TUNEL staining (Fig. 5), which revealed that there were
few apoptotic neurons in the sham-operated group. In the I/R group,
there were large numbers of apoptotic neurons in the ischemic
penumbra area; however, NRIPoC significantly decreased these
numbers (P<0.05). There were numerous apoptotic neurons in the
ischemic core areas of both the I/R and NRIPoC groups.
Effects of NRIPoC on IRI-induced Bcl-2
and Bax expression in the ischemic penumbra area
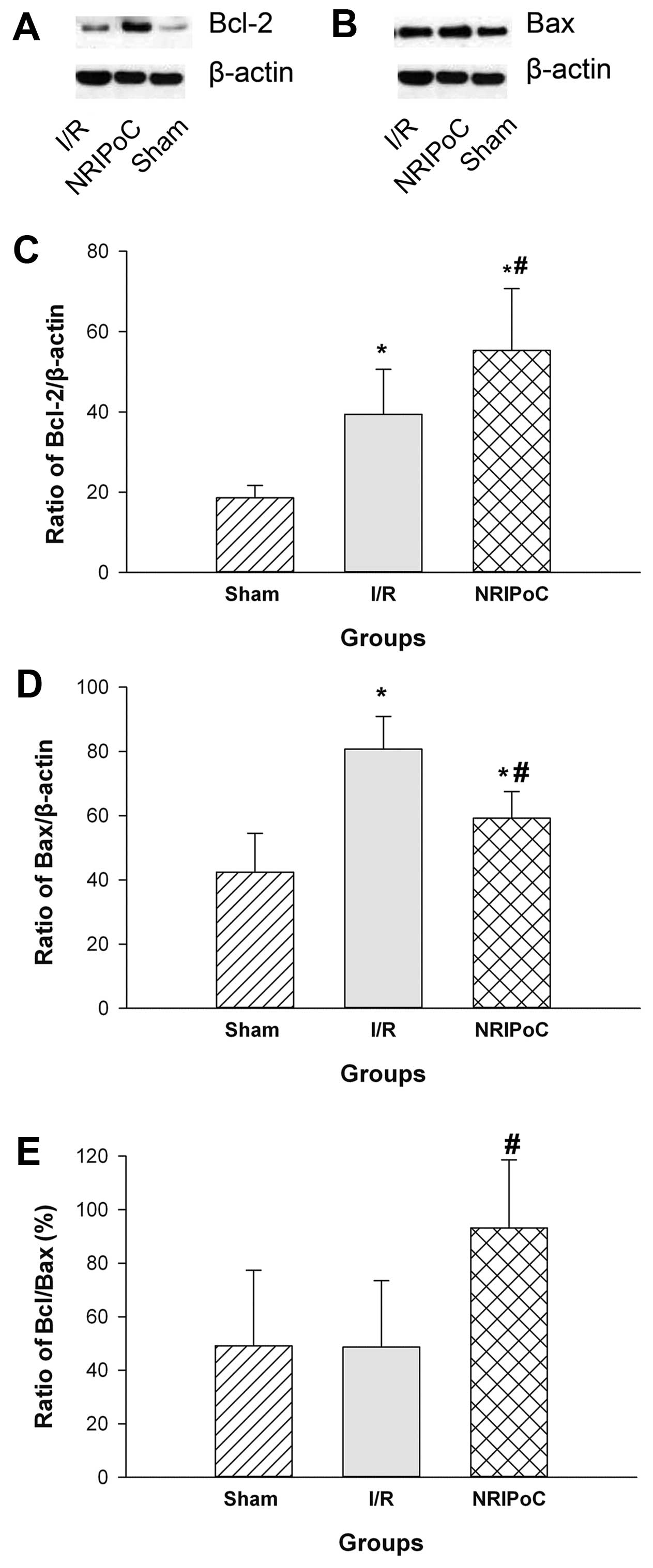
Bcl-2 and Bax protein levels were significantly
increased in the I/R and NRIPoC groups compared with the
sham-operated group (P<0.05) (Fig.
6). NRIPoC significantly increased Bcl-2 protein expression and
decreased Bax protein expression following IRI (P<0.05). There
was no difference in the Bcl-2/Bax ratio between the I/R and
sham-operated groups; however, this ratio was significantly
increased in the NRIPoC group compared with the I/R group
(P<0.05).
Effects of NRIPoC on p-STAT3 protein
expression in the ischemic penumbra area
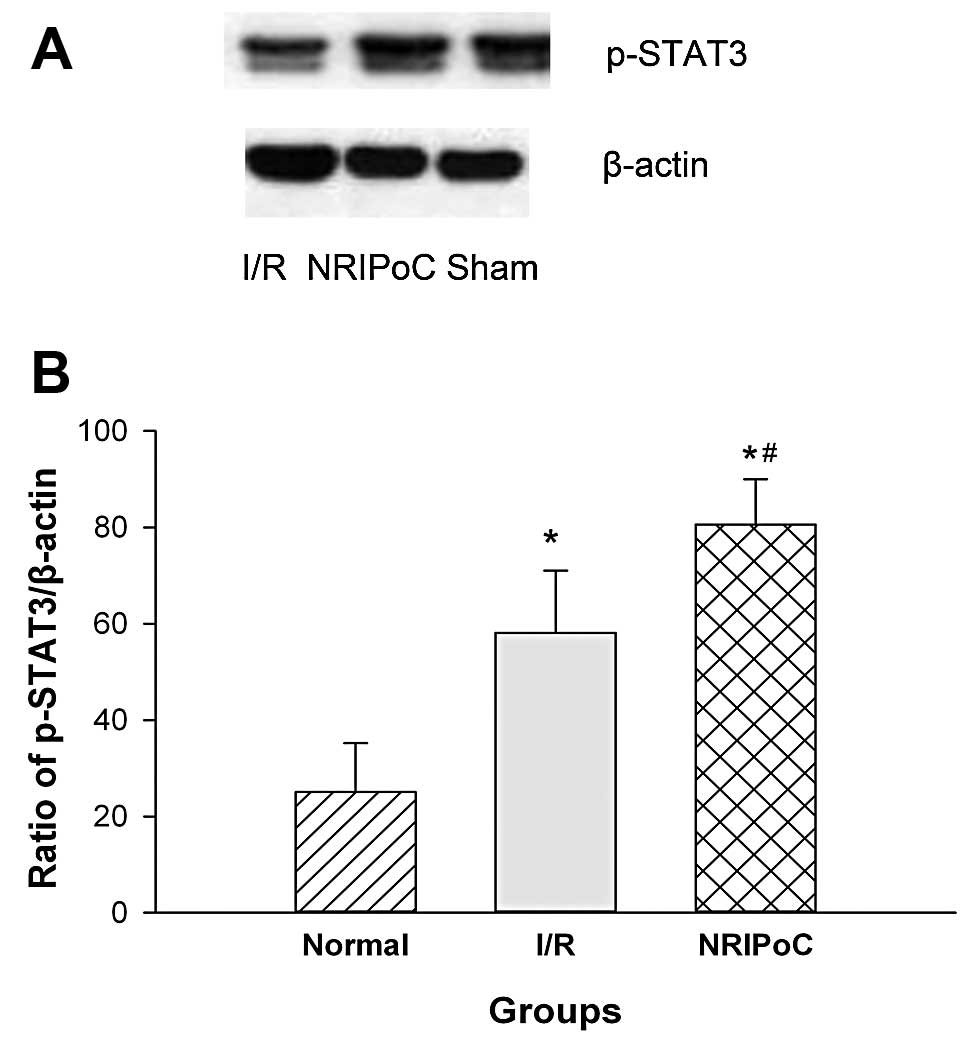
The expression of p-STAT3 protein in the ischemic
penumbra area was significantly increased following IRI (P<0.05)
(Fig. 7). NRIPoC significantly
increased p-STAT3 protein expression in the ischemic penumbra area
compared with the I/R group (P<0.05).
Effects of NRIPoC on I/R-induced NF-κB
and TNF-α expression in the ischemic penumbra area
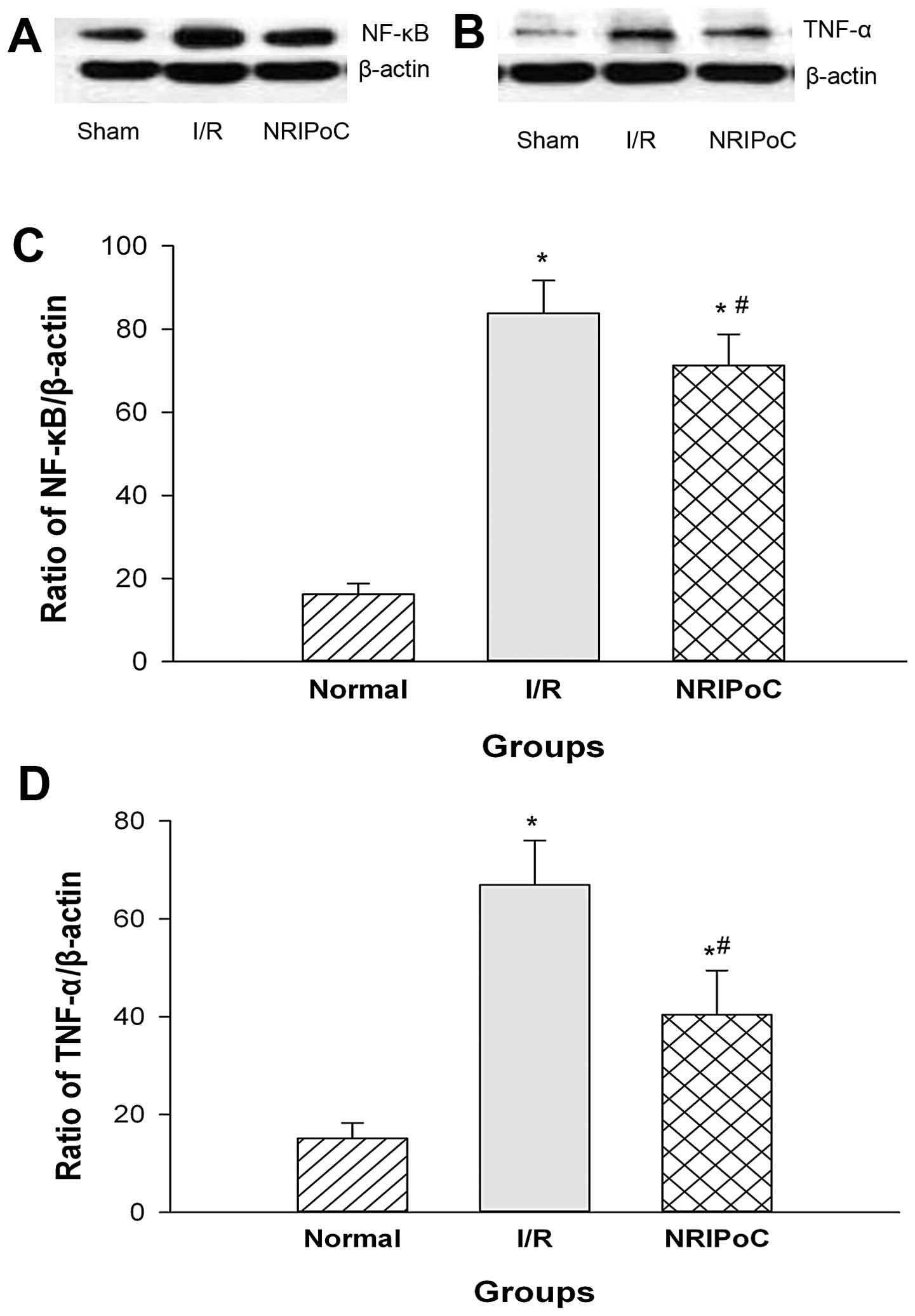
The NF-κB and TNF-α protein expression levels in the
ischemic penumbra area were significantly increased following IRI
(P<0.05) (Fig. 8). NRIPoC
significantly decreased NF-κB and TNF-α expression compared with
the I/R group (P<0.05); however, the levels did not return to
those of the sham-operated group (P<0.05).
Effects of NRIPoC on neurological
function
Neurological function in the sham-operated group was
normal (NDS =0) (Table I). The
median NDS of the NRIPoC group was 1 score lower on the scale than
that in the I/R group, but there was no statistically significant
difference between the 2 groups (P>0.05), and the median NDS in
the NRIPoC group was still higher than that of the sham-operated
group (P<0.01).
 | Table INeurological deficit score of the
rats in this study. |
Table I
Neurological deficit score of the
rats in this study.
| Group | Neurological
deficit score | Median
(minimum-maximum) |
|---|
|
|---|
| 0 | 1 | 2 | 3 | 4 |
|---|
| Sham | 15 | 0 | 0 | 0 | 0 | 0 |
| I/R | 0 | 0 | 5 | 10 | 0 | 3 (2–3) |
| NRIPoC | 0 | 3 | 6 | 6 | 0 | 2 (1–3) |
Discussion
In this study, we used an in vivo model of
MCAO-induced focal ischemia, which has the advantage of being
simple and reliable without requiring a craniectomy. MCAO has been
widely used to study the effects and mechanisms of pre- and
postconditioning on focal cerebral ischemia. Although there have
been various ischemic penumbra definitions following focal cerebral
ischemia put forward over the 30 years since ischemic penumbra was
first introduced by Astrup et al (50) in 1981, salvaging the ischemic
penumbra is the current primary therapeutic target in acute stroke
(51). Following cerebral
ischemia reperfusion, many neurons in the ischemic penumbra or
peri-infarct zone may undergo apoptosis after several hours or
days. This renders the time window of therapeutic opportunity for
salvaging those cells of the ischemic penumbra.
We hypothesized that the neuroprotective effects of
NRIPoC may be mediated through its effects on the ischemic
penumbra. The results from the present study demonstrate that
NRIPoC significantly increased the number of surviving neurons and
decreased neuronal apoptosis in the ischemic penumbra following IRI
in a rat model of focal cerebral ischemia induced by 90 min of
MCAO. Conversely, there was no difference in neuronal survival in
the ischemic core between the I/R and NRIPoC groups. The finding
that NRIPoC significantly decreased neuronal apoptosis in the
penumbra are is consistent with the results of a previous study
demonstrating that RIPoC significantly attenuated neuronal
apoptosis associated with global cerebral ischemia in the
hippocampal CA1 region (19). TTC
is a light-sensitive compound that reacts with lactate
dehydrogenase (LDH), which stains normal brain tissue red, while
ischemic brain tissue remains white. In the present study, TTC
staining revealed that NRIPoC significantly reduced the cerebral
infarct size, suggesting that NRIPoC increased cell viability in
the cerebral penumbra area. This observation is in accordance with
the results of previous studies by Ren et al (13,17).
In addition to necrosis, there is overwhelming
evidence suggesting that apoptosis significantly contributes to
cell death following cerebral IRI (27,52,53). Preventing apoptosis in the
penumbra area may reduce neuronal loss and limit cerebral IRI.
Ischemia pre- and postconditioning has been shown to decrease
neuronal apoptosis and attenuate IRI in brain tissue (6,54–57). Similarly, NRIPoC significantly
decreased neuronal apoptosis and reduced the cerebral infarct
volume after focal cerebral ischemia in the present study. These
results suggest that anti-apoptotic signaling may be an important
mechanism responsible for the neuroprotective effects of
NRIPoC.
Bcl-2 family members are key regulators of apoptosis
and modulate mitochondrial membrane permeability. Following cell
ischemia or hypoxia, the expression of mitochondrial membrane
proteins is increased, and the apoptotic protein, Bax, which
translocates from the cytoplasm into the mitochondria, promotes the
release of cytochrome c, while the anti-apoptotic protein,
Bcl-2, located in the mitochondrial wall, inhibits the release of
cytochrome c (58–60). Cytochrome c initiates the
process of apoptosis by activating the caspase cascade (59). Interactions between the pro- and
anti-apoptotic Bcl-2 family proteins on the outer mitochondrial
membrane play a key role in cell survival. Therefore, the Bcl-2/Bax
protein ratio may determine the level of cell apoptosis or survival
following apoptotic injury (61).
In the present study, NRIPoC significantly decreased both Bax and
Bcl-2 protein expression and increased the Bcl-2/Bax ratio. These
results suggest that the anti-apoptotic effects of NRIPoC may be
closely associated with changes in Bcl-2 and Bax expression. The
mechanisms through which NRIPoC modulates Bcl-2 and Bax expression
following cerebral IRI in vivo are currently unknown. The
JAK/STAT signaling pathway transduces stress-activating
extracellular chemical signals into cellular responses for a number
of pathophysiological processes, such as immunity, inflammation and
apoptosis (62). p-STAT1 and
p-STAT3 expression have been shown to be increased following focal
cerebral ischemia, and the activation of STAT1 may lead to ischemic
neuronal necrosis (42,43,63); however, the effects of activated
STAT3 following focal cerebral ischemia have not yet been
elucidated. The function of activated STAT3 is controversial; some
studies have associated it with survival (43,64), while others have related it to
cell death (65).
There is no existing evidence that STAT3 is involved
in neuronal apoptosis by the regulation of Bcl-2 and Bax expression
following cerebral ischemia. Previous studies have confirmed that
STAT3 alterations affect Bcl-2 and Bax protein expression
(decreased Bcl-2 and increased Bax) and induce inflammation and
apoptosis in many types of tumor cells (66–68). Lee et al (69) demonstrated that co-activated NF-κB
and STAT3 modulate Bax/Bcl-xL expression and promote cell survival
in head and neck squamous cell carcinoma.
In mycosis fungoides tumor cells, some
apoptosis-related genes, such as Bcl-2 and Bax, have been
identified as STAT3 target genes (66). In primary cortical neurons and
murine models of stroke, the activation of the Jak2/Stat3 pathway
by secretoneurin has been found to exert neuroprotective effects
and induce neuronal plasticity after hypoxia and ischemic insult
(70). Using a mouse model of
transient focal cerebral ischemia, Jung et al (71) demonstrated that interleukin-6
(IL-6) exerted protective effects against cerebral ischemic injury
through IL-6R-mediated STAT3 activation and antioxidative
signaling. Similar to previous study by Suzuki et al
(43), we found that p-STAT3
expression was increased following focal cerebral ischemia.
Furthermore, NRIPoC induced a more pronounced increase in p-STAT3
expression in the ischemic penumbra area. This, coupled with the
Bcl-2/Bax ratio was consistent with the decrease in the number of
apoptotic neurons and the attenuation of cerebral I/R-induced brain
injury. This result provides an experimental basis for the clinical
application of NRIPoC for reducing the cerebral infarct volume
following stroke through the rescue of the remaining surviving
neurons in the penumbra area. These results indicate that p-STAT3
may be an endogenous anti-injury factor, and that the JAK2-STAT3
signaling pathway may mediate the neuroprotective effects of RIPoC
though the regulation of Bcl-2/Bax expression in rat focal cerebral
ischemia. This assumption requires further confirmation by the use
of specific inhibitors of the JAK2-STAT3 signaling pathway in order
to clarify the effects of STAT3.
The pathogenesis of cerebral IRI involves complex
pathophysiological events, such as the excessive production of
reactive oxygen species, the excessive activation of glutamate
receptors, the overload of intracellular calcium and inflammation
(26,27,72). Early inflammatory mediators
following cerebral IR are the basis of ischemic injury that then
transforms to inflammatory injury. Accumulating evidence suggests
that the neuroinflammatory processes occurring after cerebral
ischemia involve various pathways and molecules (26). Of these, TNF-α is responsible for
the degree of inflammation. NF-κB is a critical transcription
factor for the maximal expression of several cytokines that are
involved in inflammation (73).
Whereas TNF-α and IL-1β appear to exacerbate cerebral injury,
transforming growth factor-β and IL-10 may exert neuroprotective
effects during cerebral ischemia reperfusion (26). Activated NF-κB upregulates the
transcription of TNF-α and IL-1β. An increase in TNF-α activates
NF-κB, and IL-10 inhibits the activation of NF-κB mediated by
endotoxins. The synthesis of inflammatory mediators is greatly
increased by the overexpression of TNF-α following I/R, which
results in an imbalance between inflammatory and anti-inflammatory
factors and the amplification of the inflammatory cascade. The
overexpression of TNF-α may aggravate brain damage following
cerebral I/R, which is though to be mediated by the transcriptional
regulation of inflammatory factors by NF-κB (26,74). The present study demonstrated that
TNF-α and NF-κB protein expression in the ischemic penumbra area
was significantly increased following I/R, and this increase was
reduced by NRIPoC, which suggests that the neuroprotective effects
of NRIPoC may correlate with the increased inflammatory response in
the ischemic penumbra area following IRI. The binding of TNF-α to
its receptor, TNF-RI (p55TNFR), induces the caspase cascade, which
promotes apoptosis. Several alternative mechanisms of TNF-α-induced
apoptosis have also been described (75). The downregulation of TNF-α by
NRIPoC in the ischemic penumbra area may be one of the reasons why
NRIPoC reduced neuronal apoptosis following cerebral I/R. Further
studies are required to examine the role of STAT3 signaling in
regulating TNF-α and NF-κB expression induced by cerebral I/R, as
well as the neuroprotective effects of NRIPoC.
In conclusion, the present study demonstrates that
NRIPoC attenuates cerebral IRI in an in vivo rat model
through the alleviation of inflammation, regulation of the STAT3
signaling pathway and Bcl-2 and Bax expression levels, and that
these changes are possibly due to the reduction of apoptosis.
References
|
1
|
Bonita R: Epidemiology of stroke. Lancet.
339:342–344. 1992. View Article : Google Scholar
|
|
2
|
Murray CJ and Lopez AD: Mortality by cause
for eight regions of the world: Global Burden of Disease Study.
Lancet. 349:1269–1276. 1997. View Article : Google Scholar
|
|
3
|
Gourdin MJ, Bree B and De Kock M: The
impact of ischaemia-reperfusion on the blood vessel. Eur J
Anaesthesiol. 26:537–547. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Murry CE, Jennings RB and Reimer KA:
Preconditioning with ischemia: a delay of lethal cell injury in
ischemic myocardium. Circulation. 74:1124–1136. 1986. View Article : Google Scholar
|
|
5
|
Kitagawa K, Matsumoto M, Tagaya M, et al:
‘Ischemic tolerance’ phenomenon found in the brain. Brain Res.
528:21–24. 1990.
|
|
6
|
Zhao H, Sapolsky RM and Steinberg GK:
Interrupting reperfusion as a stroke therapy: ischemic
postconditioning reduces infarct size after focal ischemia in rats.
J Cereb Blood Flow Metab. 26:1114–1121. 2006.PubMed/NCBI
|
|
7
|
Wang JY, Shen J, Gao Q, et al: Ischemic
postconditioning protects against global cerebral
ischemia/reperfusion-induced injury in rats. Stroke. 39:983–990.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Liu X, Chen H, Zhan B, et al: Attenuation
of reperfusion injury by renal ischemic postconditioning: the role
of NO. Biochem Biophys Res Commun. 359:628–634. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yellon DM and Dana A: The preconditioning
phenomenon: A tool for the scientist or a clinical reality? Circ
Res. 87:543–550. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li CM, Zhang XH, Ma XJ and Luo M: Limb
ischemic postconditioning protects myocardium from
ischemia-reperfusion injury. Scand Cardiovasc J. 40:312–317. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hong DM, Jeon Y, Lee CS, et al: Effects of
remote ischemic preconditioning with postconditioning in patients
undergoing off-pump coronary artery bypass surgery - randomized
controlled trial. Circ J. 76:884–890. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Vinten-Johansen J and Shi W: The science
and clinical translation of remote postconditioning. J Cardiovasc
Med (Hagerstown). 14:206–213. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ren C, Yan Z, Wei D, Gao X, Chen X and
Zhao H: Limb remote ischemic postconditioning protects against
focal ischemia in rats. Brain Res. 1288:88–94. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sun J, Tong L, Luan Q, et al: Protective
effect of delayed remote limb ischemic postconditioning: role of
mitochondrial K(ATP) channels in a rat model of focal cerebral
ischemic reperfusion injury. J Cereb Blood Flow Metab. 32:851–859.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang Q, Zhang X, Ding Q, et al: Limb
remote postconditioning alleviates cerebral reperfusion injury
through reactive oxygen species-mediated inhibition of delta
protein kinase C in rats. Anesth Analg. 113:1180–1187. 2011.
View Article : Google Scholar
|
|
16
|
Zhou Y, Fathali N, Lekic T, et al: Remote
limb ischemic postconditioning protects against neonatal
hypoxic-ischemic brain injury in rat pups by the opioid
receptor/Akt pathway. Stroke. 42:439–444. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ren C, Gao X, Steinberg GK and Zhao H:
Limb remote-preconditioning protects against focal ischemia in rats
and contradicts the dogma of therapeutic time windows for
preconditioning. Neuroscience. 151:1099–1103. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Qi ZF, Luo YM, Liu XR, et al:
AKT/GSK3beta-dependent autophagy contributes to the neuroprotection
of limb remote ischemic postconditioning in the transient cerebral
ischemic rat model. CNS Neurosci Ther. 18:965–973. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Peng B, Guo QL, He ZJ, et al: Remote
ischemic postconditioning protects the brain from global cerebral
ischemia/reperfusion injury by up-regulating endothelial nitric
oxide synthase through the PI3K/Akt pathway. Brain Res.
1445:92–102. 2012. View Article : Google Scholar
|
|
20
|
Koch S, Katsnelson M, Dong C and
Perez-Pinzon M: Remote ischemic limb preconditioning after
subarachnoid hemorrhage: a phase Ib study of safety and
feasibility. Stroke. 42:1387–1391. 2011. View Article : Google Scholar
|
|
21
|
Walsh SR, Nouraei SA, Tang TY, Sadat U,
Carpenter RH and Gaunt ME: Remote ischemic preconditioning for
cerebral and cardiac protection during carotid endarterectomy:
results from a pilot randomized clinical trial. Vasc Endovascular
Surg. 44:434–439. 2010. View Article : Google Scholar
|
|
22
|
Hu S, Dong H, Zhang H, et al: Noninvasive
limb remote ischemic preconditioning contributes neuroprotective
effects via activation of adenosine A1 receptor and redox status
after transient focal cerebral ischemia in rats. Brain Res.
1459:81–90. 2012. View Article : Google Scholar
|
|
23
|
Zhong H, Gao Z, Chen M, et al:
Cardioprotective effect of remote ischemic postconditioning on
children undergoing cardiac surgery: a randomized controlled trial.
Paediatr Anaesth. 23:726–733. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liu X, Sha O and Cho EY: Remote ischemic
postconditioning promotes the survival of retinal ganglion cells
after optic nerve injury. J Mol Neurosci. 51:639–646. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Dave KR, Saul I, Prado R, Busto R and
Perez-Pinzon MA: Remote organ ischemic preconditioning protect
brain from ischemic damage following asphyxial cardiac arrest.
Neurosci Lett. 404:170–175. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lakhan SE, Kirchgessner A and Hofer M:
Inflammatory mechanisms in ischemic stroke: therapeutic approaches.
J Transl Med. 7:972009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Broughton BR, Reutens DC and Sobey CG:
Apoptotic mechanisms after cerebral ischemia. Stroke. 40:e331–e339.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
MacManus JP and Linnik MD: Gene expression
induced by cerebral ischemia: an apoptotic perspective. J Cereb
Blood Flow Metab. 17:815–832. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li Y, Powers C, Jiang N and Chopp M:
Intact, injured, necrotic and apoptotic cells after focal cerebral
ischemia in the rat. J Neurol Sci. 156:119–132. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Feuerstein GZ, Wang X and Barone FC:
Inflammatory gene expression in cerebral ischemia and trauma.
Potential new therapeutic targets. Ann NY Acad Sci. 825:179–193.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Benjelloun N, Renolleau S, Represa A,
Ben-Ari Y and Charriaut-Marlangue C: Inflammatory responses in the
cerebral cortex after ischemia in the P7 neonatal Rat. Stroke.
30:1916–1924. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zeng L, Wang Y, Liu J, et al:
Pro-inflammatory cytokine network in peripheral inflammation
response to cerebral ischemia. Neurosci Lett. 548:4–9. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Saito K, Suyama K, Nishida K, Sei Y and
Basile AS: Early increases in TNF-alpha, IL-6 and IL-1 beta levels
following transient cerebral ischemia in gerbil brain. Neurosci
Lett. 206:149–152. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Maddahi A, Kruse LS, Chen QW and Edvinsson
L: The role of tumor necrosis factor-α and TNF-α receptors in
cerebral arteries following cerebral ischemia in rat. J
Neuroinflammation. 8:1072011.
|
|
35
|
Vakili A, Mojarrad S, Akhavan MM and
Rashidy-Pour A: Pentoxifylline attenuates TNF-α protein levels and
brain edema following temporary focal cerebral ischemia in rats.
Brain Res. 1377:119–125. 2011.
|
|
36
|
Latanich CA and Toledo-Pereyra LH:
Searching for NF-kappaB-based treatments of ischemia reperfusion
injury. J Invest Surg. 22:301–315. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ridder DA and Schwaninger M: NF-kappaB
signaling in cerebral ischemia. Neuroscience. 158:995–1006. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
MacDonald RL and Stoodley M:
Pathophysiology of cerebral ischemia. Neurol Med Chir (Tokyo).
38:1–11. 1998. View
Article : Google Scholar
|
|
39
|
Zhang S, Qi Y, Xu Y, et al: Protective
effect of flavonoid-rich extract from Rosa laevigata Michx
on cerebral ischemia-reperfusion injury through suppression of
apoptosis and inflammation. Neurochem Int. 63:522–532.
2013.PubMed/NCBI
|
|
40
|
Zhang L, Dong LY, Li YJ, Hong Z and Wei
WS: The microRNA miR-181c controls microglia-mediated neuronal
apoptosis by suppressing tumor necrosis factor. J
Neuroinflammation. 9:2112012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Gu JH, Ge JB, Li M, Wu F, Zhang W and Qin
ZH: Inhibition of NF-κB activation is associated with
anti-inflammatory and anti-apoptotic effects of Ginkgolide B in a
mouse model of cerebral ischemia/reperfusion injury. Eur J Pharm
Sci. 47:652–660. 2012.
|
|
42
|
Justicia C, Gabriel C and Planas AM:
Activation of the JAK/STAT pathway following transient focal
cerebral ischemia: signaling through Jak1 and Stat3 in astrocytes.
Glia. 30:253–270. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Suzuki S, Tanaka K, Nogawa S, Dembo T,
Kosakai A and Fukuuchi Y: Phosphorylation of signal transducer and
activator of transcription-3 (Stat3) after focal cerebral ischemia
in rats. Exp Neurol. 170:63–71. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Satriotomo I, Bowen KK and Vemuganti R:
JAK2 and STAT3 activation contributes to neuronal damage following
transient focal cerebral ischemia. J Neurochem. 98:1353–1368. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Xie HF, Xu RX, Wei JP, Jiang XD and Liu
ZH: P-JAK2 and P-STAT3 protein expression and cell apoptosis
following focal cerebral ischemia-reperfusion injury in rats. Nan
Fang Yi Ke Da Xue Xue Bao. 27:208–211. 2182007.(In Chinese).
|
|
46
|
Longa EZ, Weinstein PR, Carlson S and
Cummins R: Reversible middle cerebral artery occlusion without
craniectomy in rats. Stroke. 20:84–91. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Wang N, Guo QL, Ye Z, Xia PP, Wang E and
Yuan YJ: Preconditioning of intravenous parecoxib attenuates focal
cerebral ischemia/reperfusion injury in rats. Chin Med J (Engl).
124:2004–2008. 2011.PubMed/NCBI
|
|
48
|
Wei L, Yu SP, Gottron F, Snider BJ, Zipfel
GJ and Choi DW: Potassium channel blockers attenuate hypoxia- and
ischemia-induced neuronal death in vitro and in vivo. Stroke.
34:1281–1286. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Sambrook J, Fritsch EF and Maniatis T:
Molecular Cloning: a Laboratory Manual. 2nd edition. Cold Spring
Harbor Laboratory Press; New York: 1989
|
|
50
|
Astrup J, Siesjö BK and Symon L:
Thresholds in cerebral ischemia - the ischemic penumbra. Stroke.
12:723–725. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Paciaroni M, Caso V and Agnelli G: The
concept of ischemic penumbra in acute stroke and therapeutic
opportunities. Eur Neurol. 61:321–330. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Tajiri S, Oyadomari S, Yano S, et al:
Ischemia-induced neuronal cell death is mediated by the endoplasmic
reticulum stress pathway involving CHOP. Cell Death Differ.
11:403–415. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Hu BR, Martone ME, Jones YZ and Liu CL:
Protein aggregation after transient cerebral ischemia. J Neurosci.
20:3191–3199. 2000.PubMed/NCBI
|
|
54
|
Xing B, Chen H, Zhang M, et al: Ischemic
postconditioning inhibits apoptosis after focal cerebral
ischemia/reperfusion injury in the rat. Stroke. 39:2362–2369. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Liu Y, Sercombe R, Xie D, Liu K and Chen
L: Inhibition of caspase-9 activation and apoptosis is involved in
ischemic preconditioning-induced neuroprotection in rat brain.
Neurol Res. 29:855–861. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Prasad SS, Russell M and Nowakowska M:
Neuroprotection induced in vitro by ischemic preconditioning and
postconditioning: modulation of apoptosis and PI3K-Akt pathways. J
Mol Neurosci. 43:428–442. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Xia DY, Li W, Qian HR, Yao S, Liu JG and
Qi XK: Ischemia preconditioning is neuroprotective in a rat
cerebral ischemic injury model through autophagy activation and
apoptosis inhibition. Braz J Med Biol Res. 46:580–588. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Kroemer G: Mitochondrial control of
apoptosis: an introduction. Biochem Biophys Res Commun.
304:433–435. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Lim ML, Lum MG, Hansen TM, Roucou X and
Nagley P: On the release of cytochrome c from mitochondria
during cell death signaling. J Biomed Sci. 9:488–506. 2002.
|
|
60
|
Kuwana T, Mackey MR, Perkins G, et al:
Bid, Bax, and lipids cooperate to form supramolecular openings in
the outer mitochondrial membrane. Cell. 111:331–342. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Love S: Apoptosis and brain ischaemia.
Prog Neuropsychopharmacol Biol Psychiatry. 27:267–282. 2003.
View Article : Google Scholar
|
|
62
|
Planas AM, Gorina R and Chamorro A:
Signalling pathways mediating inflammatory responses in brain
ischaemia. Biochem Soc Trans. 34:1267–1270. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Planas AM, Soriano MA, Berruezo M, et al:
Induction of Stat3, a signal transducer and transcription factor,
in reactive microglia following transient focal cerebral ischaemia.
Eur J Neurosci. 8:2612–2618. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Yamaguchi K, Itoh Y, Yokomizo C, et al:
Blockade of interleukin-6 signaling enhances hepatic steatosis but
improves liver injury in methionine choline-deficient diet-fed
mice. Lab Invest. 90:1169–1178. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Wen TC, Peng H, Hata R, Desaki J and
Sakanaka M: Induction of phosphorylated-Stat3 following focal
cerebral ischemia in mice. Neurosci Lett. 303:153–156. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Nielsen M, Kaestel CG, Eriksen KW, et al:
Inhibition of constitutively activated Stat3 correlates with
altered Bcl-2/Bax expression and induction of apoptosis in mycosis
fungoides tumor cells. Leukemia. 13:735–738. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Dinasarapu AR, Gupta S, Ram Maurya M, et
al: A combined omics study on activated macrophages - enhanced role
of STATs in apoptosis, immunity and lipid metabolism.
Bioinformatics. 29:2735–2743. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Spehlmann ME, Manthey CF, Dann SM, et al:
Trp53 deficiency protects against acute intestinal inflammation. J
Immunol. 191:837–847. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Lee TL, Yeh J, Friedman J, et al: A signal
network involving coactivated NF-kappaB and STAT3 and altered p53
modulates BAX/BCL-XL expression and promotes cell survival of head
and neck squamous cell carcinomas. Int J Cancer. 122:1987–1998.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Shyu WC, Lin SZ, Chiang MF, et al:
Secretoneurin promotes neuroprotection and neuronal plasticity via
the Jak2/Stat3 pathway in murine models of stroke. J Clin Invest.
118:133–148. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Jung JE, Kim GS and Chan PH:
Neuroprotection by interleukin-6 is mediated by signal transducer
and activator of transcription 3 and antioxidative signaling in
ischemic stroke. Stroke. 42:3574–3579. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Allen CL and Bayraktutan U: Oxidative
stress and its role in the pathogenesis of ischaemic stroke. Int J
Stroke. 4:461–470. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Blackwell TS and Christman JW: The role of
nuclear factor-kappa B in cytokine gene regulation. Am J Respir
Cell Mol Biol. 17:3–9. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Ginis I, Jaiswal R, Klimanis D, Liu J,
Greenspon J and Hallenbeck JM: TNF-alpha-induced tolerance to
ischemic injury involves differential control of NF-kappaB
transactivation: the role of NF-kappaB association with p300
adaptor. J Cereb Blood Flow Metab. 22:142–152. 2002.PubMed/NCBI
|
|
75
|
Jurewicz A, Matysiak M, Tybor K, Kilianek
L, Raine CS and Selmaj K: Tumour necrosis factor-induced death of
adult human oligodendrocytes is mediated by apoptosis inducing
factor. Brain. 128:2675–2688. 2005. View Article : Google Scholar : PubMed/NCBI
|