Introduction
Renal fibrosis is the pathological response of
kidneys to chronic impairment, which is both a common pathological
basis for the progressive course of chronic kidney disease and an
essential process from chronic kidney disease to the terminal stage
of renal failure. Progressive glomerulosclerosis is a common
pathological characteristic of a number of chronic glomerular
diseases (1). In general, the
quantity, form and position of glomerular mesangial cells (GMCs)
are relatively stable and their ability to produce ground substance
is relatively weak. However, when mesangial cells (MCs) are
stimulated by damaging factors and hazardous substances and are
thus revitalized and proliferated, these cells secrete a large
amount of ground substance. In addition to the reduced degradation,
glomerular sclerosis will eventually develop.
Transforming growth factor-β1 (TGF-β1) is a
well-recognized factor contributing to inflammation and fibrosis.
It has demonstrated that TGF-β is likely to be the core factor that
leads to glomerular sclerosis. Over the past decades, studies have
revealed various molecular pathways involved in the development and
occurrence of renal fibrosis, which aids in the deeper
understanding of the pathogenesis of diseases and provides novel
strategies for effective prevention and treatment (2–6).
Prostaglandin E2 (PGE2) is the
metabolite of arachidonic acid, which exists in the body.
Membrane-associated PGE2 synthase-1 (mPGES-1) in
combination with cyclooxygenase-2 (COX-2) is the key enzyme for the
compound of PGE2 (7).
The vital role of PGE2 has been demonstrated in a number
of pathophysiological processes, such as blood pressure regulation,
inflammatory response, immunoreaction and energy metabolism
(8). Increasing evidence
indicates that PGE2 also participates in the regulation
of the contraction of vascular smooth muscle, glomerular
filtration, the release of renin and water and salt transportation
in kidney tubules (9). The
biological function of PGE2 is exerted by 4 types of
membrane receptors, including EP1, EP2, EP3 and EP4 (10), which are coded by different genes
with different signal transduction mechanisms (11). Upon activation, EP1 increases the
concentration of Ca2+ in the intracytoplasm. EP2 and EP4
couple with activated G proteins (Gs) to increase the level of
cyclic AMP (cAMP) in cells. On the contrary, EP3 is able to reduce
the cAMP level (12). Previous
studies have demonstrated that the EP1 receptor is involved in and
regulates a number of physiological and pathological processes,
such as the mediation of stress response (13,14), the promotion of chemical
carcinogen generation (15) and
the mediation of inflammation, fever and pain (16). Moreover, Makino et al
(17) found strong hybridization
signals of EP1 mRNA in the glomerulus and kidney tubules in the
renal cortex through hybridization in situ, while in the
study by Qian et al (18),
it was demonstrated that PGE2 mediates MC proliferation
and cell cycle arrest through the EP1 receptor. All the
abovementioned findings suggest that the function of the EP1
receptor is related to the damage to MCs and the production of
glomerulosclerosis.
At present, the function of the EP1 receptor in the
pathogenesis of renal fibrosis and chronic kidney diseases is not
well understood. Therefore, in this study, we cultured primary GMCs
isolated from wild-type (WT) mice and mice in which the EP1 gene
was knocked down. We specifically activated EP1 through EP1
receptor stimulation, treating the MCs for 24 h with TGF-β1. We
aimed to determine the effects of EP1 receptor, which is induced by
TGF-β1, on the proliferation of mouse MCs, the accumulation of
extracellular matrix and the expression of prostaglandin synthase.
In addition, we examined the changes occurring in the extracellular
signal-regulated kinase (ERK) signaling pathway, in order to
elucidate the possible mechanisms involved.
Materials and methods
Animals and cell culture
We used male mice, 8–12 weeks old, with a C57BL/6
background. The use of the animals was approved by the Animal
Experimentation Committee of Peking University Health Science
Center, Beijing, China. The EP1 receptor-deficient
(EP1−/−) mice were generated as previously described
(19), and the lack of functional
EP1 receptor was confirmed by quantitative reverse
transcription-polymerase chain reaction (RT-qPCR) and western blot
analysis. Primary mice GMCs were cultured. The isolation of GMCs
was performed according to the method described in the study by
Takemoto et al (20). In
brief, kidneys from 8- to 12-week-old male C57BL/6 mice were
obtained. The glomeruli were purified from minced renal cortex
tissue and the glomeruli suspension was digested for 40 min at 37°C
with type I collagenase. The glomeruli were then collected through
70 and 40 μm stainless steel sieves. The digested samples were then
centrifuged at 1,000 rpm for 5 min, and the precipitate was
resuspended in growth medium [Dulbecco’s modified Eagle’s medium
(DMEM) supplemented with 20% fetal bovine serum (FBS) (Gibco,
Invitrogen, Carlsbad, CA, USA)]. The cells were cultured at 37°C in
a humidified incubator containing 5% CO2. Cells at
passages 8 to 10 were used. Subsequently, the primary mouse GMCs
were subjected to different treatments and were divided into 5
different groups as follows: i) WT group, ii) WT + TGF-β1 group,
iii) EP1−/− group, iv) EP1−/− + TGF-β1 group
and v) WT + TGF-β1 + EP1 agonist group. Prior to the experiments,
the cells were incubated without FBS for 12 h. Each individual
experiment was repeated at least 3 times with different cell
preparations.
Reagents
The PGE2 EIA kit was obtained from Cayman
Chemical (Ann Arbor, MI, USA). 17-Phenyl trinor PGE2
ethyl amide, a selective agonist of EP1 was obtained from
Sigma-Aldrich (St. Louis, MO, USA). EP1 antibody was also from
Cayman Chemical. The following antibodies were also used (all mouse
antibodies): ERK (sc-135900; Santa Cruz Biotechnology, Santa Cruz,
CA, USA), phosphorylated ERK (p-ERK, sc-16982; Santa Cruz
Biotechnology), cyclin D1 (#2922) and proliferating cell nuclear
antigen (PCNA, #2586; both from Cell Signaling Technology, Danvers,
MA, USA), fibronectin (FN, sc-135900; Santa Cruz Biotechnology),
collagen I (ColI, ab6308; Abcam, Cambridge, MA, USA), COX-2
(#12282; Cell Signaling Technology) and mPGES-1 (ab62049; Abcam).
Other reagents were from Sigma-Aldrich unless otherwise
indicated.
RNA extraction and RT-qPCR
Total RNA from the cells in the different groups was
isolated using TRIzol reagent (Life Technologies, Carlsbad, CA,
USA). Total RNA was reverse transcribed into cDNA using the TaqMan
Reverse Transcription Reagents kit (Applied Biosystems, Foster
City, CA, USA) according to the manufacturer’s instructions.
Quantitative (real-time) PCR was performed with the use of iCycler
with the SYBR-Green I probe (Bio-Rad, Hercules, CA, USA). Each
sample was analyzed in triplicate and normalized to β-actin. The FN
PCR protocol was 95°C 5 min→(95°C 30 sec→56°C 30 sec→72°C 30
sec)x35→72°C 10 min. The ColI, COX-2, mPGES-1, cyclin D1, PCNA,
β-actin PCR protocol was 95°C 5 min→(95°C 30 sec→57°C 30 sec→72°C
30 sec) x35→72°C 10 min. The primer sequences are presented in
Table I. PCR products were
validated by electrophoresis on a 2% agarose gel.
 | Table IPrimers used for RT-qPCR. |
Table I
Primers used for RT-qPCR.
| Gene name | Chain | Sequence
(5′→3′) | Product size
(bp) |
|---|
| β-actin | FP |
TTTAATTTCACGCACGATTTC | 150 |
| RP |
CCCATCTATGAGGGTTACGC | |
| EP1 | FP |
TAACGATGGTCACGCGATGG | 291 |
| RP |
ATGCAGTAGTGGGCTTAGGG | |
| COX-2 | FP |
AGAAGGAAATGGCTGCAGAA | 194 |
| RP |
GCTCGGCTTCCAGTATTGAG | |
| mPGES-1 | FP |
CGCGGTGGCTGTCATCA | 205 |
| RP |
AGGGTTGGGTCCCAGGAAT | |
| FN | FP |
AATGGAAAAGGGGAATGGAC | 244 |
| RP |
CTCGGTTGTCCTTCTTGCTC | |
| ColI | FP |
GAGCGGAGAGTACTGGATCG | 142 |
| RP |
GTTCGGGCTGATGTACCAGT | |
| Cyclin D1 | FP |
GCGTACCCTGACACCAATCT | 219 |
| RP |
GAACCGGTCCAGGTAGTTCA | |
| PCNA | FP |
GACACATACCGCTGCGATCG | 307 |
| RP |
TCACCACAGCATCTCCAATAT | |
Western blot analysis
Following the addition of immunoprecipitation cell
lysis buffer, the cells were incubated on ice for 30 min. Following
treatment as described above, the cell lysate was removed to 1.5 ml
EP tubes and spun for 15 min. The supernatant was used for the
experiment. Protein concentrations were determined using the BCA
assay kit (Pierce, Rockford, IL, USA). Samples was diluted in
loading buffer, and then boiled for 5 min. Subsequently, 130 μg
protein of each sample were separated on a 10% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gel and
transferred onto a nitrocellulose membrane at 0.37 A for 1 h.
Following transfer, the blots were blocked at room temperature for
1 h in 5% (w/v) non-fat dried milk. This was followed by incubation
with primary antibodies (mouse anti-FN, mouse anti-ColI, mouse
anti-COX-2, mouse anti-mPGES-1, mouse anti-cyclin D1, mouse
anti-PCNA and mouse anti-ERK, 1:1,000) at 4°C overnight. The
membrane was washed with Tris-buffered saline with Tween-20 (TBST),
incubated with DyLight 800-labeled antibody to mouse IgG (1:5,000)
for 2 h, and the membrane was then scanned using the Odyssey
Infrared Imaging System for semi-quantitative analysis.
PGE2 ELISA analysis
Cell supernatant PGE2 levels were
measured with an Alpha screen PGE2 Assay kit (Perkin
Elmer, Massachusetts, SA, USA). The cells in the WT and
EP1−/− groups were either treated with or without TGF-β1
for 24 h. For PGE2 determination, supernatant was
collected and PGE2 levels were measured according to the
supplier’s instructions.
MTT assay
The cell proliferation ability was detected by MTT
assay. Briefly, MCs (1×103/well) were seeded in 96-well
culture plates and incubated in DMEM containing 20% FBS under
standard conditions until 40–50% confluence, then the medium was
changed to serum-free DMEM for 12 h. The supernatant was removed,
10% FBS DMEM was added to the cells in group 1 (WT group) and group
3 (EP1−/− group), and 10% FBS DMEM containing 10 ng/ml
TGF-β1 was added to the cells in group 2 (WT + TGF-β1 group) and
group 4 (EP1−/− + TGF-β1 group) for 24 h. Following
treatment, 10 μl MTT solution were added to each well, and the
cells were continuously incubated for 1 h. The optical densities
(OD) of the wells were examined at a wavelength of 450 nm (each
group had 6 wells).
Statistical analysis
All values are expressed as the means ± standard
error of the mean (SEM). Data were analyzed using the Student’s
t-test (paired groups) or two-way ANOVA, followed by Bonferroni’s
post hoc test (multigroup comparisons). A value of P<0.05 was
considered to indicate a statistically significant difference.
Results
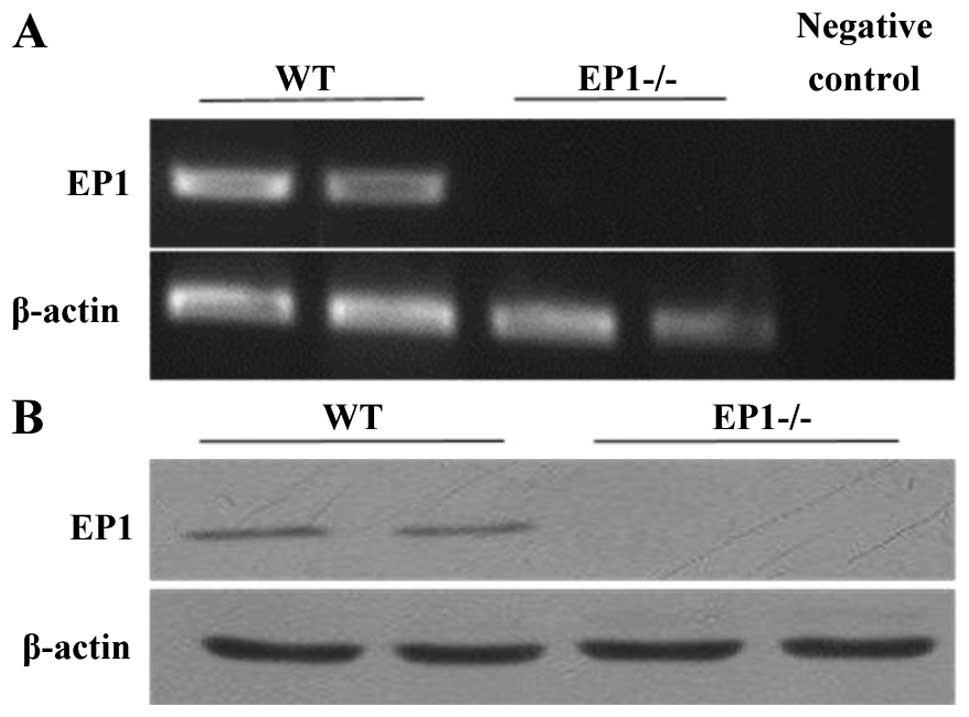
Identification of mice with deficiency of
EP1 gene
Total RNA and total protein was extracted from the
kidneys of the WT mice and (EP1−/−) mice in which the
EP1 gene had been knocked down. RT-qPCR and western blot analysis
were then performed to identify and confirm the generation of
EP1−/− mice (Fig. 1).
The results revealed that EP1 expression in the WT mice was
positive, while it was negative in the mice in which the EP1 gene
had been knocked down.
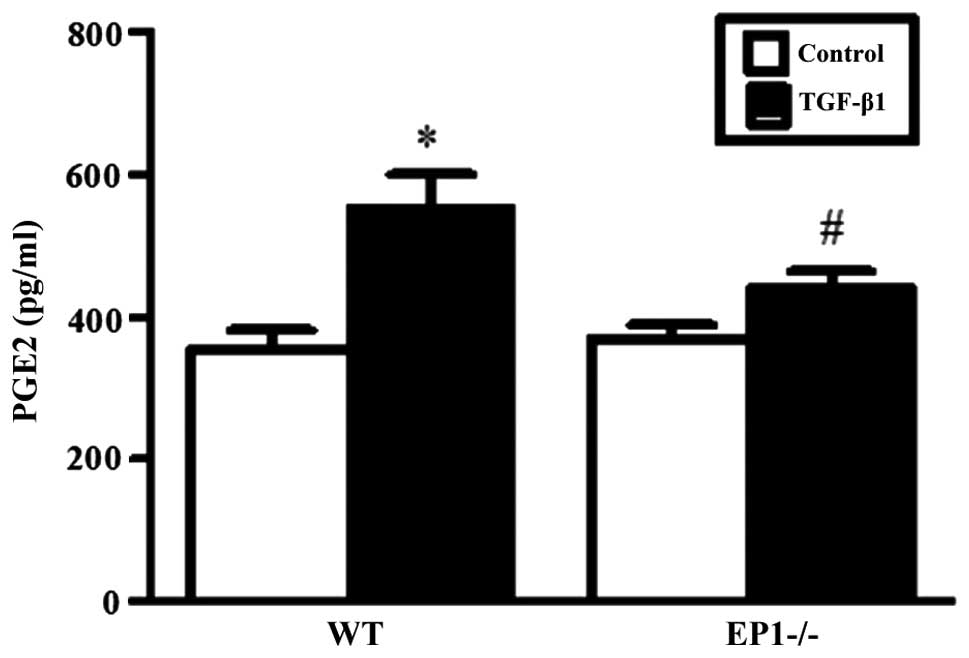
Effect of EP1 receptor on PGE2
content in MCs treated with TGF-β1
Based on our pre-experimental results, the optimal
treatment time and most effective concentration of TGF-β1 for MCs
was 24 h and 10 ng/ml, respectively (data not shown). As shown by
ELISA, treatment of the MCS in the WT and EP1−/− groups
with 10 ng/ml TGF-β1 for 24 h markedly increased the content of
PGE2 (P<0.05) compared with the relative control
untreated groups. However, compared with the WT + TGF-β1 group, the
content of PGE2 was lower in the EP1−/− +
TGF-β1 group (P<0.05) (Fig.
2).
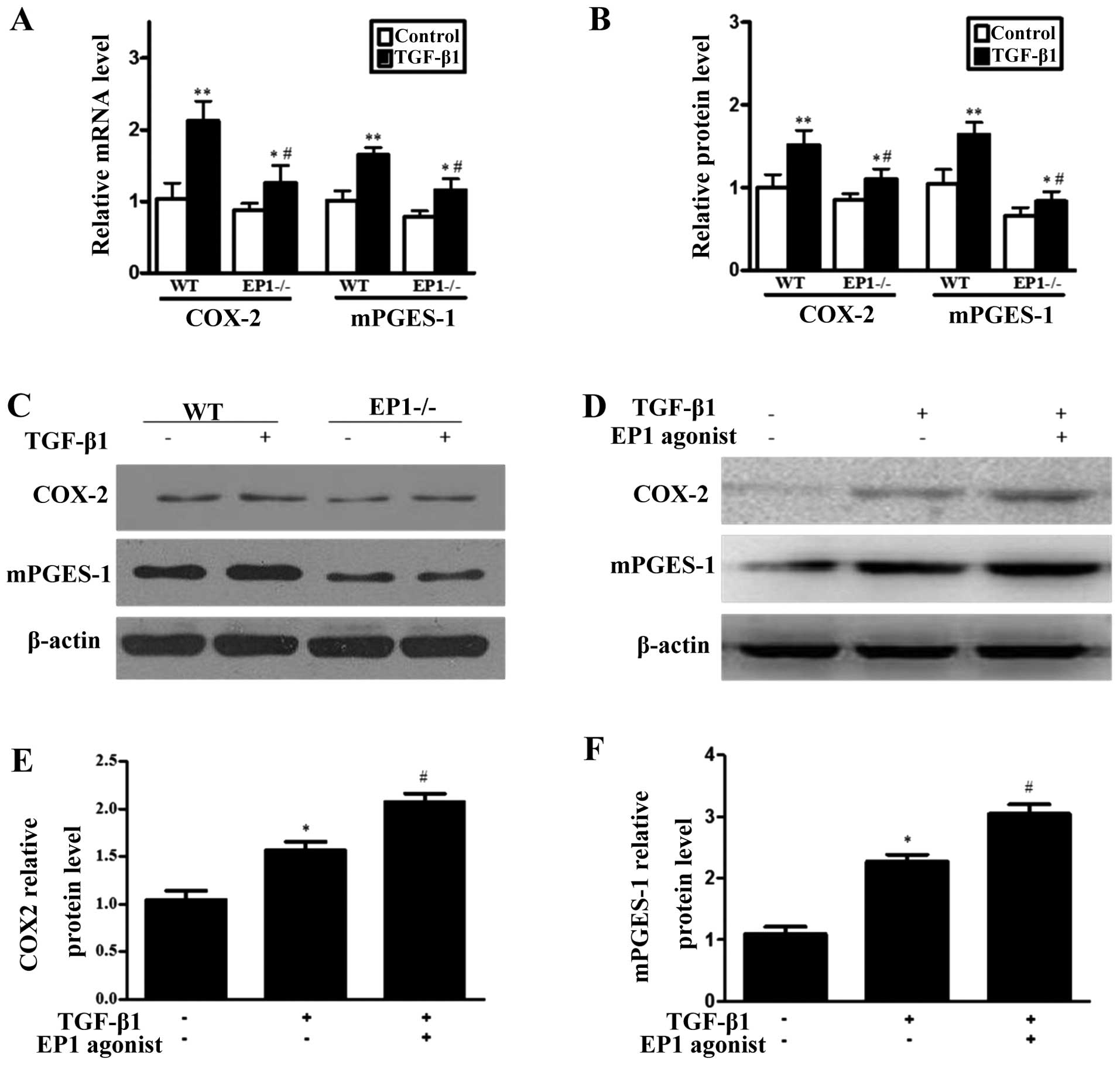
Effect of EP1 receptor on mRNA and
protein expression of COX-2 and mPGES-1 in MCs treated with
TGF-β1
The results from RT-qPCR and western blot analysis
revealed that following treatment of the MCs in the WT and
EP1−/− group with 10 ng/ml TGF-β1 for 24 h, the mRNA and
protein expression of COX-2 and mPGES-1 markedly increased compared
with the relative untreated control groups and the differences were
statistically significant (P<0.05); however, the mRNA and
protein expression of COX-2 and mPGES-1 was lower in the
EP1−/− + TGF-β1 group compared with the WT + TGF-β1
group (P<0.05) (Fig. 3A–C).
Following treatment of the TGF-β1-treated MCs with 10 μM of the EP1
receptor specific agonist, 17-phenyl trinor PGE2 ethyl
amide, the protein expression of COX-2 and mPGES-1 increased
(P<0.05) (Fig. 3D–F). These
results demonstrated that after the WT MCs were treated with
TGF-β1, the expression of COX-2 and mPGES-1 markedly increased; the
deficiency of the EP1 gene reduced the increase in the expression
of mPGES-1 induced by TGF-β1, while the activation of the EP1
receptor promoted the increase in the expression of mPGES-1 in the
MCs induced by TGF-β1.
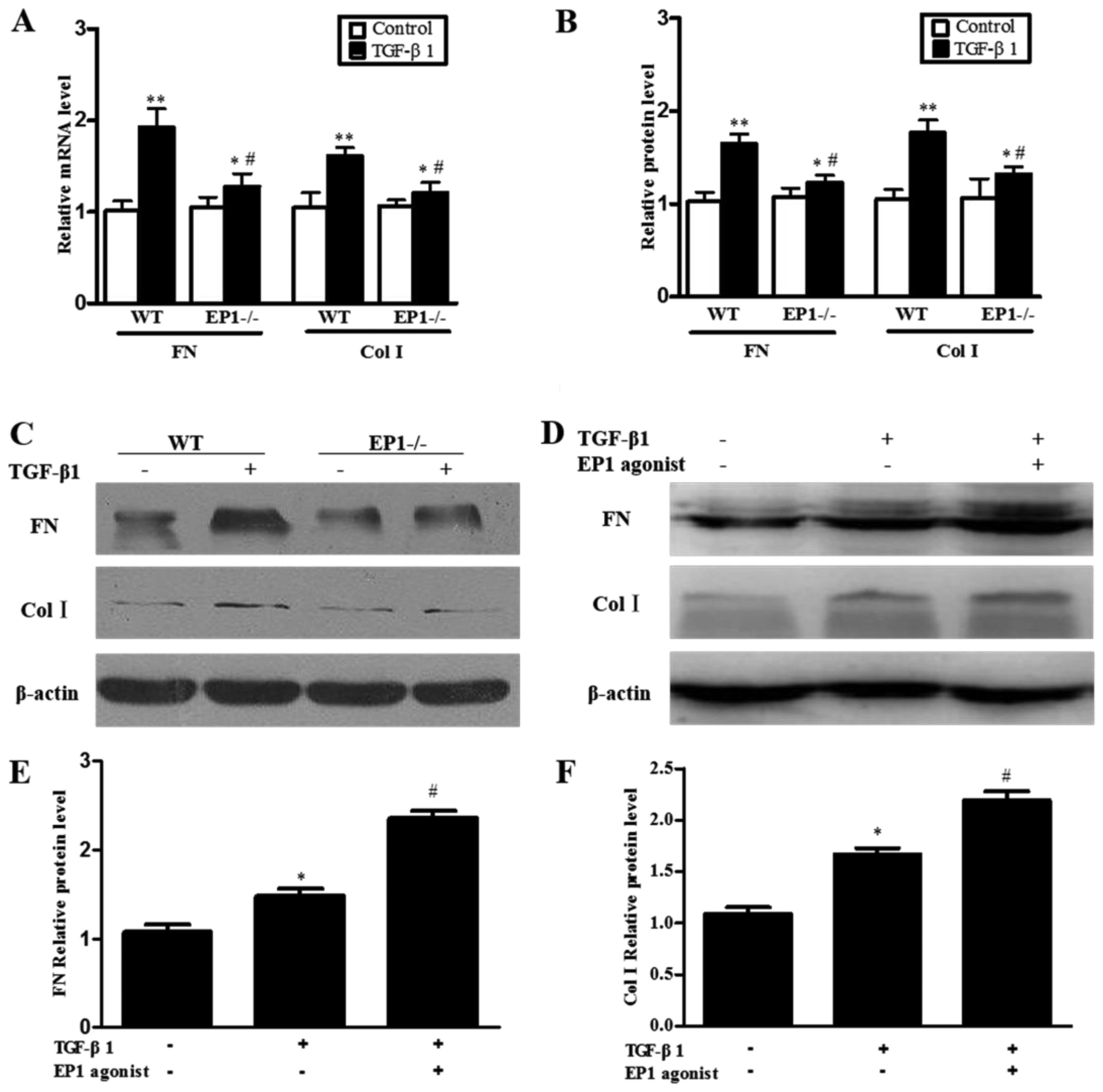
Effect of EP1 receptor on the
accumulation of extracellular matrix in MCs treated with
TGF-β1
The results of RT-qPCR and western blot analysis
revealed that following treatment of the WT and EP1−/−
MCs with 10 ng/ml TGF-β1 for 24 h, the mRNA and protein expression
of FN and ColI markedly increased compared with the relative
untreated control groups and the differences were statistically
significant (P<0.05); however, the mRNA and protein expression
of FN and ColI in the EP1−/− + TGF-β1 group was
distinctly lower than that in the WT + TGF-β1 group (P<0.05)
(Fig. 4A–C). In order to further
confirm that the EP1 receptor plays a role in the regulation of the
accumulation of extracellular matrix in MCs, we examined the
effects of TGF-β1 on the accumulation of extracellular matrix in
MCs following the specific activation of the EP1 receptor with the
EP1 agonist, 17-phenyl trinor PGE2 ethyl amide. The
results revealed that the activation of the EP1 receptor increased
the expression of FN and ColI in the cultured WT MCs treated with
TGF-β1 and 10 μM 17-phenyl trinor PGE2 ethyl amide
(Fig. 4D–F). These results
suggest that the EP1 receptor promotes the accumulation of
extracellular matrix induced by TGF-β1.
Role of EP1 receptor in the proliferation
of MCs treated with TGF-β1
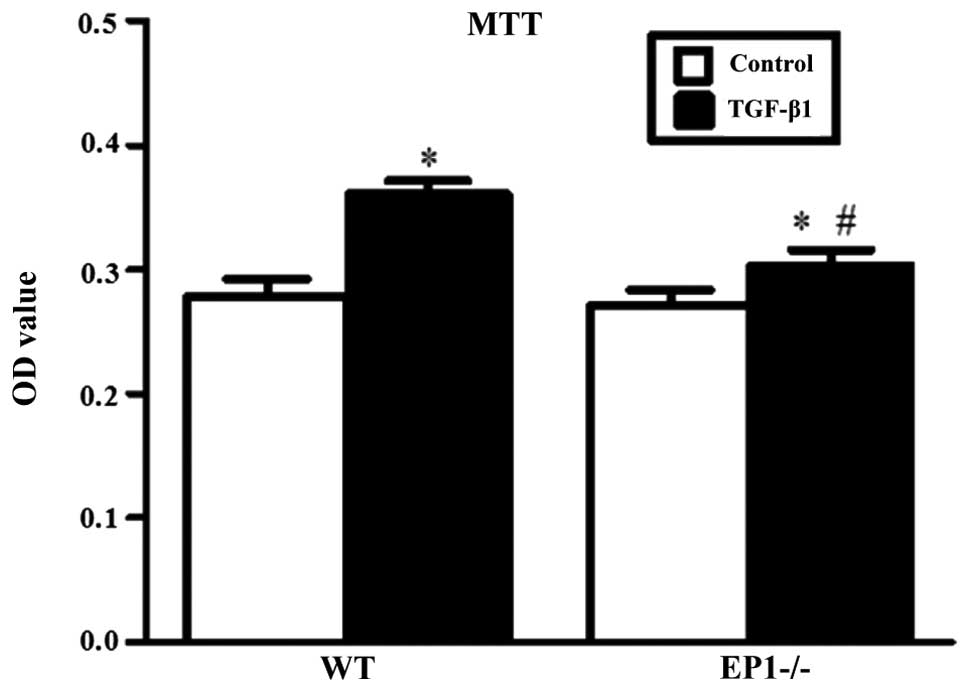
The results of MTT assay revealed that when
comparing the WT and EP1−/− MCs treated with 10 ng/ml
TGF-β1 for 24 h with each relative untreated control group, there
was a marked increase in cell proliferation and the differences
were statistically significant (P<0.05); however, when comparing
the EP1−/− + TGF-β1 group with the WT + TGF-β1 group,
cell proliferation was markedly decreased in the EP1−/−
+ TGF-β1 group, with statistically significant differences
(P<0.05) (Fig. 5). These
results suggest that following the knockdown of the EP1 gene, the
proliferation of the MCs which was induced by TGF-β1 decreased.
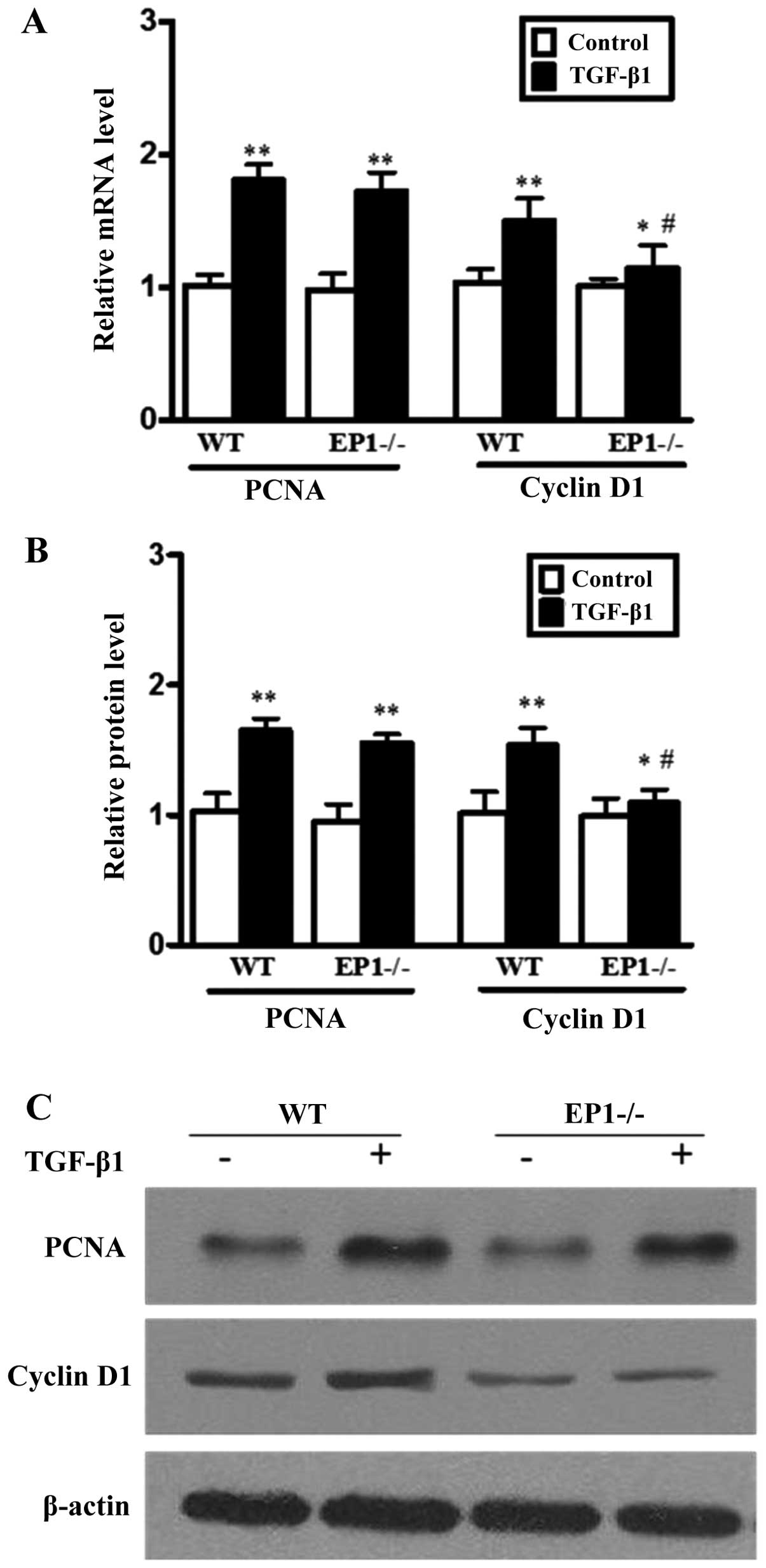
Effect of EP1 receptor on the expression
of cyclin D1 and PCNA in MCs treated with TGF-β1
In order to further elucidate the mechanisms of
action of the EP1 receptor in the regulation of MC proliferation,
we performed RT-qPCR and western blot analysis to determine the
expression levels of cyclin D1 and PCNA in the MCs treated with
TGF-β1. The results revealed that compared to the WT + TGF-β1
group, the mRNA and protein expression of cyclin D1 in the
EP1−/− + TGF-β1 group was markedly decreased
(P<0.05); there were no statistically significant differences in
PCNA expression between these 2 groups (Fig. 6). The results also indicated that
the deficiency of the EP1 receptor suppressed the expression of
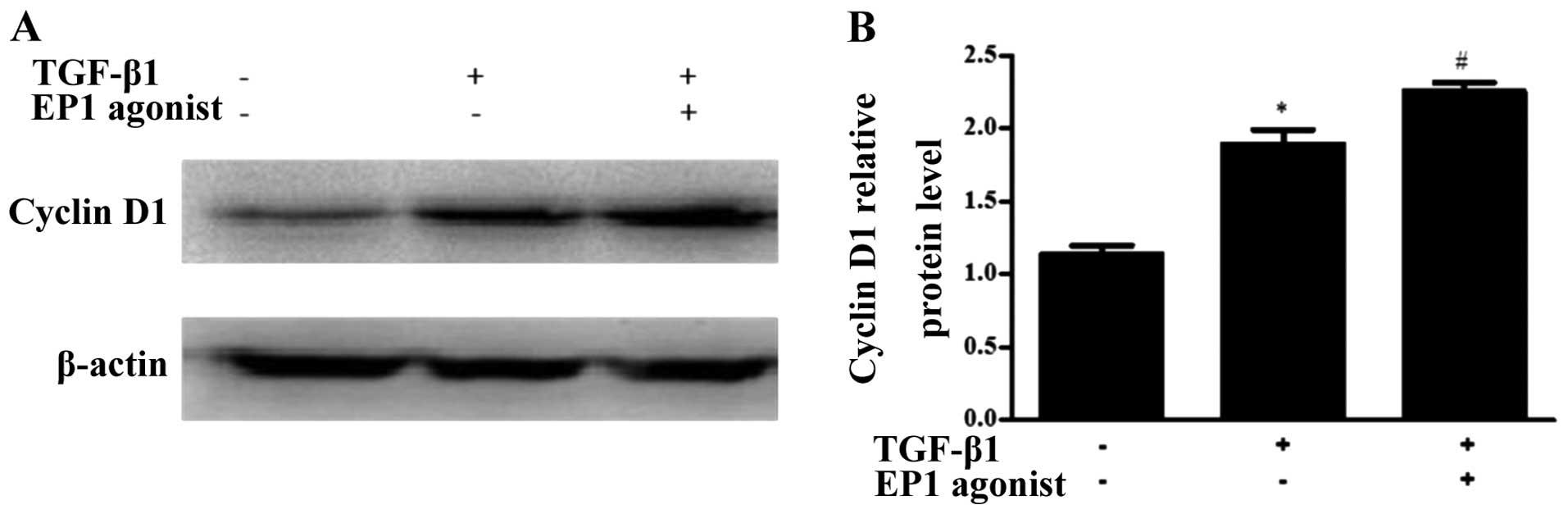
cyclin D1. Moreover, in the WT MCs treated with TGF-β1, the
expression of cyclin D1 increased following the addition of the EP1
agonist, 17-phenyl trinor PGE2 ethyl amide (P<0.05)
(Fig. 7). These results suggest
that the deficiency of the EP1 gene induces cell cycle arrest by
suppressing the expression of cyclin D1 instead of PCNA and
therefore suppressing the proliferation of MCs induced by
TGF-β1.
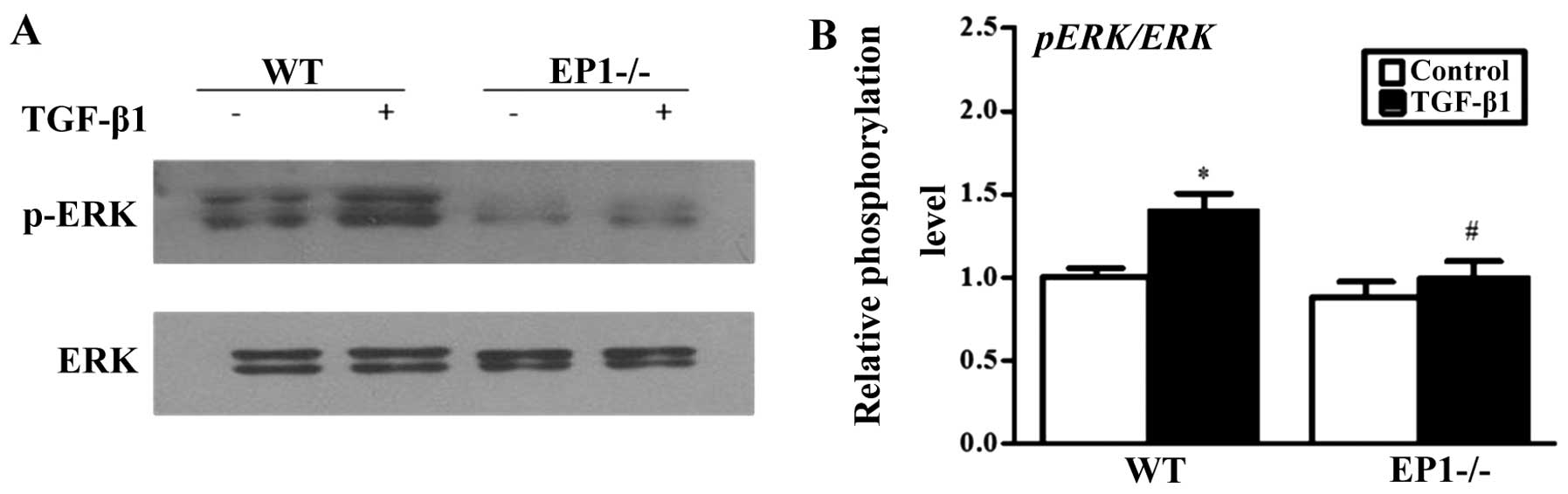
Effect of EP1 receptor on ERK
phosphorylation in MCs treated with TGF-β1
Considering the vital role of ERK in activating cell
proliferation and the accumulation of the extracellular matrix
(21), we further examined the
effects of the deficiency of the EP1 gene and EP1 receptor
activation on the phosphorylation of ERK mediated by TGF-β1. The
results revealed that the knockdown of the EP1 receptor suppressed
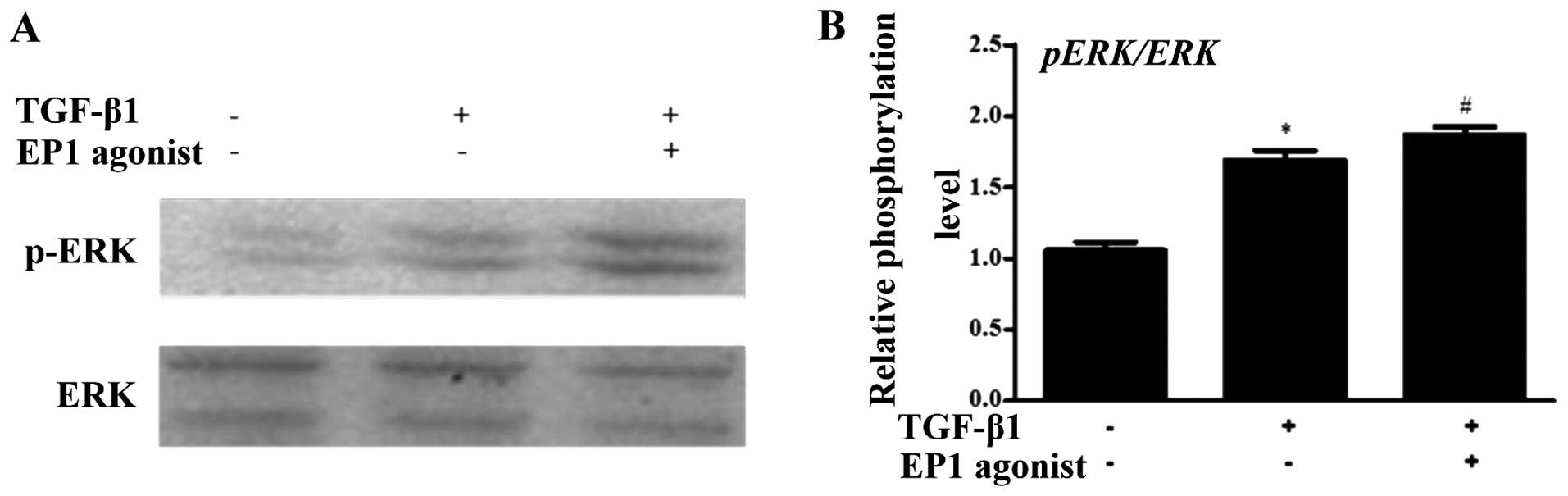
the phosphorylation of ERK which was induced by TGF-β1 (Fig. 8). However, the activation of the
EP1 activator markedly increased the phosphorylation of ERK which
was induced by TGF-β1 (Fig. 9).
Therefore, our results suggest that EP1 promotes the proliferation
of MCs and the accumulation of extracellular matrix by enhancing
ERK phosphorylation.
Discussion
With the in-depth investigation of the prostaglandin
system, the effects of PGE2 receptor have gained
widespread attention. It has been demonstrated that 4 types of EP
receptors are distributed in the kidneys and blood vascular system
(22). A number of studies have
indicated that when acute and chronic renal demage occurs, the
expression of the EP receptor undergoes marked changes (23–25). It has been reported that there
exist obvious signals of EP1 hybridization in situ in the
glomerulus mesentery. The proliferation of MCs which is induced
under high glucose conditions is almost entirely suppressed by EP1
antagonist (17). Studies
performing animal experiments have demonstrated that the selective
antagonist of prostaglandin receptor EP1 can effectively prevent
the development of diabetic nephropathy in mice induced by
streptozotocin (STZ) (17) and
reduce renal demage in mice caused by high blood pressure (26). Overall, the abovementioned data
suggest that EP1 plays a role in multiple renal pathophysiological
process. However, at present, to the best of our knowledge, there
are no related reports on the effects of EP1 on chronic renal
fibrosis.
The most important finding of this study is that
PGE2 is likely to activate EP1 receptor and thus exerts
effects on the proliferation of MCs and the accumulation of
extracellular matrix. Previous studies have indicated that although
the development of renal fibrosis involves different mechanisms,
all mechanisms involve the increase in cellular matrix compound or
the increase in extracellular matrix caused by the decreased
degradation, which are the essential factors resulting in
glomerulosclerosis and renal interstitial fibrosis (27,28). Undoubtedly, inflammatory response
plays an important role in the development of chronic kidney
diseases. Moreover, during the development of kidney disease,
PGE2, together with its 4 types of receptors, as well as
other prostaglandin substances all participate in the regulation of
the disease process throughout different stages of inflammation in
a manner of conditional dependence. They participate in the
regulation of the disease process in both inflammation and
preventions (29,30). The biological functions of
PGE2 are exerted by the 4 types of receptors from EP1 to
EP4. With the activation of one of these receptors, specific
biological process occur through specific signaling pathways
(11). Evidence indicates that
the suppression of COX-2 and mPGES-1 protects cell proliferation
(31–33). Previous studies have demonstrated
that selective COX-2 inhibitor attenuates the proliferation of MCs
and suppresses the progression of renal fibrosis. These findings
also suggest that PGE2, which is the source of
COX-2/mPGES-1 aggravates cell proliferation and the accumulation of
extracellular matrix, thus taking part in the development of renal
fibrosis. Among all enzymes involved in the production of
PGE2, COX-2 and mPGES-1 are constitutively expressed in
cultured MCs (34,35). In this study, we found that after
the MCs were stimulated with TGF-β1, the expression of COX-2
increased, as well as that of mPGES-1. Therefore, the continuous
and high-level expression of mPGES-1 plays an important role in the
production of PGE2 among damaged cells and the
activation of EP1 receptor. In addition, following stimulation with
TGF-β1, the expression of COX-2/mPGES-1/PGE2 and FN and
ColI in the MCs lacking the EP1 gene decreased and cell
proliferation decreased when compared to the WT MCs. However, the
specific activation of the EP1 receptor enhanced the expression of
PGE2, as well as that of COX-2, mPGES-1, FN and ColI
which was induced by TGF-β1. Hence, our results suggest that
following stimulation with TGF-β1, the expression of COX-2/mPGES-1
in the MCs increases, which also increases the expression of
PGE2 and activates the EP1 receptor; on the contrary,
the deficiency of the EP1 gene lowers the expression of
COX-2/mPGES-1 and alleviates damage to MCs and renal fibration.
Thus, EP1 may play an essential role in regulating the damage to
MCs induced by TGF-β1.
Cyclin D1 participates in the progression of the
cell cycle from the G1 stage to the S stage. The suppression of its
functions prevents cells entering the S stage. However, the
overexpression of this gene may shorten the G1 stage and thus lead
to incontrollable cell proliferation (36). In this study, we found that after
the EP1 receptor is expressed, the expression level of cyclin D1
increases suggesting that EP1 is a type of receptor which promotes
prolifertaion. This was supported by our results showing that in
the MCs in which the EP1 gene was knocked down, cell proliferation
and the expression of cyclin D1 induced by TGF-β1 were
suppressed.
ERK is a vital mediator in conducting signals from
the cell surface to the cell nucleus. Different extracellular
stimulations activate ERK, such as mitogens, growth factors, AngII
and oxidative stress (37). The
stimulated ERK molecule phosphorylates other substrates, such as
cytoskeletal proteins and thus exerts biological effects directly,
or transfers them to the nucleus and phosphorylates transcription
factors, such as cAMP response element-binding protein (CREB) and
nucleoprotein so as to regulate gene expression intracellularly.
Previous studies have demonstrated that the activation of ERK plays
an important role in the cell cycle and ECM accumulation (36,38–41). In our study, we found that the
deficiency of the EP1 gene markedly suppresses the phosphorylation
of ERK induced by TGF-β1; however, following the activation of EP1,
EP1 is able to strengthen the phosphorylation of ERK induced by
TGF-β1. This suggests that cell proliferation, cyclin D1 expression
and the accumulation of extracellular matrix induced by TGF-β1 may
be partially related to ERK phosphorylation mediated by EP1.
Combined with our present experimental results, we
suggest that the deficiency of the EP1 gene markedly suppresses the
proliferation of MCs and the accumulation of extracellular matrix
induced by TGF-β1. The activation of PGE2-EP1 plays an
important role in inducing damage to MCs, which is related to cell
cycle arrest caused by the reduction of cyclin D1 expression and
the phosphorylation of ERK. In order to further confirm the
abovementioned results, we used an EP1 receptor agonist (17-phenyl
trinor PGE2 ethyl amide) to specifically activate the
EP1 receptor. Our results demonstrated that the activation of the
EP1 receptor promoted the accumulation of MC extracellular matrix
which was induced by TGF-β1 and promoted the expression of cyclin
D1 and the phosphorylation of ERK. These results suggest that the
EP1 receptor may be regarded as a potential target for the
treatment of renal fibrosis. However, further studies are required
to confirm our results and determine whether this will provide a
new strategy for the treatment of chronic kidney diseases.
Acknowledgements
This study was funded by grants form the National
Natural Science Foundation of China (no. 81170656) and the Science
Foundation of Nantong City, Jiangsu, China (no. HS2011021).
References
|
1
|
Böttinger EP: TGF-beta in renal injury and
disease. Semin Nephrol. 27:309–320. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ortiz A, Ucero AC and Egido J: Unravelling
fibrosis: two newcomers and an old foe. Nephrol Dial Transplant.
25:3492–3495. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wang S, Wilkes MC, Leof EB, et al:
Noncanonical TGF-beta pathways, mTORC1 and Abl, in renal
interstitial fibrogenesis. Am J Physiol Renal Physiol. 298:142–149.
2010. View Article : Google Scholar
|
|
4
|
Yeh YC, Wei WC, Wang YK, et al:
Transforming growth factor-β1 induces Smad3-dependent β1 integrin
gene expression in epithelial-to-mesenchymal transition during
chronic tubulointerstitial fibrosis. Am J Pathol. 177:1743–1754.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zeng R, Han M, Luo Y, et al: Role of
Sema4C in TGF-beta1-induced mitogen-activated protein kinase
activation and epithelial-mesenchymal transition in renal tubular
epithelial cells. Nephrol Dial Transplant. 26:1149–1156. 2011.
View Article : Google Scholar :
|
|
6
|
Lee SB and Kalluri R: Mechanistic
connection between inflammation and fibrosis. Kidney Int Suppl.
119:S22–S26. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sankaran D, Bankovic-Calic N, Ogborn MR,
et al: Selective COX-2 inhibition markedly slows disease
progression and attenuates altered prostanoid production in Han:
SPRD-cy rats with inherited kidney disease. Am J Physiol Renal
Physiol. 293:821–830. 2007. View Article : Google Scholar
|
|
8
|
de Silva KI, Daud AN, Deng J, Jones SB,
Gamelli RL and Shankar R: Prostaglandin E2 mediates
growth arrest in NFS-60 cells by down-regulating interleukin-6
receptor expression. Biochem J. 370:315–321. 2003. View Article : Google Scholar
|
|
9
|
Yamamoto E, Izawa T, Juniantito V, et al:
Involvement of endogenous prostaglandin E2 in tubular
epithelial regeneration through inhibition of apoptosis and
epithelial-mesenchymal transition in cisplatin-induced rat renal
lesions. Histol Histopathol. 25:995–1007. 2010.PubMed/NCBI
|
|
10
|
Coleman RA, Smith WL and Narumiya S:
International Union of Pharmacology classification of prostanoid
receptors: properties, distribution, and structure of the receptors
and their subtypes. Pharmacol Rev. 46:205–229. 1994.PubMed/NCBI
|
|
11
|
Narumiya S, Sugimoto Y and Ushikubi F:
Prostanoid receptors: structures, properties, and functions.
Physiol Rev. 79:1193–1226. 1999.PubMed/NCBI
|
|
12
|
Hata AN and Breyer RM: Pharmacology and
signaling of prostaglandin receptors: multiple roles in
inflammation and immune modulation. Pharmacol Ther. 103:147–166.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Matsuoka Y, Furuyashiki T, Bito H, et al:
Impaired adrenocorticotropic hormone response to bacterial
endotoxin in mice deficient in prostaglandin E receptor EP1 and EP3
subtypes. Proc Natl Acad Sci USA. 100:4132–4137. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Matsuoka Y, Furuyashiki T, Yamada K, et
al: Prostaglandin E receptor EP1 controls impulsive behavior under
stress. Proc Natl Acad Sci USA. 102:16066–16071. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mutoh M, Watanabe K, Kitamura T, et al:
Involvement of prostaglandin E receptor subtype EP(4) in colon
carcinogenesis. Cancer Res. 62:28–32. 2002.PubMed/NCBI
|
|
16
|
Moriyama T, Higashi T, Togashi K, et al:
Sensitization of TRPV1 by EP1 and IP reveals peripheral nociceptive
mechanism of prostaglandins. Mol Pain. 1:32005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Makino H, Tanaka I, Mukoyama M, et al:
Prevention of diabetic nephropathy in rats by prostaglandin E
receptor EP1-selective antagonist. J Am Soc Nephrol. 13:1757–1765.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Qian Q, Kassem KM, Beierwaltes WH and
Harding P: PGE2 causes mesangial cell hypertrophy and
decreases expression of cyclin D3. Nephron Physiol. 113:p7–p14.
2009. View Article : Google Scholar
|
|
19
|
Kennedy CR, Zhang Y, Brandon S, et al:
Salt-sensitive hypertension and reduced fertility in mice lacking
the prostaglandin EP2 receptor. Nat Med. 5:217–220. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Takemoto M, Asker N, Gerhardt H, et al: A
new method for large scale isolation of kidney glomeruli from mice.
Am J Pathol. 161:799–805. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Morino N, Mimura T, Hamasaki K, et al:
Matrix/integrin interaction activates the mitogen-activated protein
kinase, p44erk-1 and p42erk-2. J Biol Chem. 270:269–273. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li R, Mouillesseaux KP, Montoya D, et al:
Identification of prostaglandin E2 receptor subtype 2 as
a receptor activated by OxPAPC. Circ Res. 98:642–650. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Camacho M, Gerbolés E, Escudero JR, Antón
R, Garcia-Moll X and Vila L: Microsomal prostaglandin E synthase-1,
which is not coupled to a particular cyclooxygenase isoenzyme, is
essential for prostaglandin E(2) biosynthesis in vascular smooth
muscle cells. J Thromb Haemost. 5:1411–1419. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Guan Y, Zhang Y, Wu J, et al:
Antihypertensive effects of selective prostaglandin E2
receptor subtype 1 targeting. J Clin Invest. 117:2496–2505. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Vukicevic S, Simic P, Borovecki F, et al:
Role of EP2 and EP4 receptor-selective agonists of prostaglandin
E(2) in acute and chronic kidney failure. Kidney Int. 70:1099–1106.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
González AA, Céspedes C, Villanueva S,
Michea L and Vio CP: E Prostanoid-1 receptor regulates renal
medullary alphaENaC in rats infused with angiotensin II. Biochem
Biophys Res Commun. 389:372–377. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Boor P, Ostendorf T and Floege J: Renal
fibrosis: novel insights into mechanisms and therapeutic targets.
Nat Rev Nephrol. 6:643–656. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang Q, Usinger W, Nichols B, et al:
Cooperative interaction of CTGF and TGF-β in animal models of
fibrotic disease. Fibrogenesis Tissue Repair. 4:42011. View Article : Google Scholar
|
|
29
|
Biswas S, Bhattacherjee P, Paterson CA,
Tilley SL and Koller BH: Ocular inflammatory responses in the EP2
and EP4 receptor knockout mice. Ocul Immunol Inflamm. 14:157–163.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yuhki K, Ueno A, Naraba H, et al:
Prostaglandin receptors EP2, EP3, and IP mediate exudate formation
in carrageenin-induced mouse pleurisy. J Pharmacol Exp Ther.
311:1218–1224. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang K, Tarakji K, Zhou Z, et al:
Celecoxib, a selective cyclooxygenase-2 inhibitor, decreases
monocyte chemoattractant protein-1 expression and neointimal
hyperplasia in the rabbit atherosclerotic balloon injury model. J
Cardiovasc Pharmacol. 45:61–67. 2005. View Article : Google Scholar
|
|
32
|
Wang M, Ihida-Stansbury K, Kothapalli D,
et al: Microsomal prostaglandin E2 synthase-1 modulates
the response to vascular injury. Circulation. 123:631–639. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yang HM, Kim HS, Park KW, et al:
Celecoxib, a cyclooxygenase-2 inhibitor, reduces neointimal
hyperplasia through inhibition of Akt signaling. Circulation.
110:301–308. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Cheng HF, Wang CJ, Moeckel GW, et al:
Cyclooxygenase-2 inhibitor blocks expression of mediators of renal
injury in a model of diabetes and hypertension. Kidney Int.
62:929–939. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang JL, Cheng HF, Shappell S, et al: A
selective cyclooxygenase-2 inhibitor decreases proteinuria and
retards progressive renal injury in rats. Kidney Int. 57:2334–2342.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ramakrishnan M, Musa NL, Li J, Liu PT,
Pestell RG and Hershenson MB: Catalytic activation of extracellular
signal-regulated kinases induces cyclin D1 expression in primary
tracheal myocytes. Am J Respir Cell Mol Biol. 18:736–740. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chambard JC, Lefloch R, Pouysségur J and
Lenormand P: ERK implication in cell cycle regulation. Biochim
Biophys Acta. 1773:1299–1310. 2007. View Article : Google Scholar
|
|
38
|
Chen XL, Chen ZS, Ding Z, Dong C, Guo H
and Gong NQ: Antisense extracellular signal-regulated kinase-2 gene
therapy inhibits platelet-derived growth factor-induced
proliferation, migration and transforming growth factor-beta(1)
expression in vascular smooth muscle cells and attenuates
transplant vasculopathy. Transpl Int. 21:30–38. 2008.
|
|
39
|
Graf K, Xi XP, Yang D, Fleck E, Hsueh WA
and Law RE: Mitogen-activated protein kinase activation is involved
in platelet-derived growth factor-directed migration by vascular
smooth muscle cells. Hypertension. 29:334–339. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lavoie JN, Rivard N, L’Allemain G and
Pouyssegur J: A temporal and biochemical link between growth
factor-activated MAP kinases, cyclin D1 induction and cell cycle
entry. Prog Cell Cycle Res. 2:49–58. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Meloche S, Seuwen K, Pagès G and
Pouysségur J: Biphasic and synergistic activation of p44mapk (ERK1)
by growth factors: correlation between late phase activation and
mitogenicity. Mol Endocrinol. 6:845–854. 1992.PubMed/NCBI
|