Introduction
Regenerative therapies for the treatment of
cartilage injury have become an important focus for the field of
stem cell therapy. Although pluripotent embryonic stem cells (ESCs)
are the ideal seeds for cell therapy, there are ethical concerns on
the use of donated, early human embryos and problems with tissue
rejection in patients following transplantation (1–3).
In order to overcome these issues, somatic cells have been
reprogrammed to yield induced pluripotent stem (iPS) cells by
introduction of four essential transcription factors;
Oct3/4, Sox2, c-Myc and Klf4 (3–5).
The iPS cells closely resemble ESCs; they can differentiate into
cellular derivatives of all three germ layers and possess the
capacity of unlimited replication (3,6).
The iPS cells have the potential to provide an abundant cell source
for tissue engineering, as well as generating patient-matched in
vitro models to study genetic and environmental factors in
cartilage repair and osteoarthritis (7). ESCs and iPS cells have been induced
to differentiate into osteoblasts in vitro. Differentiation
of ESCs towards the osteoblast lineage is enhanced by supplementing
serum-containing media with ascorbic acid, β-glycerophosphate,
dexamethasone and retinoic acid, or by co-culture with fetal murine
osteoblasts (8). Recently, two
studies have reported that mouse iPS cells could be induced to
differentiate into osteoblasts in vitro (7,9).
The latter study also demonstrated the potential use of mouse iPS
cells in the repair of cartilage defects and in the generation of
tissue models of cartilage that were matched to specific genetic
backgrounds (7). Finally, it has
been shown that human iPS cells can be cultured on microchannel
polycaprolactone scaffolds prepared using a robotic dispensing
technique, with osteogenesis promoted by the addition of exogenous
osteogenic factors (3). However,
despite the success in facilitating the differentiation of iPS
cells or ESCs into osteoblasts in vitro, the process is
inefficient and time-consuming, and this has hindered their
development and therapeutic potential.
Mesenchymal stem cells can differentiate into mature
and functional osteoblasts that produce extracellular matrix
proteins and proteins that regulate matrix mineralization (10). Runx2, also known as
Cbfa1/AmI3, is a runt domain family protein, which acts as an
osteoblast-specific transactivator essential for osteoblast
differentiation and bone formation (10–14). Zhang et al (11) showed that commitment of
undifferentiated mesenchymal stem cells to an osteoprogenitor
lineage was associated with the expression of Runx2, and
Runx2 protein levels were significantly upregulated in
quiescence (G0) or during proliferative arrest induced
by serum deprivation or by contact inhibition of osteoblast cells
(15). Notably,
Runx2−/− mice show a complete lack of
intramembranous and endochondral ossification, due to the
maturation arrest of osteoblasts (10). Runx2 regulates the
expression of osteoblast-specific genes, including ALP, type
I collagen and osteocalcin (10),
and is necessary throughout life to promote the differentiation of
new osteoblasts during bone remodeling (10). The study by Hu et al
(16) reported that histone
deacetylation suppresses Runx2 transcription. The study
showed that several histone deacetylase (HDAC) inhibitors promoted
osteoblast maturation and the expression of specific genes through
upregulation of Runx2 gene expression in bone marrow stem
cells (16). This result clearly
indicated that epigenetic regulation, particularly histone
acetylation, was an important factor in regulating Runx2
expression. Recently, epigenetic regulation has been considered as
a significant mechanism for influencing stem cell differentiation.
The unique patterns of DNA methylation and histone modifications
have been found to play important roles in the induction of
lineage-specific differentiation of bone marrow stem cells
(16). HDAC enzymes alter the
transcription of numerous genes involved in the control of
proliferation, cell survival, differentiation and genetic stability
(17). Previous studies revealed
that a microRNA, miR-449a, interferes specifically with the
expression of HDAC1 (9,17–19). microRNAs (miRNAs) are small [~22
nucleotides (nt)] endogenous non-coding RNAs, which function at the
post-transcriptional level by annealing to the 3′-untranslated
region (3′-UTR) of target mRNAs to inhibit translation. They have
emerged as key regulatory factors in development, organogenesis,
apoptosis, cell proliferation, differentiation and tumorigenesis
(1,2,9).
Several groups have reported that miR-449a targets
HDAC1 and induces growth arrest of tumor cells derived from
hepatocellular, prostate and lung carcinoma (17–19). In an independent study, Okamoto
et al (9) demonstrated
that six miRNAs, including miR-10a/b, miR-19b,
miR-9, miR-124a and miR-181a, were crucial
regulatory factors in the osteoblast differentiation of mouse iPS
cells.
In view of this evidence, the present study
attempted to clarify whether overexpression of miR-449a
suppressed HDAC1 expression, increased Runx2
expression and stimulated the differentiation of human iPS cells
into osteoblasts. Therefore, if successful this procedure may
improve the efficiency of generating osteoblasts from human iPS
cells in vitro, and provide a reliable source for cell
therapy.
Materials and methods
Preparation of human amniotic epithelial
cells (HuAECs)
Human placentas were obtained with written and
informed consent from pregnant females who were negative for human
immunodeficiency virus-I, hepatitis B and hepatitis C. The study
was recognized for the appropriate use of human amnion by the
Institutional Ethics Committee. This study was approved (Permit
THTJHE20130018) by the Medical Ethics Committee of Tongji
University, in compliance with the Experimental Medical Regulations
of the National Science and Technology Commission, China. Amnion
membranes were mechanically peeled from the chorines of placentas
obtained from females with an uncomplicated Cesarean section. The
epithelial layers with the basement membrane attached were obtained
and used to harvest HuAECs as previously described with some
modification (1,20). Briefly, the membrane was placed in
a 250-ml flask containing Dulbecco’s modified Eagle’s medium (DMEM)
and cut with a razor to yield 0.5–1.0-cm2 segments. The
segments were digested with 0.25% trypsin-EDTA at 37°C for 45 min.
The resulting cell suspension were seeded in a six-well plate in
DMEM medium supplemented with 10% fetal calf serum (PAA, Pasching,
Austria), penicillin (100 U/ml) and glutamine (0.3 mg/ml), and
incubated in a humidified tissue culture incubator containing 5%
CO2 at 37°C. The HuAECs grown to a density of ~100% were
used as feeder layers for human iPS culture following Mitomycin C
(Sigma-Aldrich, St. Louis, MO, USA) treatment.
Co-culture of human iPS cells with
HuAECs
The human iPS cells were generated as previously
described (1). iPS cultures were
separated from the feeder cells by treatment of 0.125% trypsin-EDTA
solution and plated onto, and co-cultured with, HuAECs. The cells
were cultured in DMEM:F12 (1:1) medium supplemented with 15%
KnockOut™ Serum Replacement, 1 mM sodium pyruvate, 2 mM
L-glutamine, 0.1 mM non-essential amino acids, 0.1 mM
β-mercaptoethanol and penicillin (25 U/ml)-streptomycin (925
mg/ml), and mixed, but without leukemia inhibitory factor (LIF).
These cells were incubated in a humidified tissue culture incubator
containing 5% CO2 at 37°C. All the cells had been
cultured on the same feeder until the 5th passage prior to
undergoing the ulterior experiments.
Recombinant lentivirus generation vector
construction and cell transfection
All the steps of the recombinant lentivirus package
were as previously described (2,17–21). The Lv2-miR-449a and
Lv2-miR-mut lentivirus were constructed by Genepharma Corporation
(Shanghai, China) and the methods of lentivirus transfected were
according to the manufacturer’s instructions. In brief,
co-transfection of human iPS cells was conducted using
4×107 PFU/ml Lv2-miR-449a or Lv2-miR-mut
lentivirus, respectively, according to the manufacturer’s
instructions. The iPS cells were seeded in a six-well plate and
cultured in DMEM:F12 (1:1) medium supplemented with 15% KnockOut™
Serum Replacement, 1 mM sodium pyruvate, 2.0 mM L-glutamine, 0.1 mM
non-essential amino acids, 0.1 mM β-mercaptoethanol and penicillin
(25 U/ml)-streptomycin (925 mg/ml), and mixed, but without LIF.
These cells were incubated in a humidified tissue culture incubator
containing 5% CO2 at 37°C until 80% confluent.
Embryoid body (EB) formation and
induction of differentiation into the osteoblasts-like cells
All the steps were as previously described (3,6–9,16).
Briefly, for EB formation the iPS cells were dissociated with
0.125% trypsin-EDTA solution and suspended onto Petri dishes with
DMEM:F12 (1:1) medium supplemented with 15% KnockOut™ Serum
Replacement, 1 mM sodium pyruvate, 2 mM L-glutamine, 0.1 mM
non-essential amino acids, 0.1 mM β-mercaptoethanol, penicillin (25
U/ml)-streptomycin (925 mg/ml), and mixed, but without LIF for 6
days. Subsequently, to induce EB cell differentiation, day 6 EB
cells were cultured in induced cell-conditioned medium (DMEM: F12
(1:1) supplemented with 1% KnockOut™ Serum Replacement, 1 mM sodium
pyruvate, 2 mM L-glutamine, 0.1 mM non-essential amino acids,
penicillin (25 U/ml)-streptomycin (925 mg/ml), 10 mg/ml insulin, 10
ng/ml human epidermal growth factor, 10 ng/ml human basic
fibroblast growth factor, 50 mg/ml ascorbic acid, 50 mM
β-glycerophosphate, 1 mM dexamethasone, 1 mM all-trans-retinoic
acid and 100 ng/ml human recombinant BMP-4, mixed and incubated in
a humidified tissue culture incubator containing 5% CO2
at 37°C for 12 days until differentiated completely.
RNA extraction and analysis by reverse
transcription-quantitative PCR (RT-qPCR)
Total RNA was isolated with Trizol reagent
(Invitrogen Life Technologies Corporation, Grand Island, NY, USA),
according to the manufacturer’s instructions (21). Briefly, cells
(3×108/ml) were collected and 0.8 ml Trizol reagent was
added. After incubating the sample for 5 min, 0.2 ml chloroform was
added and the tubes were agitated vigorously and incubated for 2
min. The samples were centrifuged at 17,000 × g for 15 min at 4°C.
Subsequently, the aqueous phases were transferred to clean tubes,
and 0.4 ml isopropanol was added and mixed. Following another
centrifugation, the total RNA was collected. The RNA samples were
treated with DNase I (Sigma-Aldrich), quantified by a regular UV
spectrophotometer and reverse-transcribed into cDNA with the
ReverTra Ace-α First Strand cDNA Synthesis kit [Toyobo (Shanghai)
Biotech Co., Ltd., Shanghai, China]. qPCR was conducted with a
RealPlex4 real-time PCR detection system from Eppendorf (Hamburg,
Germany), with the SyBR Green RealTime PCR Master mix [Toyobo
(Shanghai) Biotech Co., Ltd.] as the detection dye. qPCR
amplification was performed over 40 cycles with denaturation at
95°C for 15 sec and annealing at 58°C for 45 sec. Target cDNA was
quantified with the relative quantification method. A comparative
threshold cycle (Ct) was used to determine the gene expression
relative to a control (calibrator), and steady-state mRNA levels
are reported as an n-fold difference relative to the calibrator.
For each sample, the marker gene Ct values were normalized with the
formula: ΔCt = Ct_genes − Ct_18S RNA. To determine relative
expression levels, the following formula was used: ΔΔCt =
ΔCt_all_groups − ΔCt_control_group. The values used to plot
relative expressions of genes were calculated with the expression
as 2−ΔΔCt. The mRNA levels were calibrated on the basis
of 18S rRNA levels. The cDNA of each gene was amplified with
primers as described in Table
I.
 | Table IRT-qPCR primers used in the
study. |
Table I
RT-qPCR primers used in the
study.
| Gene name | RT-qPCR primers
(5′→3′) |
|---|
| HDAC1 | F:
TATTATGGACAAGGCCACCC
R: CATCTCCTCAGCATTGGCTT |
| Runx2 | F:
ACAGTAGATGGACCTCGGGA
R: ATACTGGGATGAGGAATGCG |
| Bmp4 | F:
TGAGCCTTTCCAGCAAGTTT
R: GCATTCGGTTACCAGGAATC |
| Osterix | F:
CTCAGCTCTCTCCATCTGCC
R: GGGACTGGAGCCATAGTGAA |
|
Osteopontin | F:
GTGATTTGCTTTTGCCTCCT
R: GCCACAGCATCTGGGTATTT |
|
Osteocalcin | F:
CTCACACTCCTCGCCCTATT
R: TTGGACACAAAGGCTGCAC |
| 18S
rRNA | F:
CAGCCACCCGAGATTGAGCA
R: TAGTAGCGACGGGCGGTGTG |
Luciferase reporter gene assay
All the steps of the luciferase report assay were as
previously described (2,20–22). NIH-3T3 cells were seeded at
3×104/well in 48-well plates and co-transfected with 400
ng Lv2-miR-449a or Lv2-miR-mut vectors, 20 ng
pGL3-HDAC1-3UTR-WT or pGL3-HDAC1-3UTR-Mut, and pGL-TK (Promega,
Madison, WI, USA) using the Lipofectamine 2000 reagent according to
the manufacturer’s instructions. After 48 h transfection,
luciferase activity was measured using the dual-luciferase reporter
assay system (Promega) according to the manufacturer’s
instructions. The results were expressed as relative luciferase
activity (Firely LUC/Renilla LUC).
RNA extraction and northern blotting
analysis
All the steps of northern blotting were as
previously described (2,20–22). For all groups, 20 μg of good
quality total RNA was analyzed on a 7.5 M urea 12% polyacrylamide
denaturing gel and transferred to a Hybond N+ nylon
membrane (Amersham, Freiburg, Germany) with electrotransfer. The
membranes were crosslinked using UV light for 30 sec at 1200
mJ/cm2. Hybridization was performed with the
miR-17-3p antisense starfire probe,
5′-ACCAGCTAACAATACACTGCCA-3′; to detect the 22-nt miR-17-3p
fragments according to the manufacturer’s instructions. Following
washing, the membranes were exposed for 20–40 h to Kodak XAR-5
films (Sigma-Aldrich). As a positive control, all the membranes
were hybridized with a human U6 snRNA probe, 5′-GCAGGG
GCCATGCTAATCTTCTCTGTATCG-3′. Exposure times for the U6
control probe varied between 15 and 30 min.
Western blot analysis
All group cells were seeded at 3×106/well
in 6-well plates and cultured until 85%, which were lysed using a
2× loading lysis buffer [50 mM Tris-HCl (pH 6.8), 2% sodium dodecyl
sulfate, 10% β-mercaptoethanol, 10% glycerol and 0.002% bromophenol
blue]. The total cellular proteins from the cultured cells was
subjected to 12% SDS-PAGE and transferred onto hybrid-PVDF
membranes (Millipore, Bedford, MA, USA). Following blocking with 5%
(w/v) skimmed dried milk in Tris-buffered saline containing
Tween-20 [TBST; 25 mM Tris/HCl (pH 8.0), 125 mM NaCl and 0.05%
Tween-20], the PVDF membranes were washed four times (15 min each)
with TBST at room temperature and incubated with antibody dilution
solution (Beyotime Institute of Biotechnology, Jiangsu, China) to
dilute the following primary antibodies: Rabbit anti-human osterix
polyclonal antibody (sc-133871), rabbit anti-human osteocalcin
polyclonal antibody (sc-30044), rabbit anti-human osteopontin
polyclonal antibody (sc-20788; all 1:1000; Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA), rabbit anti-human HDAC1
polyclonal antibody (no. 2062), rabbit anti-human RUNX2
polyclonal antibody (no. 8486), rabbit anti-human BMP4 polyclonal
antibody (no. 4680), rabbit anti-human H3K27Me3 polyclonal antibody
(no. 9733), rabbit anti-human H3Ac polyclonal antibody (no. 7627)
and rabbit anti-human GAPDH polyclonal antibody (no. 5174; all
1:1000; Cell Signaling Technology, MA, USA). Following extensive
washing, membranes were incubated with horseradish peroxidase
(HRP)-conjugated goat anti-rabbit immunoglobulin G (IgG) secondary
antibody (sc-2768; 1:1000; Santa Cruz Biotechnology, Inc.) for 1 h.
Following washing four times (15 min each) with TBST at room
temperature, the immunoreactivity was visualized by enhanced
chemiluminescence (ECL) using the ECL kit from Perkin-Elmer Life
Science (Norwalk, CT, USA). Subsequently, the chemisluminescent
signals were detected by using a chemiluminescence detection system
(GE Typhoon 9400; GE Healthcare, Shanghai, China).
ELISA assay
The alkaline phosphatase (ALP) ELISA kit (Hermes
Criterion Biotechnology, Vancouver, BC, Canada) was used according
to the manufacturer’s instructions (16,23) to determine the level of ALP in
cells. Briefly, all the cells were harvested and dissociated in 0.1
M Tris (pH 7.4) containing 1% Triton X-100 and 5 mM
MgCl2 by sonication. ALP concentration was measured and
the data were normalized against the protein concentration and
expressed as nanogram of ALP per milligram of total protein. All
the samples were added to anti-ALP antibody precoated microtest
wells and incubated for 60 min. Three washes were performed and the
HRP-conjugated detection antibodies were added followed by the
substrate solution. The absorbance was determined at a wavelength
of 450 nm using the enzyme-linked immunosorbent assay reader (Model
680; Bio-Rad, Hercules, CA, USA).
Hematoxylin and eosin (H&E)
staining
The histopathological analysis of each group was
stained with H&E staining. Briefly, all fresh tissues were
washed 3 times with PBS and fixed with 4% paraformaldehyde
(Sigma-Aldrich) for 30 min, dehydrated through a graded series of
ethanol, vitrified in xylene, and embedded in paraffin. Next,
serial 6-μm-thick sections were cut, and stained with H&E.
Alkaline phosphatase-positive cell
staining
All the steps were as previously described (6). Briefly, all the cells were washed
twice with 0.01 M phosphate-buffered saline (PBS) and fixed in 4%
paraformaldehyde for 20 min. Subsequent to fixing, the cells were
rinsed with PBS twice, and PBS was replaced with 1 ml of the
staining solution [Naphthol AS-MX phosphate (0.1 mg/ml) and fast
blue BB salt (0.6 mg/ml)] dissolved in Tris-HCl buffer [0.1 M (pH
8.8)] containing N,N-dimethylformamide (0.5%) and MgCl2
(2 mM) and the cells were incubated at 37°C for 60 min until the
ALP-positive cells stained deep blue. The staining reaction was
stopped by PBS solution. Digital images were captured with the
Olympus IX71 microscope (Olympus Optical Co., Ltd., Tokyo,
Japan).
Alizarin red histochemical staining
All the steps were as previously described (3,6–9,16).
Briefly, cell culture plates were fixed in 4% paraformaldehyde for
20 min, washed in 0.01 M PBS and stained for 10 min with a 1%
solution of Alizarin Red (Sigma-Aldrich). The plates were washed in
running tap water and left to air dry. Manual counts of bone nodule
colonies identified by Alizarin Red were subsequently
performed.
Von Kossa assay for calcium
determine
All the steps were as previously described (14). Briefly, cell culture plates were
fixed with 95% ethanol for 20 min, and washed in distilled water.
Subsequently, the cells were covered with a 1.0% AgNO3
solution and maintained for 60 min under UV light until the calcium
turned black. Following this, a 5.0% NaS2O3
solution was added for 2 min, and plates were washed in running tap
water and left to air dry. Digital images were captured with the
Olympus IX71 microscope.
Co-immunoprecipitation assay
All group cells were seeded at 3×105/well
in 6-well plates and cultured until 85% confluent (24). The cells were lysed (500 μl per
plate) in a modified cell lysis buffer for western blotting and
immunoprecipitation [20 mM Tris (pH 7.5), 150 mM NaCl, 1% Triton
X-100, 1 mM EDTA, sodium pyrophosphate, β-glycerophosphate,
Na3VO4 and leupeptin] (Beyotime institute of
Biotechnology). Subsequent to lysis, each sample was centrifuged to
remove the insoluble debris from the lysate and preincubated with
20 μg protein A agarose beads (Beyotime Institute of Biotechnology)
by agitating for 30 min at 4°C, followed by centrifugation and
transfer to a fresh 1.5-ml tube. The rabbit anti-human histone H3
polyclonal antibody (Cell Signaling Technology) was incubated for
90 min before re-addition of 20 μg protein A agarose beads to
capture the immune complexes. The pelleted beads were washed three
times with 500 μl cell lysis buffer, dissolved in 4× SDS-PAGE
samples loading buffer and heated for 10 min at 95°C.
Chromatin immunoprecipitation (ChIP)
assays
ChIP experiments were carried out using the
anti-acetylated histone H3 antibody (Cell Signaling Technology),
anti-trimethylated H3-K27 antibody (Cell Signaling Technology) and
normal rabbit IgG (Upstate Biotechnology, Lake Placid, NY, USA) as
a negative control. In brief, all the steps were performed as
previously described (1,12,16). The cells were fixed by 1%
formaldehyde for 30 min at 37°C and were quenched by 125 mM glycine
for 10 min at room temperature to form DNA-protein cross-links. The
samples were sonicated on ice until chromatin fragments became
200–1000 basepairs in size and incubated with antibodies at 4°C
overnight. The PCR amplification was performed under the following
conditions: 33 cycles of denaturation at 95°C for 30 sec, annealing
at 55°C for 30 sec and extension at 72°C for 30 sec.
Statistical analysis
Each experiment was performed as least three times,
and data are shown as the mean ± standard error where applicable.
The differences were evaluated with Student’s t-tests. A
P-value of <0.05 was considered to indicate a statistically
significant difference.
Results
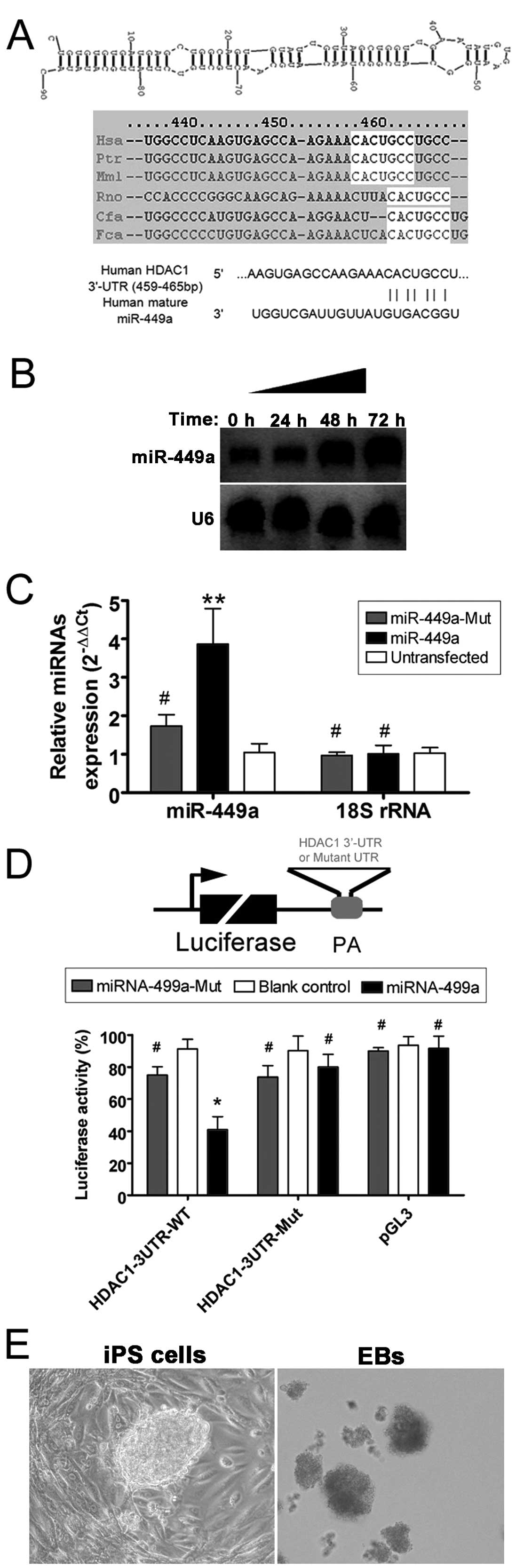
miR-449a binding sites in the
3′-untranslated region (3′-UTR) of HDAC1 mRNA
The sequences of the miR-449a precursor,
mature miRNA and the proposed target gene, HDAC1, were
analyzed in multiple species using online research tools and the
miRBase database (http://www.mirbase.org). In the present study, the
potential human miR-449a target residues in the human
HDAC1 3′-UTR were focused on, and seven consecutive putative
miRNA target residues were identified that are conserved across
species (Fig. 1). A luciferase
activity assay was used to identify whether mature miR-449a
binds to this site within the HDAC1 mRNA 3′-UTR and
regulates its expression. The luciferase activity of the
HDAC1 3′-UTR was significantly inhibited by wild-type
miR-449a (Fig. 1),
suggesting that miR-449a interacts with HDAC1.
Mutation of either the HDAC1 3′-UTR construct or
miR-449a to disrupt the potential interaction yielded
results similar to the pGL3 control, indicating that the binding
was specific. Detection of miR-449a miRNA by northern
blotting and RT-qPCR confirmed that miR-449a expression was
significantly increased 48–72 h following transfection of human iPS
cells (Fig. 1).
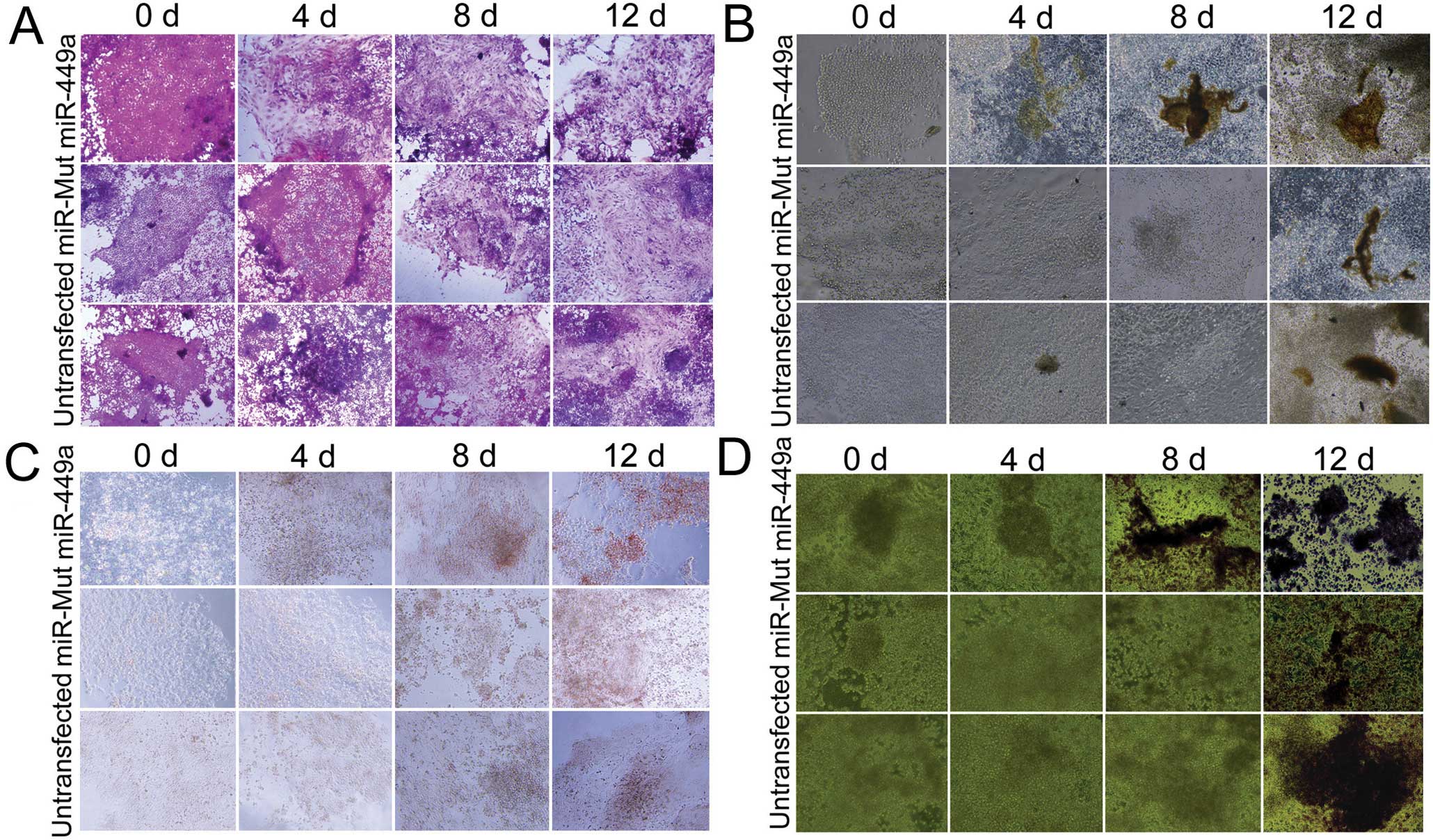
miR-449a-transfected iPS cells
efficiently differentiate into osteoblasts
Human iPS cells were transfected with a wild-type
miR-449a, a mutant miR-449a or were untransfected.
Subsequent to the formation of EB, differentiation was induced in
the three groups of cells and examined over 12 days. The
miR-449a-transfected iPS cells were the first to show the
morphology of osteoblasts only four days after induction of
differentiation; a proportion of the cell population showed
quadrilateral or dwarf columnar morphology, and a honeycomb
arrangement and neat rules, which was not evident in the other
groups at that time (Fig. 2).
Furthermore, at the early time-points there were more
positively-stained cells in the miR-449a-transfected iPS
cells compared to the miR-mut-transfected iPS cells or the
untransfected group by several different assays (including the Von
Kossa assay, Fig. 2B; Alizarin
red staining, Fig. 2C; and
staining for ALP, Fig. 2D). These
experiments indicated that although osteoblasts were present in all
three groups 12 days after induction of differentiation, they were
already detectable after only four days in iPS cells transfected
with miR-449a.
miR-449a-transfected iPS cells
significantly express osteoblast markers upon differentiation
ELISA assay, RT-qPCR and western blotting were used
to quantify the increase in expression of various markers. The
concentration of ALP was increased in all cells in a time-dependent
manner. However, the amount of ALP in miR-449a-transfected
iPS cells was significantly higher at all time-points after the
induction of differentiation compared to the other two groups
(Fig. 3). The mRNA levels of
several important cell markers were analyzed in each group by
RT-qPCR, and the relative mRNA levels were normalized to 18S
rRNA, which served as an internal control. The results showed
that osteopontin, osteocalcin, BMP4, osterix and
Runx2 were expressed at significantly higher levels in the
miR-449a-transfected iPS group compared to the
miR-mut-transfected iPS or untransfected groups at each
time-point following induction of differentiation (Fig. 3). However, HDAC1 was
expressed at significantly lower levels in the
miR-449a-transfected iPS group compared to the
miR-mut-transfected iPS or untransfected group. The results
of the RT-qPCR assays were confirmed by western blotting (Fig. 3), which revealed that the levels
of osteocalcin, osterix and Runx2 were all higher in the
miR-449a-transfected group, compared to the two control
groups, whereas the levels of HDAC1 expression were lower in the
miR-449a-transfected iPS group.
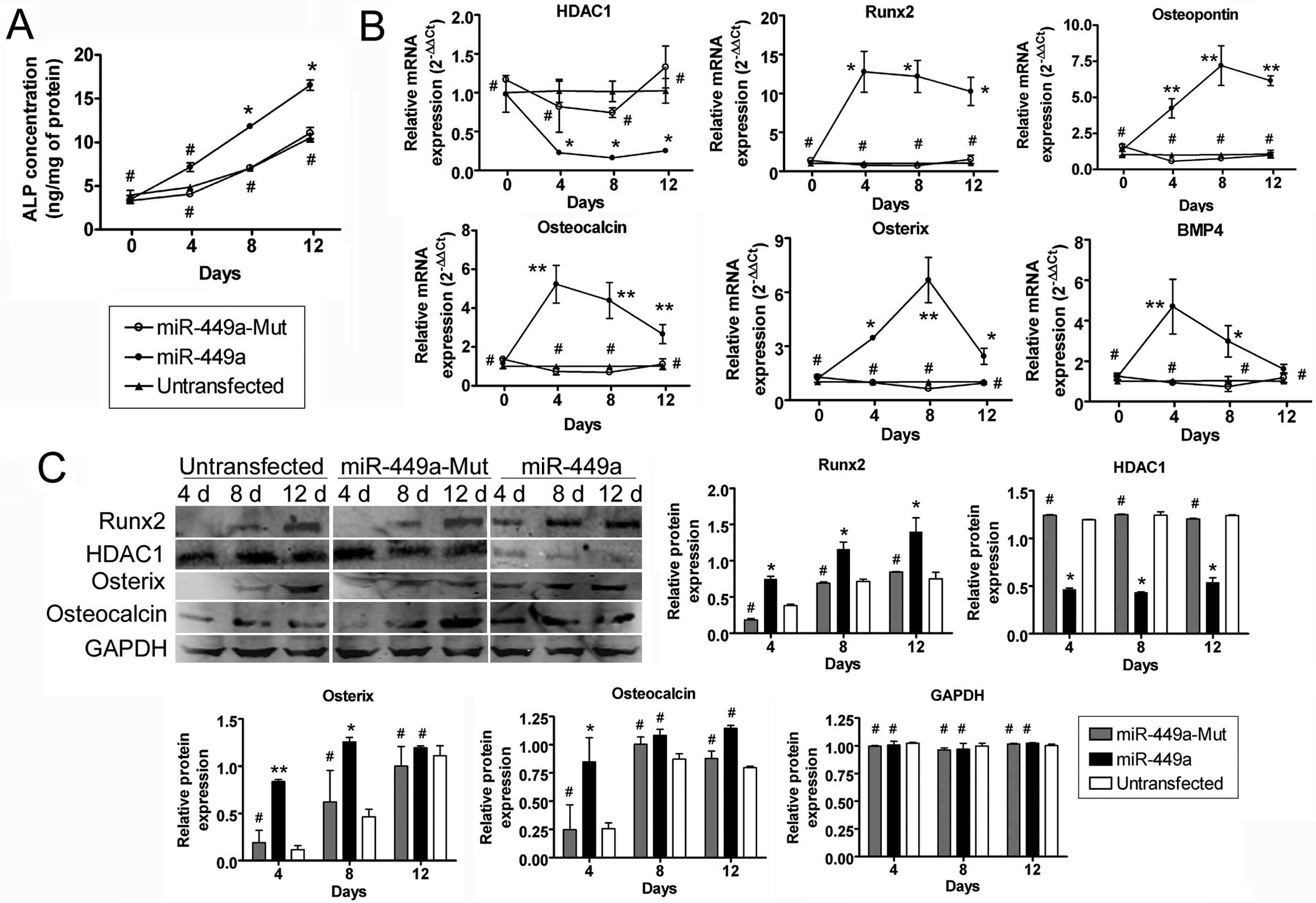 | Figure 3Osteoblast markers expression was
assayed by the ELISA assay, reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) and western blotting. (A) The
concentration of alkaline phosphatase (ALP) at each time-point
after induction of differentiation was investigated by the ELISA
assay. The results indicated that the rate of increase in ALP was
significantly higher in the miR-449a-transfected
human-induced pluripotent (iPS) cells compared to the other two
groups. *P<0.05, vs. untransfected group;
#P>0.05, vs. untransfected group; n=3. (B)
RT-qPCR was used to compare the transcriptional levels of several
important cell markers, including osteoblast markers and associated
transcription factors following the induction of iPS cell
differentiation. Relative mRNA expression was normalized to
18S rRNA, which served as an internal control. The results
showed that osteopontin, osteocalcin, BMP4, osterix and
Runx2 were expressed at significantly higher levels in the
miR-449a-transfected iPS group compared to the other two
groups at each time-point after induction of differentiation.
However, HDAC1 was expressed at significantly lower levels
in the miR-449a-transfected iPS group compared to the other
groups. **P<0.01, vs. untransfected group;
*P<0.05, vs. untransfected group;
#P>0.05, vs. untransfected group; n=3. (C)
Western blotting revealed that the levels of osteocalcin, osterix
and Runx2 were significantly higher in the
miR-449a-transfected cells compared to the
miR-mut-transfected or untransfected groups. By contrast,
the levels of HDAC1 were much lower in the
miR-449a-transfected iPS group compared to the
miR-mut-transfected or untransfected group.
**P<0.01, vs. untransfected group;
*P<0.05, vs. untransfected group;
#P>0.05, vs. untransfected group; n=3. d,
day. |
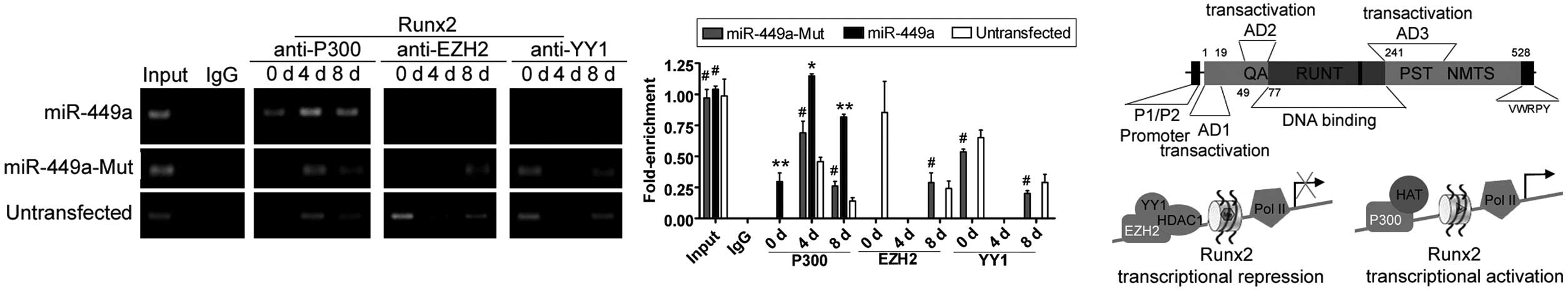
miR-449a improves the transcriptional
activity of Runx2 via chromatin re-configuration
ChIP assays showed the acetylation and
trimethylation levels of histone H3 in the Runx2 promoter
were similar in all three groups prior to differentiation. However,
only four days after induction of differentiation, the acetylation
levels of histone H3 (H3Ac) in the Runx2 promoter were
higher compared to the miR-mut-transfected iPS or untransfected
groups (Fig. 4). However, K27
trimethylation levels of histone H3 in the Runx2 promoter
were elevated in the miR-mut-transfected iPS and
untransfected groups, but not in the miR-449a-transfected
iPS group following induction of differentiation. In addition, the
results of western blotting indicated that the level of H3Ac was
maintained at a high level in the miR-449a-transfected iPS
group; by contrast, the expression level of H3Ac rapidly declined
in the miR-mut-transfected iPS or untransfected groups
following induction of differentiation (Fig. 4). However, in the
miR-449a-transfected iPS group during four and eight days
after induction of differentiation, the binding efficiency of the
Runx2 gene promoter in combination with the P300/CBP protein
was much higher compared to the combination with EZH2 and YY1
(Fig. 5). However, in the
miR-mut-transfected iPS or untransfected groups during four
and eight days after induction of differentiation, the binding
efficiency of the Runx2 gene promoter in combination with
the EZH2 and YY1 proteins was higher compared to the combination
with P300/CBP (Fig. 5).
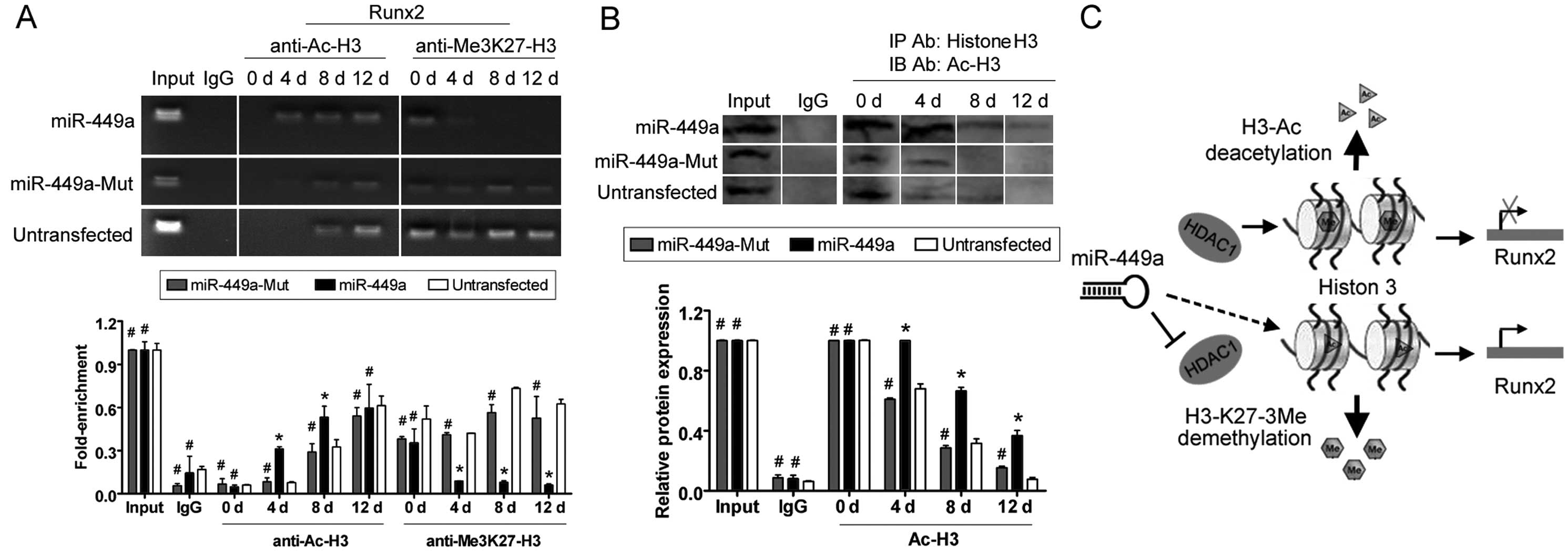 | Figure 4Histone acetylation and Runx2
transcriptional activity. (A) Chromatin immunoprecipitation (ChIP)
assays were performed to evaluate histone H3 acetylation levels in
the Runx2 promoter at each time-point after induction of
differentiation. Four, eight and 12 days after induction of
differentiation, histone H3 was acetylated in the
miR-449a-transfected human-induced pluripotent (iPS) cells
group, compared to the same region in the
miR-mut-transfected iPS or untransfected groups. By
contrast, histone H3K27 was methylated in the
miR-mut-transfected iPS and untransfected groups following
induction, compared to the miR-449a-transfected iPS group.
*P<0.05, vs. untransfected group;
#P>0.05 vs. untransfected group; n=3. (B) The
results of co-IP western blotting indicated that the level of
endogenous H3Ac was maintained at a higher level in the
miR-449a-transfected iPS group during differentiation.
*P<0.05, vs. untransfected group;
#P>0.05, vs. untransfected group; n=3. (C)
Model showing that miR-449a was capable of maintaining the
Runx2 locus in an active transcriptional state through
silencing HDAC1 expression and covalent histone
modifications. IgG, immunoglobulin G. d, day. |
Discussion
Osteoblast differentiation is a complex process
involving the precise regulation of the activation and suppression
of genes in response to physiological signals (9). At a biochemical level, the primary
pathway involves bone morphogenetic protein (BMP)-Smad signaling,
which leads to the activation of osteoblast-essential genes,
including Runx2 and osterix (9). Previous studies have showed that
Runx2 is highly expressed in osteoblasts and osteosarcoma
cells (11–16). In our preliminary experiments, the
expression level of Runx2 increased when human iPS cells
were induced to differentiate into osteoblasts, indicating that the
expression of Runx2 is involved in osteoblast
differentiation. However, the mechanism for regulating the
expression of Runx2 was unclear. Although it has previously
been possible to induce iPS cells and ESCs to differentiate into
osteoblasts in vitro, this was a long and inefficient
process, requiring ~16 days to achieve mature osteoblasts (3,7–9,16).
In these studies, the Runx2 gene expression was induced but
remained at a low level. In addition, our preliminary experiments
showed that histone acetylation decreased during the process of iPS
cell differentiation into osteoblasts, and HDAC expression
increased. With regards to the knowledge that histone acetylation
is closely linked with the activation of gene transcription, and
that overexpression of HDAC induced histone deacetylation, it can
be speculated that an increase in HDAC expression during
differentiation would lead to a decrease in the transcription of
Runx2, which would be counter-productive to osteoblast
differentiation. However, the mechanism behind HDAC overexpression
during the induction of differentiation into osteoblasts is not
understood. The overexpression of HDAC1 was possibly due to
miR-449a downregulation, and forced overexpression of
miR-449a may promote osteogenesis. The experiments of the
present study demonstrated that silencing of endogenous
HDAC1 expression by overexpression of miR-449a
maintained histone acetylation levels, induced Runx2 gene
transcription and stimulated osteoblast differentiation in human
iPS cells more efficiently and rapidly compared to previous
studies.
By contrast, in the present study miR-449a
improved the transcriptional activity of Runx2 via chromatin
re-configuration during osteoblast differentiation. In previous
studies, chromatin conformation led to differences in gene
transcription activity. A number of genes adopt a conformation that
is repressive for transcription, owing to the recruitment of the
Polycomb group (PcG) methyltransferase EZH2, which silences
transcription by catalysing trimethylation of H3-K27 (25–27). EZH2 was recruited to the histone
regulatory regions via interaction with Yin Yang 1 (YY1), and
further association with HDAC1 formed a repressive complex
(25–27). In addition, YY1 was a
multifunctional transcription factor. Certain transcription
co-repressory complexes containing nuclear HDACI and EZH2 together
with YY1 were recruited to chromatin to silence transcription of
the target genes. The above process suppressed the hyperacetylation
of histone-specific lysines and promoted di- and tri-methylation of
specific lysines (such as K9 and K27) to generate a chromatin
conformation that is repressive for transcription of the target
genes (28,29). YY1 also directly interacts with a
number of proteins with important epigenetic modifications,
including P300/CBP and HDAC1 for protein acetylation and
deacetylation, and EZH2 for histone methylation. As a member of the
PcG proteins, it not only directly interacted with DNA at a
consensus binding site to establish and maintain gene repression,
but also together with EZH2-mediated H3-K27 trimethylation, in
order to mediate histone modification and chromatin remodeling
(27–29). These enzymes catalyzed histone
modifications, including H3-K27 trimethylation that established a
chromatin conformation that is repressive for transcription. In
view of the above evidence, it can be speculated that the
transcriptional activity of Runx2 was closely associated
with co-repressory complexes or the transcription activator
complex. The present study showed that the binding efficiency of
the Runx2 gene promoter in combination with P300/CBP protein
was much higher than the combination with EZH2 and YY1 in the
miR-449a-transfected iPS group during four and eight days
after induction of differentiation. However, the binding efficiency
of the Runx2 gene promoter in combination with EZH2 and YY1
proteins was higher compared to the combination with P300/CBP in
the miR-mut-transfected iPS or untransfected groups during
four and eight days after induction of differentiation. These
results indicated that the transcription of the Runx2 gene
was activated via chromatin reconfiguration into a conformation
permissive for transcription in the miR-449a-transfected iPS
group, but not in the other two groups. Therefore, miR-449a
improved the transcriptional activity of Runx2 via chromatin
re-configuration.
Acknowledgements
The present study was supported by a grant from the
National Natural Science Foundation of China (no. 81202811), and
the project funded by the China Postdoctoral Science Foundation
(grant no. 2014M550250), and Shanghai Municipal Health Bureau Fund
(grant no. 20124320) to Te Liu.
References
|
1
|
Liu T, Zou G, Gao Y, et al: High
efficiency of reprogramming CD34(+) cells derived from human
amniotic fluid into induced pluripotent stem cells with Oct4. Stem
Cells Dev. 21:2322–2332. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Liu T, Cheng W, Huang Y, Huang Q, Jiang L
and Guo L: Human amniotic epithelial cell feeder layers maintain
human iPS cell pluripotency via inhibited endogenous microRNA-145
and increased Sox2 expression. Exp Cell Res. 318:424–434. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jin GZ, Kim TH, Kim JH, et al: Bone tissue
engineering of induced pluripotent stem cells cultured with
macrochanneled polymer scaffold. J Biomed Mater Res A.
101:1283–1291. 2012.PubMed/NCBI
|
|
4
|
Takahashi K and Yamanaka S: Induction of
pluripotent stem cells from mouse embryonic and adult fibroblast
cultures by defined factors. Cell. 126:663–676. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Takahashi K, Tanabe K, Ohnuki M, et al:
Induction of pluripotent stem cells from adult human fibroblasts by
defined factors. Cell. 131:861–872. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kuroda S, Sumner DR and Virdi AS: Effects
of TGF-beta1 and VEGF-A transgenes on the osteogenic potential of
bone marrow stromal cells in vitro and in vivo. J Tissue Eng.
3:20417314124597452012. View Article : Google Scholar
|
|
7
|
Diekman BO, Christoforou N, Willard VP, et
al: Cartilage tissue engineering using differentiated and purified
induced pluripotent stem cells. Proc Natl Acad Sci USA.
109:19172–19177. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Buttery LD, Bourne S, Xynos JD, et al:
Differentiation of osteoblasts and in vitro bone formation from
murine embryonic stem cells. Tissue Eng. 7:89–99. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Okamoto H, Matsumi Y, Hoshikawa Y, Takubo
K, Ryoke K and Shiota G: Involvement of microRNAs in regulation of
osteoblastic differentiation in mouse induced pluripotent stem
cells. PLoS One. 7:e438002012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hou X, Shen Y, Zhang C, et al: A specific
oligodeoxynucleotide promotes the differentiation of osteoblasts
via ERK and p38 MAPK pathways. Int J Mol Sci. 13:7902–7914. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang X, Ting K, Bessette CM, et al:
Nell-1, a key functional mediator of Runx2, partially rescues
calvarial defects in Runx2(+/−) mice. J Bone Miner Res. 26:777–791.
2010. View Article : Google Scholar
|
|
12
|
Nishimura R, Wakabayashi M, Hata K, et al:
Osterix regulates calcification and degradation of chondrogenic
matrices through matrix metalloproteinase 13 (MMP13) expression in
association with transcription factor Runx2 during endochondral
ossification. J Biol Chem. 287:33179–33190. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Iwasaki M, Piao J, Kimura A, et al: Runx2
haploinsufficiency ameliorates the development of ossification of
the posterior longitudinal ligament. PLoS One. 7:e433722012.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li X, Huang M, Zheng H, et al: CHIP
promotes Runx2 degradation and negatively regulates osteoblast
differentiation. J Cell Biol. 181:959–972. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lucero C, Vega O, Osorio M, et al: The
cancer-related transcription factor Runx2 modulates cell
proliferation in human osteosarcoma cell lines. J Cell Physiol.
228:714–723. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hu X, Zhang X, Dai L, et al: Histone
deacetylase inhibitor trichostatin A promotes the osteogenic
differentiation of rat adipose-derived stem cells by altering the
epigenetic modifications on Runx2 promoter in a BMP
signaling-dependent manner. Stem Cells Dev. 22:248–255. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Buurman R, Gürlevik E, Schäffer V, et al:
Histone deacetylases activate hepatocyte growth factor signaling by
repressing microRNA-449 in hepatocellular carcinoma cells.
Gastroenterology. 143:811–820. e811–e815. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jeon HS, Lee SY, Lee EJ, et al: Combining
microRNA-449a/b with a HDAC inhibitor has a synergistic effect on
growth arrest in lung cancer. Lung Cancer. 76:171–176. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Noonan EJ, Place RF, Pookot D, et al:
miR-449a targets HDAC-1 and induces growth arrest in prostate
cancer. Oncogene. 28:1714–1724. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liu T, Chen Q, Huang Y, Huang Q, Jiang L
and Guo L: Low microRNA-199a expression in human amniotic
epithelial cell feeder layers maintains human-induced pluripotent
stem cell pluripotency via increased leukemia inhibitory factor
expression. Acta Biochim Biophys Sin (Shanghai). 44:197–206. 2012.
View Article : Google Scholar
|
|
21
|
Cheng W, Liu T, Wan X, Gao Y and Wang H:
MicroRNA-199a targets CD44 to suppress the tumorigenicity and
multidrug resistance of ovarian cancer-initiating cells. FEBS J.
279:2047–2059. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang L, Liu T, Huang Y and Liu J:
microRNA-182 inhibits the proliferation and invasion of human lung
adenocarcinoma cells through its effect on human cortical
actin-associated protein. Int J Mol Med. 28:381–388.
2011.PubMed/NCBI
|
|
23
|
Mukherjee A and Rotwein P: Selective
signaling by Akt1 controls osteoblast differentiation and
osteoblast-mediated osteoclast development. Mol Cell Biol.
32:490–500. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cheng W, Liu T, Jiang F, et al:
microRNA-155 regulates angiotensin II type 1 receptor expression in
umbilical vein endothelial cells from severely pre-eclamptic
pregnant women. Int J Mol Med. 27:393–399. 2011.PubMed/NCBI
|
|
25
|
Guasconi V and Puri PL: Chromatin: the
interface between extrinsic cues and the epigenetic regulation of
muscle regeneration. Trends Cell Biol. 19:286–294. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Forcales SV and Puri PL: Signaling to the
chromatin during skeletal myogenesis: novel targets for
pharmacological modulation of gene expression. Semin Cell Dev Biol.
16:596–611. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Caretti G, Di Padova M, Micales B, Lyons
GE and Sartorelli V: The Polycomb Ezh2 methyltransferase regulates
muscle gene expression and skeletal muscle differentiation. Genes
Dev. 18:2627–2638. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang Q, Stovall DB, Inoue K and Sui G:
The oncogenic role of Yin Yang 1. Crit Rev Oncog. 16:163–197. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Affar el B, Gay F, Shi Y, et al: Essential
dosage-dependent functions of the transcription factor yin yang 1
in late embryonic development and cell cycle progression. Mol Cell
Biol. 26:3565–3581. 2006. View Article : Google Scholar : PubMed/NCBI
|