Introduction
Cardiac hypertrophy is a pathological characteristic
common to numerous forms of heart disease, such as hypertension,
ischemic myocardial injury, diabetic cardiomyopathy, valvular
dysfunction and aortic stenosis (1). Persistent hypertrophy can ultimately
lead to ventricular dilatation, arrhythmia, fibrotic disease, heart
failure and even sudden death (2,3).
Cardiac hypertrophy is a major risk factor in the development of
heart failure, and its therapeutic reversal is associated with
decreased mortality (4,5). Previous studies have indicated that
microRNAs (miRNAs or miRs) are essential in a number of biological
processes, including differentiation, apoptosis, proliferation and
development (6,7). The dysregulation of miRNAs has been
linked to several human diseases (7), including cardiovascular disease
(8,9). miRNAs are a class of highly
conserved, small non-coding RNAs (approximately 23 nucleotides in
length) that regulate gene expression at the post-transcriptional
level (10). miRNAs inhibit gene
expression by forming partial duplexes with the 3′-untranslated
region (3′-UTR) of mRNAs (11,12). miRNAs function by either
inhibiting mRNA translation or promoting mRNA degradation (13). Each miRNA can have numerous mRNA
targets (14). In addition, a
single mRNA can be targeted by different miRNAs, thereby increasing
the complexity of gene regulation by miRNAs. miRNAs have been found
in various organisms and are regarded as powerful regulators of
gene expression and cellular phenotype. Moreover, their roles in
cardiovascular biology and diseases have been an area of intense
investigation. Previous studies have identified the expression
patterns of miRNAs associated with cardiovascular diseases. For
example, miRNA-21, miRNA-23a, miRNA-24, miRNA-133,
miRNA-208/miRNA-195 and miRNA-199 have been shown to be involved in
cardiac hypertrophy (15-17), miRNA-1 in arrhythmia (18), miRNA-29 and miRNA-21 in cardiac
fibrosis (19,20), miRNA-210 and miRNA-494 in ischemic
heart disease (21) and miRNA-129
in heart failure (22). However,
the association between miRNA-455 (miR-455) and cardiac hypertrophy
remains unclear. In this study, using a target prediction algorithm
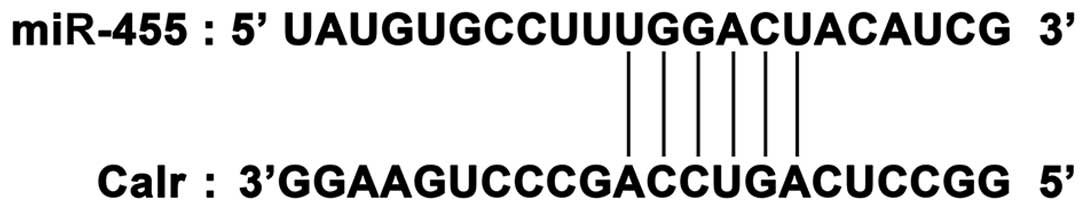
(23), we identified calreticulin
(Calr) as the putative target gene of miR-455. The mRNA sequence of
Calr was predicted to contain a conserved ‘seed’ sequence
complementary to miR-455 in the 3′-UTR (Fig. 1). Calr is closely associated with
myocardial hypertrophy (24).
Thus, miR-455 may be important in myocardial hypertrophy. In the
present study, we established a mouse model of hypertrophy by
transverse aortic constriction (TAC) in order to investigate the
effects of the aberrant expression of miR-455 on cardiac
hypertrophy induced by pressure overload and to elucidate the
potential cellular and molecular mechanisms of action of this
miRNA.
Materials and methods
Animal models
At 10 weeks after birth, 18 Kunming male mice were
provided by the Experimental Animal Center of Hebei Province,
China. All experiments were carried out in compliance with the
Guide for the Care and Use of Laboratory Animals (National Research
Council, 1996) and were reviewed and approved by the Ethics
Committee for the Use of Experimental Animals at Hebei Medical
University, Shijiazhuang, China. The mice (weighing 16–18 g) were
anesthetized and then subjected to either TAC or sham operation.
Briefly, the transverse aorta of the anesthetized mice was
constricted using a 7-0 nylon suture and was ligated using a
blunted 27-gauge needle, which was later removed. The mice were
examined at 2 and 4 weeks following surgery.
Ad-13 containing miR-455 precursor
The precursor of the miRNA, mmu-miR-455 (miRBase
accession no. MI0004679) was produced by Invitrogen (Shanghai,
China). The green fluorescent protein (GFP)-expressing vector
(Invitrogen) was used as a control.
Viral delivery protocol
The mice were randomly selected to receive a tail
vein injection of either miR-455 (5.0×109 ifu/ml, n=12)
or GFP (1.0×109 ifu/ml, n=24) at 0.1 ml (viral genomes)
per animal at 1, 8, 15 and 21 days following surgery.
Echocardiographic measurements were taken at baseline and at 2 and
4 weeks after TAC. Invasive hemodynamic measurements were also
obtained at 2 and 4 weeks after TAC, and the the animals were then
sacrificed by cervical dislocation.
Echocardiography and hemodynamic
measurements
Transthoracic echocardiography was performed using a
30 MHz high-frequency scan head (VisualSonics Vevo770; VisualSonics
Inc., Toronto, ON, Canada). All measurements were averaged for 5
consecutive cardiac cycles. Aortic blood pressure (BP), left
ventricular end-systolic pressure (LVESP) and left ventricular
end-diastolic pressure (LVEDP) were measured. Briefly, a
micromanometer catheter (Millar 1.4F, SPR-835; Millar Instruments,
Inc., Houston, TX, USA) was inserted through the right common
carotid artery into the aorta and carefully introduced into the
left ventricle (LV). The transducer was connected to a Power
Laboratory system (ADInstruments, Castle Hill, Australia) and BP,
LVESP and LVEDP were recorded.
Morphological and histological
analyses
The mice were sacrificed by cervical dislocation,
and their hearts were excised at 2 and 4 weeks after TAC. For
global morphometry, the hearts were perfused with
phosphate-buffered saline followed by 4% paraformaldehyde. For
histological analysis, the heart tissues were fixed in 10%
formalin, embedded in paraffin or frozen in liquid nitrogen,
sectioned at 4 mm thickness, and then stained with hematoxylin and
eosin and Masson’s trichrome (Senbeijia Co., Ltd., Jiangsu, China).
The heart tissue morphological characteristics and the differences
between the 3 experimental groups (sham-operated group, the group
subjected to TAC and injected with GFP and the group subjected to
TAC and injected with miR-455) were observed under a microscope
(Nikon, Tokyo, Japan). The extent of fibrosis was evaluated by
measuring the Masson’s trichrome-stained area in the entire left
ventricular wall. All measurements were acquired using an automated
image analysis system (Motic6.0; Motic, Xiamen, China).
In situ detection of myocardial
apoptosis
The apoptotic cells were fixed and permeabilized.
Subsequently, the cells were incubated with 50 μl terminal
deoxynucleotidyl transferase-mediated dUTP nick-end labeling
(TUNEL) reaction mixture (In Situ Cell Death Detection kit, POD;
Roche, Shanghai, China) and kept for 60 min in a wet box. After
washing, the label incorporated at the damaged sites of the DNA was
marked by an anti-fluorescein antibody conjugated with the reporter
enzyme, peroxidase. After washing to remove the unbound enzyme
conjugate, the POD retained in the immune complex was visualized by
a substrate reaction. The TUNEL-positive cells were imaged under a
microscope at a magnification of x400 (Nikon) and 3 horizons were
randomly selected in each slice. The TUNEL-positive cells were
counted using a digital medical image analysis system (Motic6.0;
Motic). The results were expressed as the number of TUNEL-positive
cells/102 cardiomyocytes.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) of mature miRNAs
Total RNA was extracted from the LV tissues of the
mice using TRIzol reagent (Invitrogen) and reverse transcribed into
complementary DNA (cDNA) using EasyScript First-Strand cDNA
Synthesis SuperMix (TransGen, Beijing, China) following the
manufacturer’s instructions. Mature miR-455 expression was
quantified by RT-qPCR using the All-in-One miRNA qRT-PCR Detection
system following the manufacturer’s instructions (GeneCopoeia,
Guangdong, China). The primers used for PCR were as follows:
miR-455 forward, ATGTGCCTTTGGACTACATCGAA; and U6 forward,
TCGTGAAGCGTTCCATATTTTTAA; consensus primer sequence,
TTACTACGTCATGACTAGTAA. The program was initially run for 10 min at
95°C, followed by 40 cycles of 10 sec at 95°C, 27 sec at 60°C and
27 sec at 72°C. Gene expression levels were normalized to the U6
rRNA endogenous control and fold changes were calculated using the
ΔΔCt method.
For qPCR, the transcribed complementary DNA was then
subjected to qPCR analysis using a Bio-Rad IQ5 multicolor detection
system (Bio-Rad, Hercules, CA, USA). A comparative cycle threshold
method was used to determine the relative quantification of RNA
expression. All PCR reactions were performed at least in
triplicate. Atrial natriuretic factor (Anf), skeletal muscle
alpha-actin (Acta1), β-myosin heavy chain (Myh7), transforming
growth factor β-1 (Tgfβ1), connective tissue growth factor (Ctgf),
Calr, glucose-regulated protein 78 (GRP78) and β-actin were
amplified using their specific primers (Table I). The program was initially run
for 30 sec at 95°C, followed by 40 cycles of 5 sec at 95°C, 20 sec
at 60°C and 20 sec at 72°C.
 | Table IPrimers for quantitative polymerase
chain reaction. |
Table I
Primers for quantitative polymerase
chain reaction.
| Gene names | Primer
sequences |
|---|
| Anf | F:
GCTCCTTCTCCATCACCCTG
R: ACCGGCATCTTCTCCTCCA |
| Acta1 | F:
TGAGCGTGGCTATTCCTTCG
R: CCGCAGACTCCATACCGATAA |
| Myh7 | F:
GCCAACACCAACCTGTCCAAGTTC
R: TTCAAAGGCTCCAGGTCTCAGGGC |
| Tgfβ1 | F:
CCCGAGTGTGGAAGATGAGAA
R: AACCTGAAAGCAGCCCTTCTG |
| Ctgf | F:
AATCGCCAAGCCTGTCAAGT
R: CCCAGGACAGTTGTAATGGCA |
| Calr | F:
TCTGTCCCTCCCTTTCTCCA
R: AGCTGTGCTAGAACTGGCTGC |
| GRP78 | F:
TCTGGTTGGCGGATCTACTC
R: TCTTTTGTCAGGGGTCGTTC |
| β-actin | F:
AGCGTGGCTACAGCTTCACC
R: CCGCTCGTTGCCAATAGTG |
Western blot analysis
Total protein was obtained from the ventricular
myocardial tissues using tissue homogenates, centrifugation and
heat denaturation. The protein lysates were electrophoresed and
separated by 6–12% sodium dodecyl sulfate-polyacrylamide gel
electrophoresis (SDS-PAGE) and transferred onto PVDF membranes
(Millipore Immobion-P; BioSharp, Anhui, China). The membranes were
blocked with 5% skim milk at room temperature for 1 h, and then
incubated overnight at 4°C with primary antibodies, including
rabbit anti-Bcl-2 (BA0412; 1:500), rabbit anti-Bax (BA0315-2;
1:500), rabbit anti-Calr (BM1798; 1:500), rabbit anti-GRP78
(BA2042; 1:500) (all from Boster Biotechnology, Inc., Wuhan,
China), and rabbit anti-glyceraldehyde 3-phosphate dehydrogenase
(GAPDH) (AP0063; 1:5,000; BioWorld, Inc., Jiangsu, China). The
membranes were then incubated with IRDye800-conjugated secondary
antibodies (1:20,000; Rockland, Inc., Gilbertsville, PE, USA) at
room temperature for 1 h. The Odyssey double color infrared laser
imaging system (LI-COR; Lincoln, NE, USA) was used to detect the
antigen-antibody complexes in a western blotting detection system
(Bio-Rad). The results were expressed as density values normalized
to GAPDH.
Statistical analysis
Data are expressed as the means ± standard error of
the mean. The underlying assumption of normal distribution was
investigated by performing a Kolmogorov-Smirnoff normality test and
a normal probability plot test. Statistical significance between 2
groups was examined by a t-test for normal distribution and by
one-way ANOVA for multi-group comparisons. When the ANOVA results
were significant, the differences among individual groups were
determined using the Bonferroni post hoc test. A value of P<0.05
was considered to indicate a statistically significant
difference.
Results
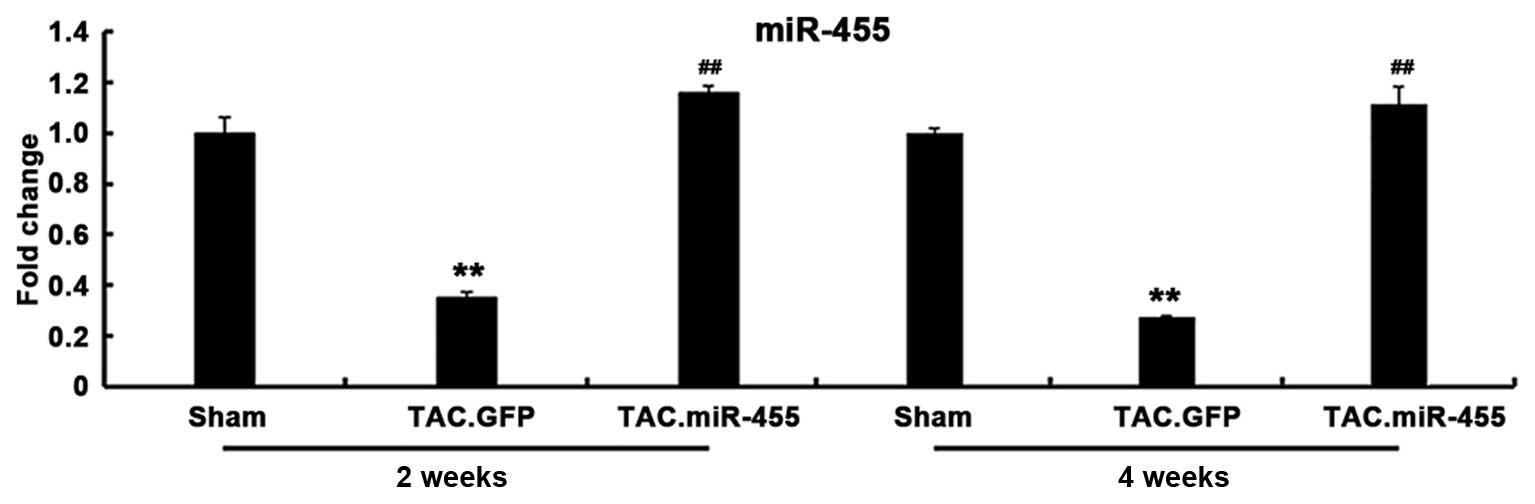
miR-455 gene expression in vivo
The gene expression of miR-455 was significantly
decreased in the GFP-treated hearts, but was significantly
increased in the miR-455-treated hearts after TAC. The expression
of the mature miR-455 in the hypertrophied hearts induced by
pressure overload was restored by Ad-mediated miR-455 gene transfer
in vivo (Fig. 2).
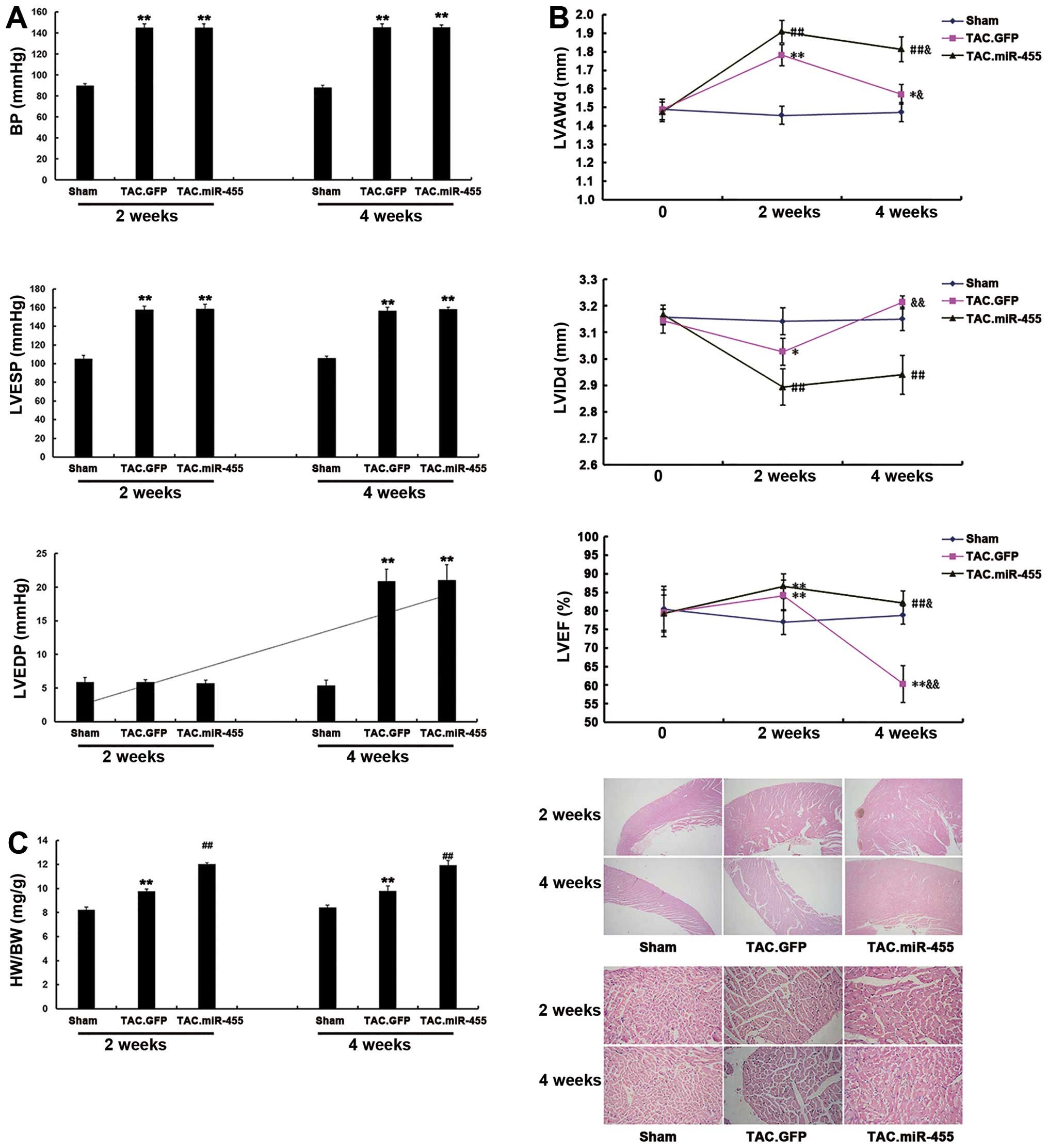
miR-455 aggravates cardiac hypertrophy in
mice 2 weeks after TAC
The effects of miR-455 on cardiac hypertrophy at 2
weeks after TAC were investigated. At this time point, all the
experimental mice survived, and some were subjected to hemodynamic
and echocardiographic examinations. BP and LVESP were similarly
elevated, whereas LVEDP was not altered in all the mice examined.
TAC induced significant cardiac hypertrophy, as characterized by an
increased left ventricular wall thickness [left ventricular
anterior wall thickness at end diastole (LVAWd)], decreased left
ventricular cavity dimension [left ventricular internal dimension
at end diastole (LVIDd)], increased heart-to-body weight ratio and
an expanded cross-sectional area of cardiomyocytes in the
GFP-treated mice. All these characteristics were significantly
aggravated by treatment with miR-455 2 weeks after TAC (Fig. 3).
miR-455 gene transfer preserves cardiac
adaptation and function at 4 weeks after TAC
In the GFP-treated mice, BP, LVESP, the
heart-to-body weight ratio and the cardiomyocyte cross-sectional
area were not altered at 4 weeks after TAC as compared with the
levels observed at 2 weeks after TAC. However, at 4 weeks after
TAC, GFP treatment elevated LVEDP, reduced left ventricular wall
thickness (LVAWd), enlarged left ventricular cavity dimension
(LVIDd) and lowered left ventricular contractility (ejection
fraction) (Fig. 3). These results
indicate significant cardiac remodeling with impaired cardiac
function in the mice at 4 weeks after TAC. Treatment with miR-455
reduced LVAWd, but did not enlarge left ventricular cavity
dimension (LVIDd) and did not lower LV contractility. Thus, the
miR-455-treated hearts maintained a state of myocardial
hypertrophy. These results suggest that miR-455 gene transfer is
effective in the prevention of cardiac remodeling and dysfunction
during a 4-week period of pressure overload.
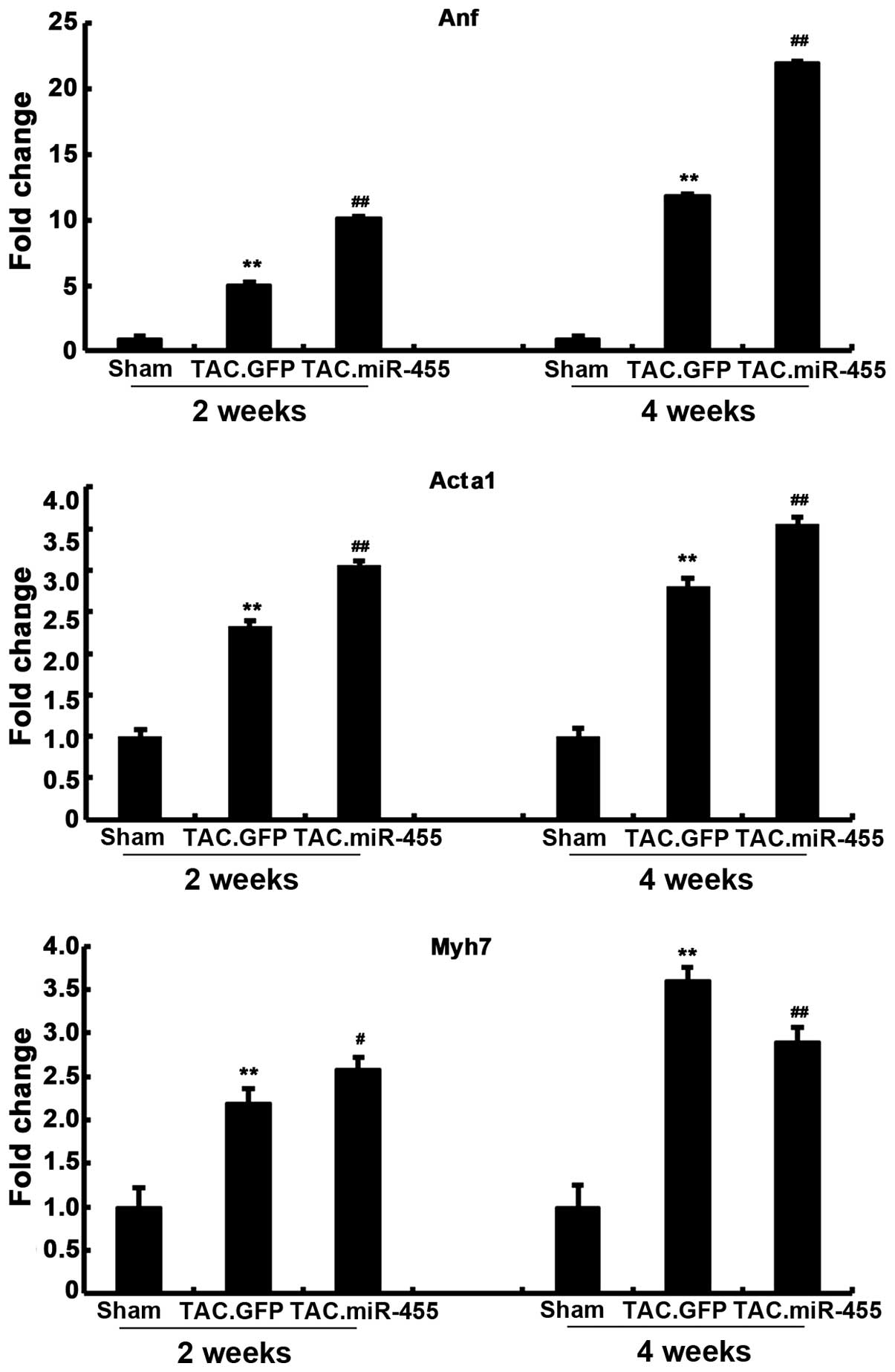
miR-455 modulates the expression of
molecular markers of cardiac hypertrophy
The effects of miR-455 treatment on molecular
abnormalities associated with pathological hypertrophy were
investigated. We assessed the expression of the hypertrophic fetal
genes, Anf, Acta1 and Myh7 at 2 and 4 weeks after gene transfer
(Fig. 4). The pressure
overload-induced hypertrophy in the GFP-treated group was
associated with the re-induction of the ‘fetal gene program’, which
was characterized by a significant increase in the mRNA expression
levels of Anf, Myh7 and Acta1, as compared with those in the
sham-operated group. Furthermore, the expression levels of these 3
genes significantly increased in the miR-455-treated mouse hearts
compared with the GFP-treated mouse hearts. That is, the expression
levels of Anf, Myh7 and Acta1 were significantly upregulated by the
pressure overload in the GFP-treated hearts, and these hypertrophic
responses were significantly aggravated after gene transfer.
However, the expression of Myh7 was higher in the GFP-treated mice
than in the miR-455-treated mice at 4 weeks.
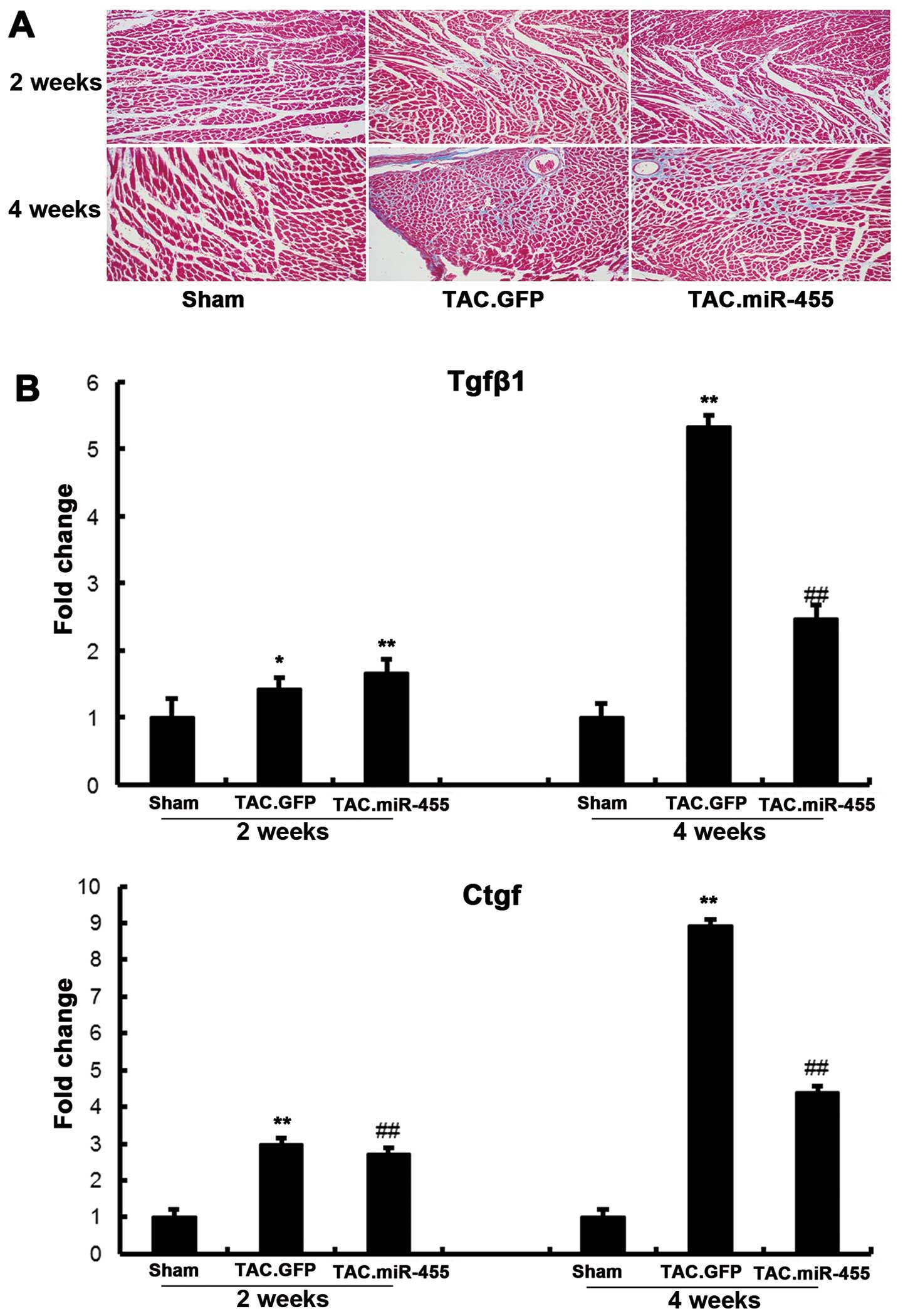
miR-455 inhibits myocardial fibrosis and
decreases the expression of molecular markers of cardiac fibrosis
at 4 weeks after TAC
Considering that cardiac fibrosis and apoptosis are
prominent features in the transition from compensatory hypertrophy
to heart failure, we investigated the potential involvement of the
restoration of miR-455 in the regulation of cardiac ECM remodeling
and apoptosis. Fibrosis is a pathological characteristic of cardiac
adaptation to stress, where the proliferation of fibroblasts and
the increased deposition of extracellular matrix (ECM) components
result in myocardial stiffness and diastolic dysfunction (24). Previous studies have demonstrated
that miRNAs play central roles in controlling cardiac fibrosis and
pathological LV remodeling (20,25,26). Histological examination of the
left ventricular sections by Masson’s trichrome staining and the
subsequent quantification of the fibrotic area revealed that TAC
significantly increased interstitial fibrosis in the GFP-treated
hearts as compared with the sham-operated hearts. By contrast,
treatment with miR-455 significantly decreased fibrosis (Fig. 5).
The mRNA expression levels of Tgfβ1 and Ctgf
significantly increased in the GFP-treated hearts as compared with
the sham-operated hearts (Fig.
6). However, these increased levels of myocardial
fibrosis-related genes following pressure overload were
significantly decreased in the mice treated with miR-455.
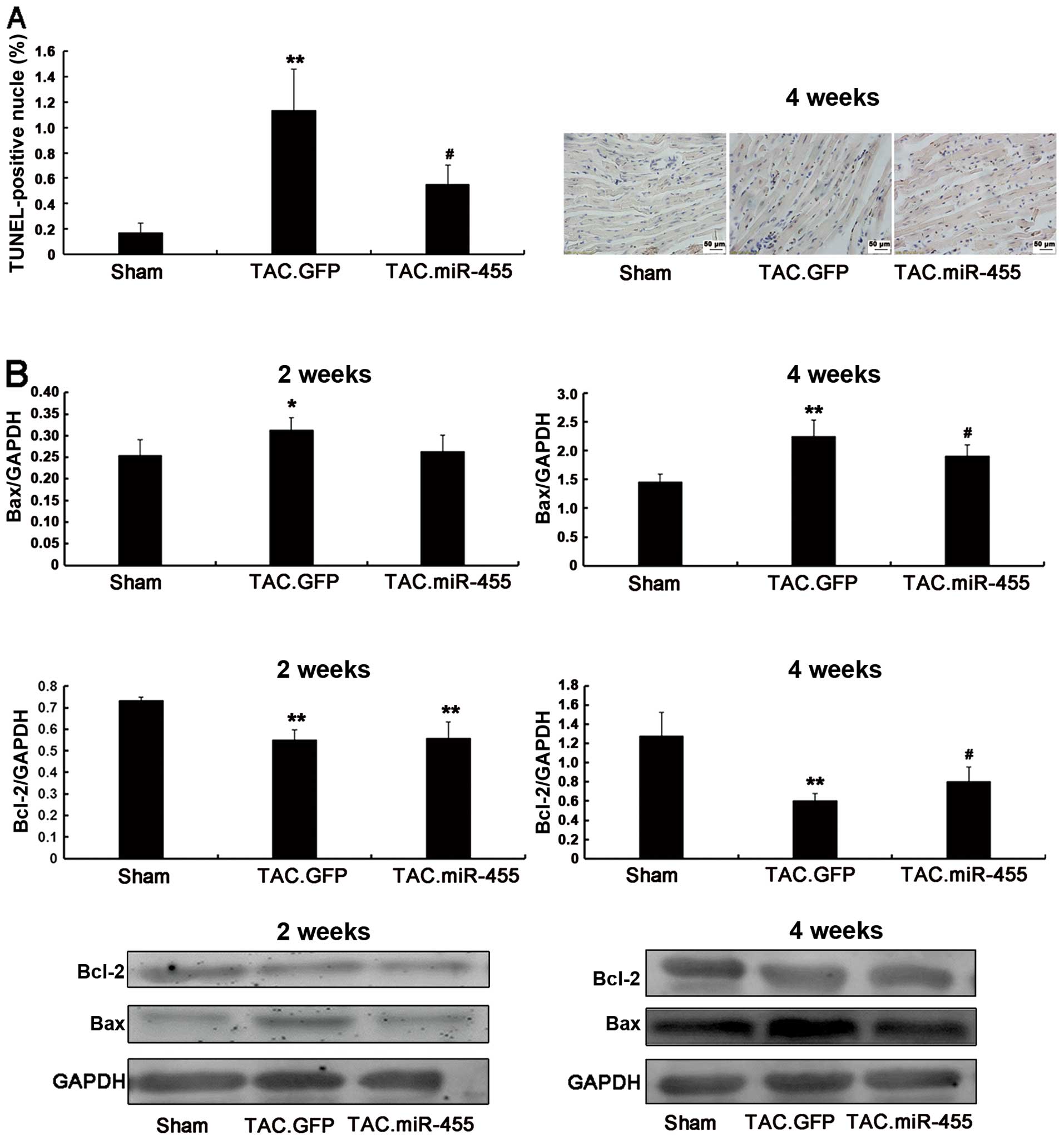
miR-455 inhibits heart myocardial
apoptosis
In response to pressure overload, cardiomyocyte
apoptosis may further contribute to the transition from left
ventricular hypertrophy to heart failure (27). Using western blot analysis, we
quantified the protein expression of the anti-apoptotic gene,
Bcl-2, and the pro-apoptotic gene, Bax (Fig. 6). Compared to treatment with GFP,
treatment with miR-455 significantly increased Bcl-2 expression and
decreased Bax expression. Consequently, the Bcl-2/Bax ratio, an
important marker of myocardial cell survival probability (27), was significantly increased in the
miR-455-treated hearts as compared with the GFP-treated hearts
(Fig. 6).
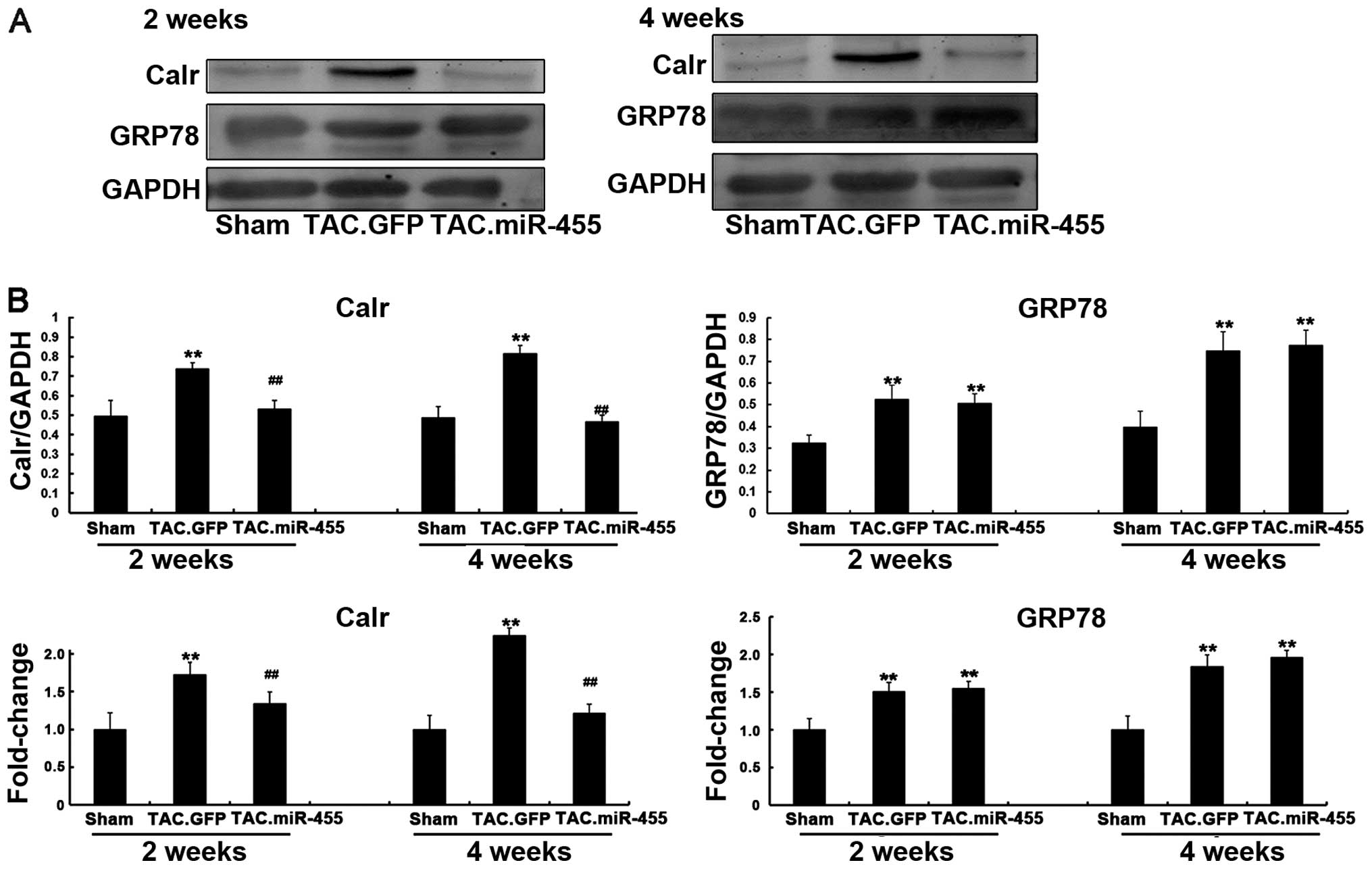
Calr is a direct target of miR-455 that
is involved in miR-455-mediated effects in mouse hearts after
TAC
Endoplasmic reticulum (ER) stress occurs during
myocardial hypertrophy (28,29). The sarcoplasmic reticulum is a
principal subcellular organelle that regulates the calcium
homeostasis, protein synthesis and the apoptosis of cardiomyocytes.
ER stress triggers calcium homeostasis imbalance and abnormal
functional protein formation; these phenomena are accompanied by
upregulated ER chaperones, such as Calr and GRP78 (30-32). However, the association between
miRNA-455 and Calr during cardiac hypertrophy remains unclear.
Therefore, we evaluated whether the expression levels of Calr and
GRP78 are regulated by miR-455 in vivo in hypertrophy
induced by pressure overload. Western blot analysis revealed that
the protein expression levels of both Calr and GRP78 significantly
increased in the left ventricular tissue of the GFP-treated mice as
compared with the sham-operated animals. However, Calr was
down-regulated and GRP78 was upregulated with the upregulation of
miR-455 in the left ventricular tissue of the miR455-treated mice
(Fig. 7). These results indicate
that Calr and not GRP78 is the target of miR-455. miRNAs can also
degrade the mRNA of their targets (33,34). The results of RT-qPCR revealed
that miR-455 significantly decreased the mRNA expression level of
Calr, but did not alter the mRNA expression level of GRP78 as
compared with GFP (Fig. 7).
Discussion
To the best of our knowledge, this study is the
first to demonstrate that miR-455 is downregulated in pressure
overload-induced cardiac hypertrophy in vivo and that this
downregulation increases the mRNA and protein levels of Calr, the
predicted target of miR-455. This study assessed the short-term and
long-term effects of miR-455 gene transfer in pressure
overload-induced cardiac hypertrophy in vivo using a
cardiotropic Ad-13 vector that efficiently transduced cardiac
tissues. This study is also the first to reveal the short-term and
long-term effects of miR-455 on pressure overload-induced cardiac
hypertrophy. A number of fetal genes, such as Anf, Acta1 and Myh7,
are re-expressed during the cardiac hypertrophic response (35). These 3 genes were upregulated in
the mice following aortic coarctation, particularly in the
miR-455-treated mice. A transition occurred from left ventricular
hypertrophy to heart failure in response to long-term pressure
overload. Cardiac fibrosis and apoptosis are prominent
characteristics in the transition from compensatory hypertrophy to
heart failure. Cardiac fibrosis and apoptosis were alleviated in
the miR-455-treated mice. The normalization of miR-455 gene
expression levels, which were downregulated during hypertrophy,
aggravated cardiac hypertrophy in the short term, but attenuated
pathological remodeling in the long term.
The different effects of miR-455 on myocardial
hypertrophy in the short and long term may be related to its target
gene, Calr. The conditions in the ER must be optimal to facilitate
the efficient synthesis and folding of most secreted membrane
proteins; suboptimal conditions lead to improper protein folding
and eventual ER stress (36).
Initially, ER stress triggers protective aspects of the conserved
signaling program known as the unfolded protein response (UPR), and
these aspects are oriented toward restoring the ER environment
(37-39). However, apoptotic aspects of the
UPR will ensue if the stress continues and ER protein folding is
not restored (40,41). Calr, a Ca2+-binding
protein of the ER, is an important chaperone that works in
conjunction with calnexin and protein disulfide isomerase. It
affects intracellular Ca2+ homeostasis through its
Ca2+ storage capacity and its effects on both the SERCA
pumps and inositol 1,4,5-trisphosphate receptors (42,43). Our findings on the different
effects of miR-455 are in agreement with other data demonstrating
the effects of Calr on cardiomyocytes (28,30). Thus, the myocardial state during
the application of miR-455 for the treatment of myocardial
hypertrophy is highly important. This study found no evidence to
prove that miR-455 directly influences the different genes. The
change observed may be due to an indirect effect of miR-455 on the
different genes. Furthermore, this change may differ when
hypertrophy results from other causes than TAC with or without
miR-455 transfection.
In conclusion, the Ad-13-mediated normalization of
miR-455 expression aggravates the hypertrophic phenotype, but
attenuates the progressive deterioration of left ventricular
function. The restoration or downregulation of miR-455 at different
time periods may lead to a pioneering therapeutic strategy to
reverse cardiac hypertrophy and alleviate function
deterioration.
References
|
1
|
Ho YL, Wu CC, Lin LC, et al: Assessment of
the coronary artery disease and systolic dysfunction in
hypertensive patients with the dobutamine-atropine stress
echocardiography: effect of the left ventricular hypertrophy.
Cardiology. 89:52–58. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Aaronson KD and Sackner-Bernstein J: Risk
of death associated with nesiritide in patients with acutely
decompensated heart failure. JAMA. 296:1465–1466. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Catalucci D, Latronico MV, Ellingsen O and
Condorelli G: Physiological myocardial hypertrophy: how and why.
Front Biosci. 13:312–324. 2008. View
Article : Google Scholar
|
|
4
|
Koren MJ, Devereux RB, Casale PN, Savage
DD and Laragh JH: Relation of left ventricular mass and geometry to
morbidity and mortality in uncomplicated essential hypertension.
Ann Intern Med. 114:345–352. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
McKinsey TA and Kass DA: Small-molecule
therapies for cardiac hypertrophy: moving beneath the cell surface.
Nat Rev Drug Discov. 6:617–635. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bartel DP and Chen CZ: Micromanagers of
gene expression: the potentially widespread influence of metazoan
microRNAs. Nat Rev Genet. 5:396–400. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kloosterman WP and Plasterk RH: The
diverse functions of microRNAs in animal development and disease.
Dev Cell. 11:441–450. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Divakaran V and Mann DL: The emerging role
of microRNAs in cardiac remodeling and heart failure. Circ Res.
103:1072–1083. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
van Rooij E and Olson EN: MicroRNAs:
powerful new regulators of heart disease and provocative
therapeutic targets. J Clin Invest. 117:2369–2376. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bartel DP: MicroRNAs: target recognition
and regulatory functions. Cell. 136:215–233. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
van Rooij E: The art of microRNA research.
Circ Res. 108:219–234. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bartel DP: MicroRNAs: genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
van Rooij E, Marshall WS and Olson EN:
Toward microRNA-based therapeutics for heart disease: the sense in
antisense. Circ Res. 103:919–928. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Small EM, Frost RJ and Olson EN: MicroRNAs
add a new dimension to cardiovascular disease. Circulation.
121:1022–1032. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Care A, Catalucci D and Felicetti F:
MicroRNA-133 controls cardiac hypertrophy. Nat Med. 13:613–618.
2007. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
van Rooij E, Sutherland LB, Liu N, et al:
A signature pattern of stress-responsive microRNAs that can evoke
cardiac hypertrophy and heart failure. Proc Natl Acad Sci USA.
103:18255–18260. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
van Rooij E, Sutherland LB, Qi X,
Richardson JA, Hill J and Olson EN: Control of stress-dependent
cardiac growth and gene expression by a microRNA. Science.
316:575–579. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yang B, Lin H and Xiao J: The
muscle-specific microRNA miR-1 regulates cardiac arrhythmogenic
potential by targeting GJA1 and KCNJ2. Nat Med. 13:486–491. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Thum T, Gross C and Fiedler J: MicroRNA-21
contributes to myocardial disease by stimulating MAP kinase
signalling in fibroblasts. Nature. 456:980–984. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
van Rooij E, Sutherland LB, Thatcher JE,
et al: Dysregulation of microRNAs after myocardial infarction
reveals a role of miR-29 in cardiac fibrosis. Proc Natl Acad Sci
USA. 105:13027–13032. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hu S, Huang M and Li Z: MicroRNA-210 as a
novel therapy for treatment of ischemic heart disease. Circulation.
122:S124–S131. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Thum T, Galuppo P and Wolf C: MicroRNAs in
the human heart: A clue to fetal gene reprogramming in heart
failure. Circulation. 116:258–267. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Betel D, Wilson M, Gabow A, Marks DS and
Sander C: The microRNA.org resource: targets and expression.
Nucleic Acids Res. 36:D149–D153. 2008. View Article : Google Scholar :
|
|
24
|
Papp S, Dziak E, Kabir G, Backx P, Clement
S and Opas M: Evidence for calreticulin attenuation of cardiac
hypertrophy induced by pressure overload and soluble agonists. Am J
Pathol. 176:1113–1121. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Creemers EE and Pinto YM: Molecular
mechanisms that control interstitial fibrosis in the
pressure-overloaded heart. Cardiovasc Res. 89:265–272. 2011.
View Article : Google Scholar
|
|
26
|
Duisters RF, Tijsen AJ, Schroen B, et al:
miR-133 and miR-30 regulate connective tissue growth factor:
implications for a role of microRNAs in myocardial matrix
remodeling. Circ Res. 104:170–178. 2009. View Article : Google Scholar
|
|
27
|
Condorelli G, Morisco C, Stassi G, et al:
Increased cardio-myocyte apoptosis and changes in proapoptotic and
antiapoptotic genes bax and bcl-2 during left ventricular
adaptations to chronic pressure overload in the rat. Circulation.
99:3071–3078. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Okada K, Minamino T and Tsukamoto Y:
Prolonged endoplasmic reticulum stress in hypertrophic and failing
heart after aortic constriction: possible contribution of
endoplasmic reticulum stress to cardiac myocyte apoptosis.
Circulation. 110:705–712. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Brostrom MA, Mourad F and Brostrom CO:
Regulated expression of GRP78 during vasopressin-induced
hypertrophy of heart-derived myocytes. J Cell Biochem. 83:204–217.
2001. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lee KH, Lee N, Lim S, et al: Calreticulin
inhibits the MEK1,2-ERK1,2 pathway in alpha 1-adrenergic
receptor/Gh-stimulated hypertrophy of neonatal rat cardiomyocytes.
J Steroid Biochem Mol Biol. 84:101–107. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Xu FF, Liu XH and Zhu XM: Calreticulin
upregulation induced by hypoxic preconditioning relieves oxidative
stress injury in rat cardiomyocytes. Sheng Li Xue Bao. 60:29–37.
2008.In Chinese. PubMed/NCBI
|
|
32
|
Liu XH: Endoplasmic reticulum stress and
myocardial hypertrophy. Sheng Li Xue Bao. 61:9–14. 2009.In Chinese.
PubMed/NCBI
|
|
33
|
Guo H, Ingolia NT, Weissman JS and Bartel
DP: Mammalian microRNAs predominantly act to decrease target mRNA
levels. Nature. 466:835–840. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lim LP, Lau NC, Garrett-Engele P, et al:
Microarray analysis shows that some microRNAs downregulate large
numbers of target mRNAs. Nature. 433:769–773. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Heineke J and Molkentin JD: Regulation of
cardiac hypertrophy by intracellular signalling pathways. Nat Rev
Mol Cell Biol. 7:589–600. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
36
|
Glembotski CC: Endoplasmic reticulum
stress in the heart. Circ Res. 101:975–984. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kaufman RJ: Stress signaling from the
lumen of the endoplasmic reticulum: coordination of gene
transcriptional and translational controls. Genes Dev.
13:1211–1233. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Austin RC: The unfolded protein response
in health and disease. Antioxid Redox Signal. 11:2279–2287. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Glembotski CC: The role of the unfolded
protein response in the heart. J Mol Cell Cardiol. 44:453–459.
2008. View Article : Google Scholar
|
|
40
|
Kim I, Xu W and Reed JC: Cell death and
endoplasmic reticulum stress: disease relevance and therapeutic
opportunities. Nat Rev Drug Discov. 7:1013–1030. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Xu C, Bailly-Maitre B and Reed JC:
Endoplasmic reticulum stress: cell life and death decisions. J Clin
Invest. 115:2656–2664. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
42
|
John LM, Lechleiter JD and Camacho P:
Differential modulation of SERCA2 isoforms by calreticulin. J Cell
Biol. 142:963–973. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Camacho P and Lechleiter JD: Calreticulin
inhibits repetitive intracellular Ca2+ waves. Cell.
82:765–771. 1995. View Article : Google Scholar : PubMed/NCBI
|