Introduction
Angiogenesis refers to the formation of new blood
vessels from existing vessels (1). As tissues require blood vessels to
supply nutrients and oxygen, angiogenesis is required for solid
tumor growth and metastasis. Tumor angiogenesis is initiated when
tumor cells secrete growth factors, such as vascular endothelial
growth factor (VEGF), which bind to the receptors on endothelial
cells (2,3). Once activated, the endothelial cells
go through a process involving proliferation, migration,
reorganization of the extracellular matrix, tube formation and
maturation, and eventually form a functional vascular plexus
(2,4). Endothelial cell proliferation is the
key initial step of angiogenesis and is tightly regulated by a
number of intersecting pathways (5). The essential components of these
signaling pathways may offer targets for anti-angiogenesis
therapeutic intervention (6).
Mitogen-activated protein kinase (MAPK) signaling
cascades participate in the regulation of numerous fundamental
cellular processes (7). In
mammals, MAPKs have three major subfamilies: Extracellular
signal-regulated kinase (ERK1/2 or p44/42 MAPK), p38 MAPK and c-Jun
N-terminal kinase (JNK) (8). The
ERK signaling pathway regulates cell proliferation and survival. In
endothelial cells, the ERK pathway is initiated by the binding of
VEGF or basic fibroblast growth factor (bFGF) to their receptors,
which leads to the activation of Ras proteins anchored in the
plasma membrane (9).
Subsequently, the Ras proteins exchange their bound guanosine
diphosphate for guanosine triphosphate, which triggers a
conformational change in Ras and activation of Raf. Raf
subsequently phosphorylates MAPK/ERK1/2 (MEK1/2), which in turn
activates ERK1/2 through phosphorylation of a threonine and a
tyrosine residue, such as Thr202/Tyr204 of ERK1 and Thr183/Tyr185
of ERK2. Activated ERK1/2 subsequently phosphorylates the
serine/threonine residues of >50 downstream substrates, leading
to modification of gene expression profiles and an increase in
endothelial cell proliferation and survival (9).
Artemisinin is an antimalarial drug isolated from
Artemisia annua L. (10).
Artemisinin and its derivatives have also been found to inhibit the
growth of lung, prostate, breast, colon and pancreatic cancer
(11). Dihydroartemisinin (DHA),
a semi-synthetic derivative of artemisinin, has exhibited potent
anticancer and anti-angiogenesis activities (12). For example, DHA inhibited
angiogenesis in a rat embryo development model (13) and a chicken chorioallantoic
membrane model (12). Through
in vitro assays, DHA decreased endothelial cell
proliferation, migration and tube formation (14–16). Despite the emergence of DHA as
novel component of cancer chemotherapies, the mechanisms of its
anti-angiogenic effects have not been fully understood.
Previous studies reported that MAPK signaling
cascades are differentially regulated by artemisinin and its
derivatives depending on the cell type and experimental settings.
Artemisinin upregulates phosphorylation of ERK1/2 and p38 MAPK to
promote neurite outgrowth in p12 cells (17). In human monocytic THP-1 cells,
artemisinin blocked the phosphorylation of JNK, ERK1/2 and p38 MAPK
(18). In HL-60 leukemia cells,
DHA induced apoptosis through activation of p38 MAPK, but not JNK
or ERK1/2 (19). In endothelial
cells cultured with VEGF, DHA failed to increase the activation of
p38 pathway (20). Thus far, the
effects of the artemisinin family of drugs on the ERK signaling
pathway have not been studied in endothelial cells.
In the present study, the role of the ERK signaling
pathway in the DHA-induced reduction of endothelial cell
proliferation was investigated. At a concentration of 20 μM,
DHA inhibits proliferation of human umbilical vein endothelial
cells (HUVECs), and suppresses the expression and phosphorylation
of ERK1/2, as well as their downstream effectors, c-Fos and c-Myc.
In addition, the ERK pathway inhibitor PD90859 comprises the
anti-proliferative effects of DHA. Thus, DHA inhibits endothelial
cell proliferation by suppressing the ERK signaling pathway.
Materials and methods
Cell culture
HUVECs were purchased from Lonza (Basel,
Switzerland) and were cultured in endothelial basal cell medium-2
supplemented using the EGM-2-MV bullet kit (Lonza) and antibiotics
(100 IU/ml penicillin and 100 μg/ml streptomycin). The cells
were cultured in humidified air at 37°C with 5% CO2. DHA
(Sigma-Aldrich, St. Louis, MO, USA) and PD98059 (Cell Signaling
Technology, Beverly, MA, USA) were dissolved in dimethyl
sulfoxide.
Cell proliferation assay
Cell proliferation was evaluated with the MTT assay
kit (Cayman Chemical, Ann Arbor, MI, USA). Briefly, the cells were
seeded at a density of 5×103 cells/well in a 96-well
plate. The following day, DHA (0, 1, 5, 10, 20, 50 and 100
μM) was added to the culture media. After 24 h, the cells
were washed with phosphate-buffered saline (PBS) and MTT solution
(10 μl of 5 mg/ml) was added to each well for 2 h. Formazan
crystals were solubilized and colorimetric intensity was analyzed
using a 96-well plate reader (Molecular Devices, Sunnyvale, CA,
USA) at a wavelength of 570 nm.
Cell viability assay
Cell viability was assessed at different time points
after DHA treatment. Cultures were washed and incubated in 0.05%
trypsin for 2 min at 37°C. Following disaggregation, cell
suspensions were diluted 1:1 in 0.4% trypan blue (w/v in 0.9% NaCl)
(Santa Cruz Biotechnology, Inc., Dallas, TX, USA) and the
percentage of dye-free cells was calculated.
Annexin V-fluorescein isocyanate
(FITC)/propidium iodide (PI) analyses
The apoptosis of HUVECs treated with DHA was
detected by Annexin V-FITC and PI staining according to the
manufacturer’s instructions (NeoBiosciences, Shenzhen, China).
Briefly, cells were trypsinized, pelleted, washed with PBS and
resuspended in binding buffer containing Annexin V-FITC (0.25%) and
PI (1 μg/ml). Detection of positive staining from
1×105 cells was obtained using a FACSAria II flow
cytometer (BD Biosciences, San Jose, CA, USA) and analyzed using
the FACSDiva acquisition and analysis software.
Western blotting
Cells were washed with PBS and lysed in
radioimmunoprecipitation assay buffer [20 mM Tris (pH 7.5), 150 mM
NaCl, 50 mM NaF, 1% nonidet P-40, 0.1% deoxycholic acid, 0.1%
sodium dodecyl sulfate (SDS), 1 mM EDTA, 1 mM phenylmethylsulfonyl
fluoride and 1 μg/ml leupeptin]. Protein concentrations of
cleared lysates were determined using the bicinchoninic assay
(Bio-Rad, Hercules, CA, USA). Equal amounts of protein were
separated by SDS-polyacrylamide gel (8% polyacrylamide gel) and
transferred to a polyvinylidene fluoride (PVDF) membrane. PVDF
membranes were blocked with 5% skimmed milk and incubated overnight
with the primary antibody in PBS-Tween at 4°C. Immunoreactivity was
detected with horseradish peroxidase-conjugated secondary
antibodies and chemiluminescence. The primary antibodies included
proliferating cell nuclear antigen (PCNA; PC10), phospho-p44/42
MAPK (ERK1/2) (Thr202/Tyr204) (D13.14.4E), p44/42 MAPK (ERK1/2)
(137F5), c-Fos and c-Myc antibodies (Cell Signaling Technology) and
anti-β-actin antibody (Sigma-Aldrich). β-actin levels were used as
controls for protein loading.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
RNA isolation and cDNA synthesis were performed
using the RNeasy mini kit (Qiagen, Hilden, Germany) and High
Capacity RNA-to-cDNA Master mix (Applied Biosystems, Foster City,
CA, USA), respectively. RT-qPCR was performed using a ViiA 7
Real-Time PCR system (Life Technologies, Carlsbad, CA, USA). All
the PCR reactions were repeated in triplicate. Relative expression
was calculated using β-actin as an endogenous internal control. The
primer sequences were summarized in Table I.
 | Table IRT-qPCR primer sequences. |
Table I
RT-qPCR primer sequences.
| Gene | Sequence | Size, bp | Tm, °C |
|---|
| ERK1
(MAPK3) |
| Forward |
CTACACGCAGTTGCAGTACAT | 157 | 58.66 |
| Reverse |
CAGCAGGATCTGGATCTCCC | | 59.31 |
| ERK2
(MAPK1) |
| Forward |
TACACCAACCTCTCGTACATCG | 169 | 59.58 |
| Reverse |
CATGTCTGAAGCGCAGTAAGATT | | 59.38 |
| c-fos |
| Forward |
TAGTTAGTAGCATGTTGAGCCAGG | 333 | 60.14 |
| Reverse |
ACCACCTCAACAATGCATGA | | 57.71 |
| c-myc |
| Forward |
AATGAAAAGGCCCCCAAGGTAGTTATCC | 112 | 64.87 |
| Reverse |
GTCGTTTCCGCAACAAGTCCTCTTC | | 64.41 |
| β-actin |
| Forward |
GGCACCACACCTTCTACAATG | 352 | 59.19 |
| Reverse |
GTGGTGGTGAAGCTGTAGCC | | 60.96 |
Electric cell-substrate impedance sensing
(ECIS) analysis
The transcellular resistance across a monolayer of
endothelial cells was measured using the ECIS technique (ECIS model
1600; Applied BioPhysics, Troy, NY, USA). Briefly, HUVECs were
plated in ECIS arrays and allowed to grow into 50% confluence. DHA
and/or PD98059 were added to the wells and the resistance across
the EC layer was acquired every 8 sec. Data plots are
representative of triplicate experiments, with each graph showing
impedance readings from a separate well, at 40 distinct electrodes
per well.
Results
Dose-response effects of DHA on HUVEC
proliferation
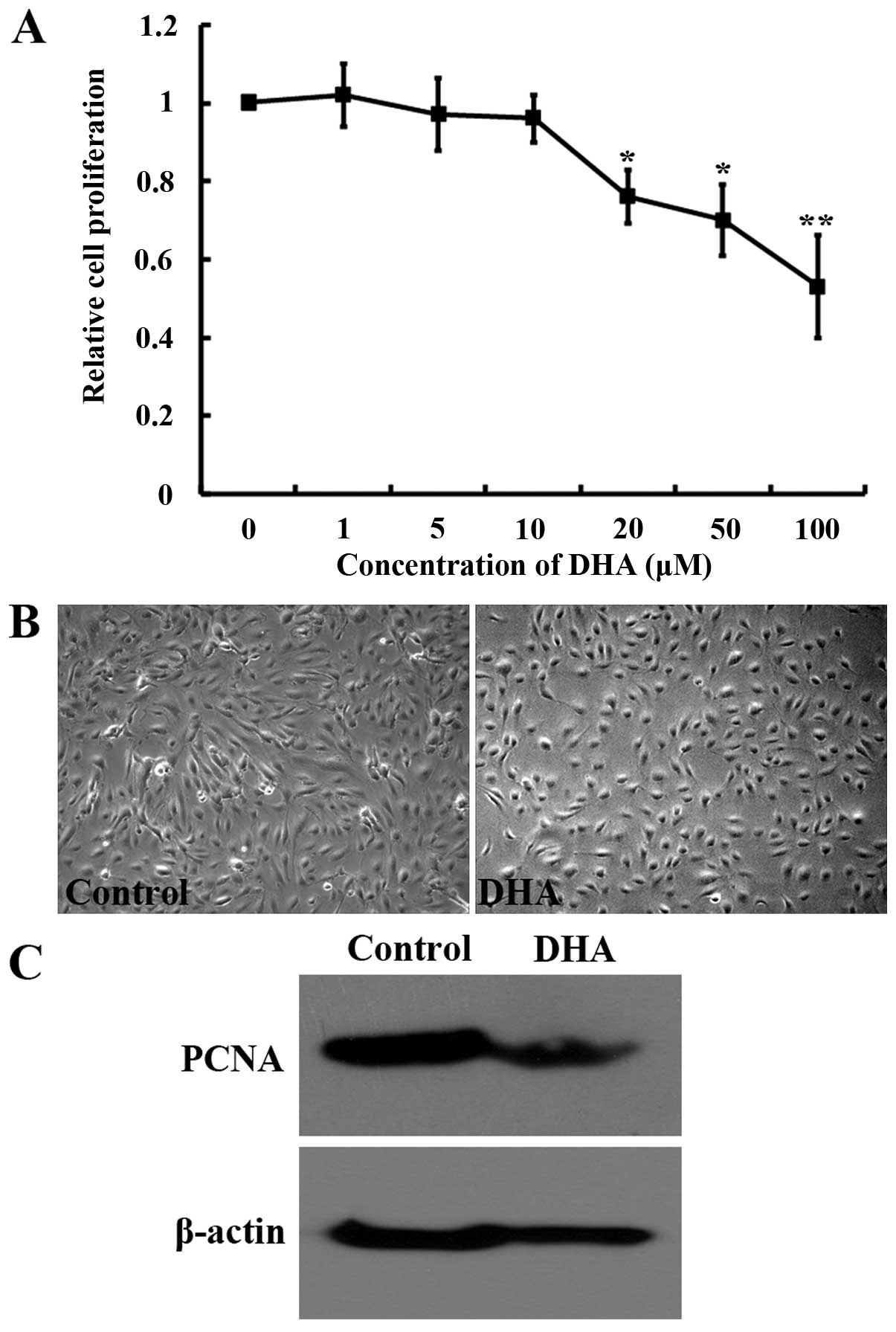
Endothelial cell proliferation is an essential
process for angiogenesis (6). To
examine the effects of DHA on endothelial cell proliferation, the
MTT assay was performed for HUVECs treated with different
concentrations of DHA. DHA significantly reduced HUVEC growth at a
concentration of ≥20 μM after 24 h incubation (P<0.05)
(Fig. 1A). Treatment with 20
μM DHA induced a marked reduction of cell density (Fig. 1B) and the expression of PCNA, an
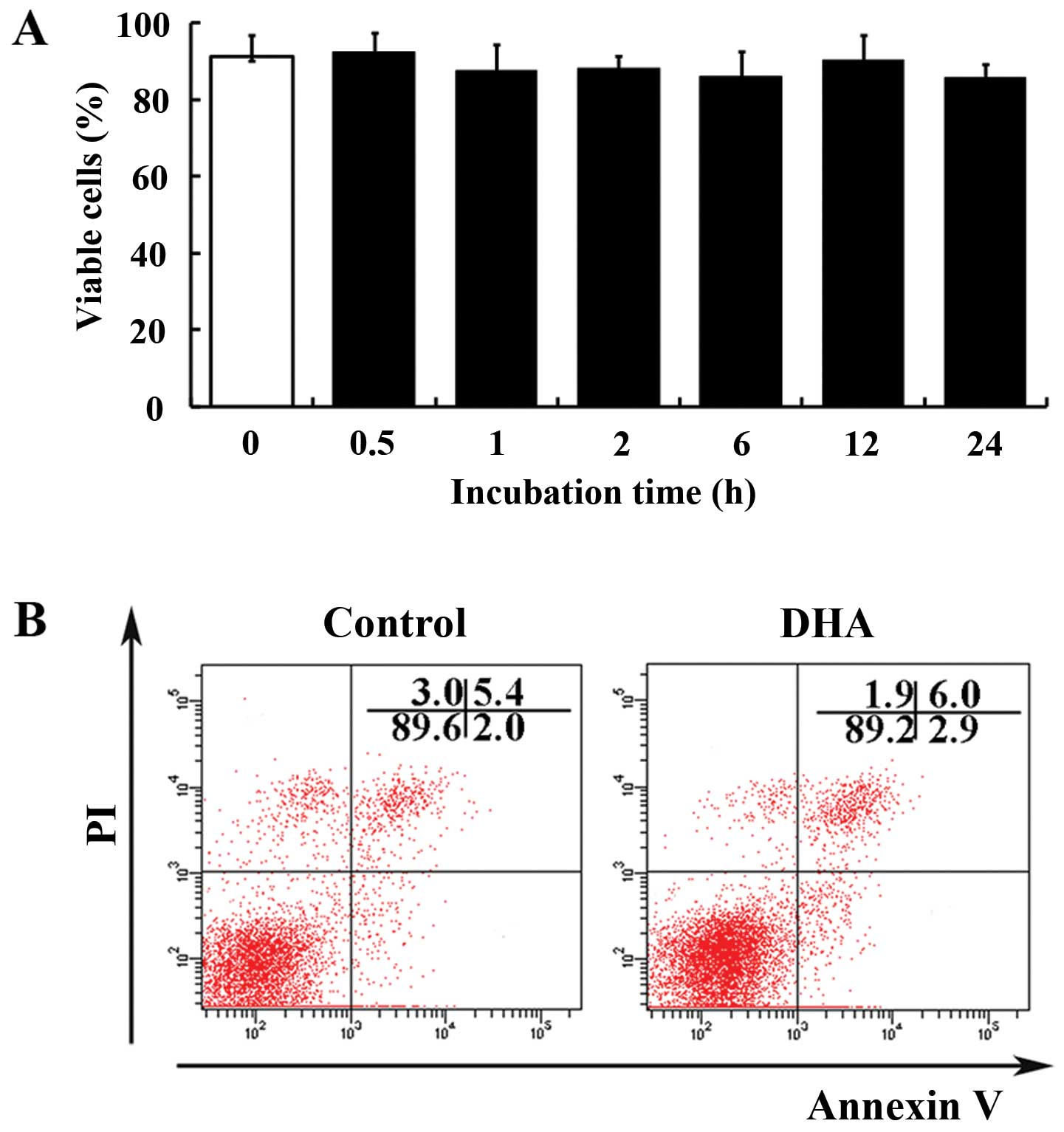
essential factor for DNA replication and repair (Fig. 1C). The effects of DHA on cell
viability were evaluated by the trypan blue exclusion assay. At 20
μM, DHA did not significantly affect the percentage of
viable cells <24 h (Fig. 2A).
Subsequently, the apoptosis of HUVECs treated with DHA was examined
by double-staining of Annexin V and PI. Flow cytometry analyses
showed that the percentage of Annexin V/PI positive cells and
viable cells remain unchanged in the absence and presence of 20
μM DHA (5.4 vs. 6.0%; 89.6 vs. 89.2%) (Fig. 2B). Therefore, at a concentration
of 20 μM, DHA inhibits proliferation without induction of
apoptosis. For the following experiments, the concentration of 20
μM was used to delineate the mechanisms of
anti-proliferative effects of DHA.
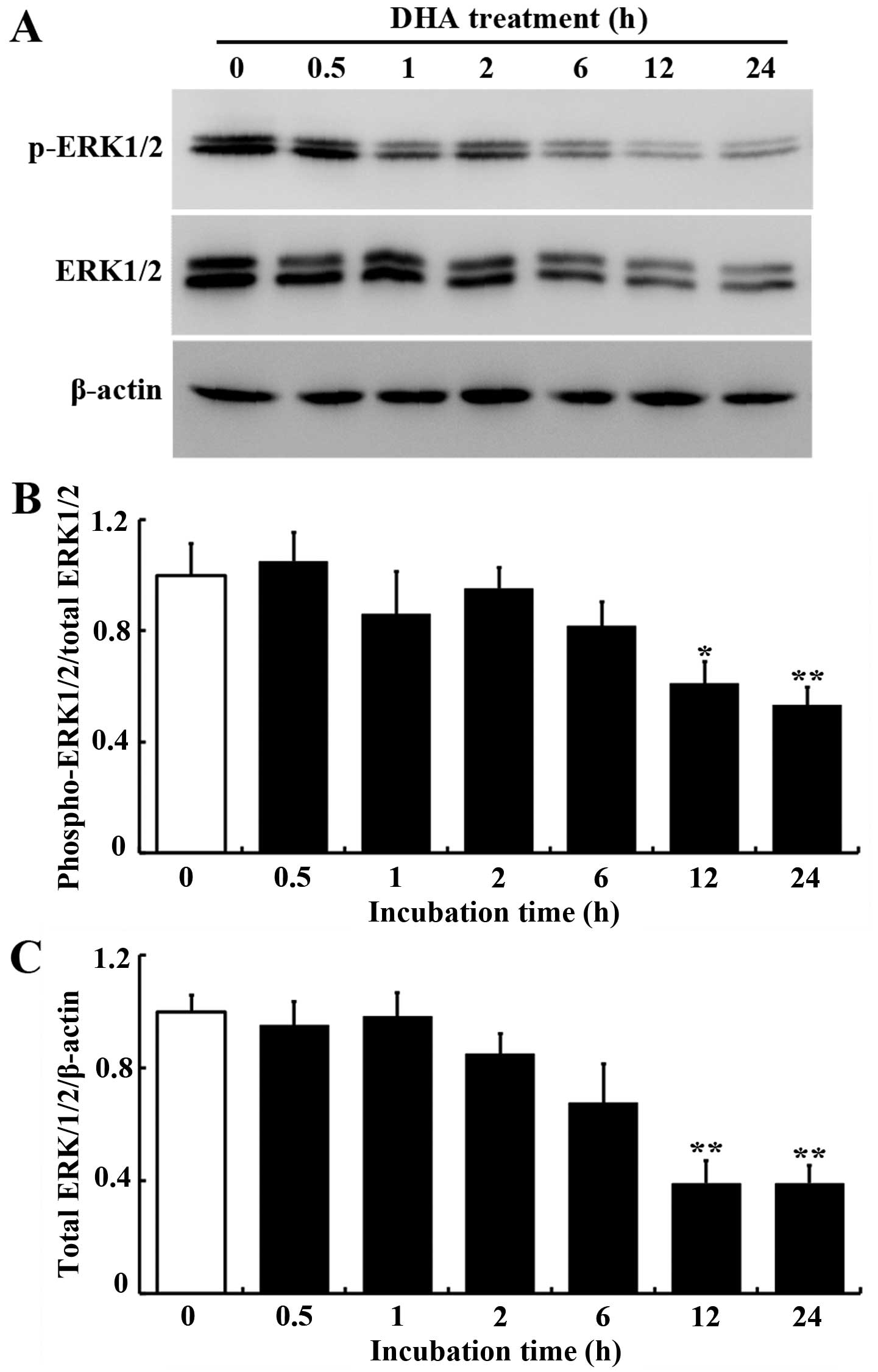
DHA inhibits ERK1/2 expression and
phosphorylation in HUVECs
The ERK pathway mediates cellular responses to
growth factors and promotes endothelial cell proliferation
(21). The effects of DHA on ERK
signaling in endothelial cells were examined by western blot
analysis. The protein levels of total and phospho-ERK1/2 were
significantly decreased in HUVECs treated with 20 μM DHA
(Fig. 3A). Densitometry analysis
showed that the ratios of total ERK1/2/β-actin and
phospho-ERK1/2/total-ERK1/2 were significantly reduced after 12 h
incubation with DHA (P<0.05, Fig.
3B; P<0.01, Fig. 3C),
suggesting that ERK1/2 protein expression and phosphorylation were
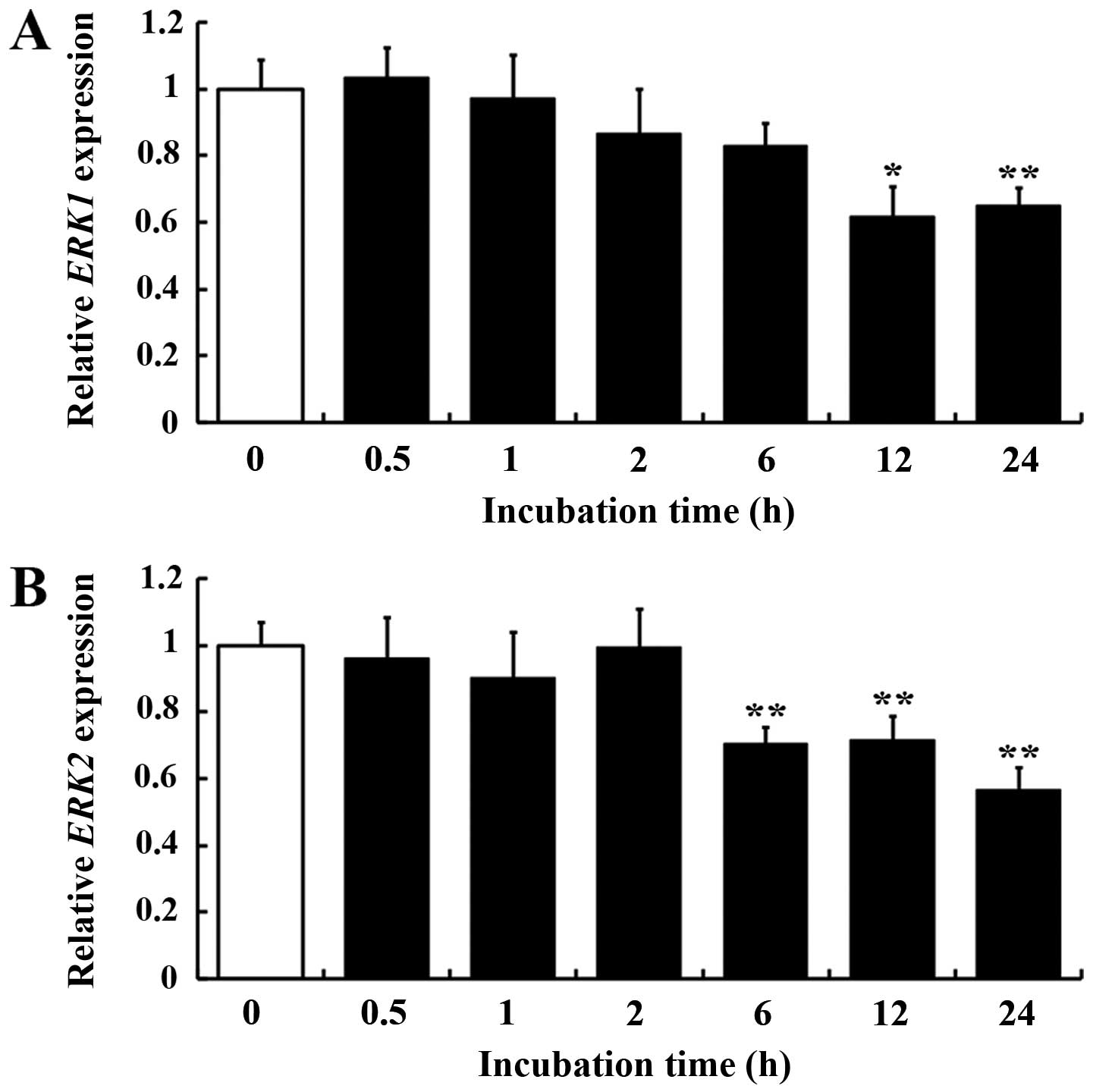
inhibited by DHA. Subsequently, the mRNA expression of ERK1/2 was
examined in HUVECs with DHA treatment. ERK1 (MAPK3)
expression was downregulated at 12 h incubation with DHA
(P<0.01) (Fig. 4A), while ERK2
(MAPK1) expression was downregulated as early as 6 h
incubation (P<0.05) (Fig. 4B).
These data indicated that DHA inhibits the ERK signaling pathway in
endothelial cells by suppression of ERK1/2 transcription and
phosphorylation.
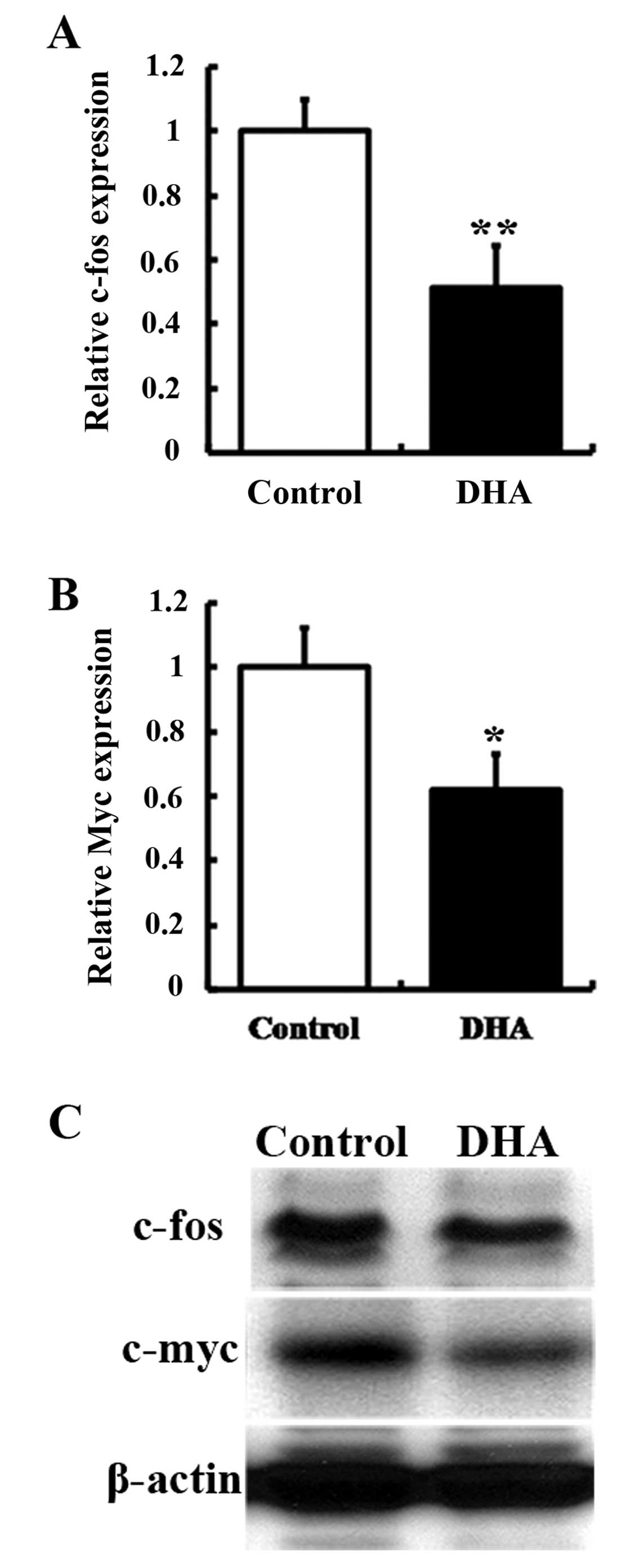
DHA downregulates downstream effectors of
ERK1/2 in HUVECs
ERK signaling regulates transcription factors, such
as c-Fos and c-Myc, which participate in the regulation of cell
proliferation (22). In addition,
c-fos and c-myc are immediate early genes that are
expressed rapidly in response to extracellular stimuli (23). The effects of DHA were examined on
c-fos and c-myc expression in HUVECs by RT-qPCR and
western blot analysis. The mRNA levels of c-fos and
c-myc were significantly decreased in HUVECs treated with 20
μM DHA (P<0.01, Fig.
5A; P<0.05, Fig. 5B). The
levels of c-Fos and c-Myc protein were also reduced by DHA
treatment (Fig. 5C). Therefore,
the downstream effectors of ERK1/2 were downregulated by DHA.
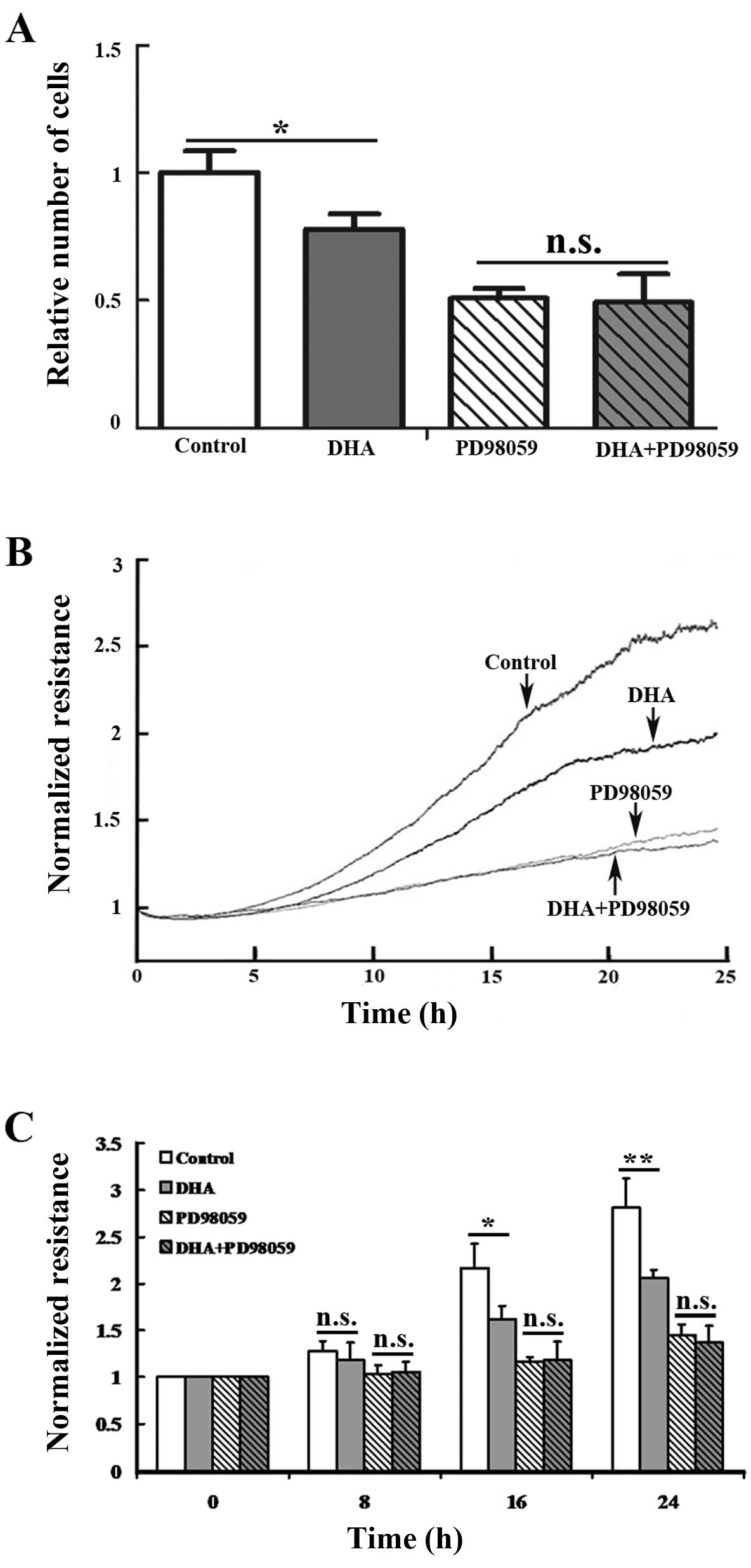
Inhibitor of the ERK signaling
compromises DHA-induced reduction of endothelial cell
proliferation
PD98059 is a non-adenosine triphosphate competitive
inhibitor that does not directly bind to ERK1 or ERK2, but
specifically binds to the inactive forms of MEK1/2 and blocks the
phosphorylation by upstream activators, such as Raf (24). As MEK1/2 are the only known direct
activators of ERK1/2, PD98059 substantially inhibits ERK signaling.
To validate the role of the ERK signaling in mediating the
anti-proliferative effects of DHA, HUVECs were pretreated with 10
μM PD98059 prior to the addition of DHA and after 24 h cell
proliferation was examined by the MTT assay. PD98059 alone and the
combination of PD98059 and DHA induced a similar reduction of the
cell number (P=0.29) (Fig. 6A).
In addition, real-time proliferation assays were performed using
the ECIS system. In this setting, electrical resistance was
measured over a monolayer of endothelial cells on electrodes, which
is proportional to the cell density in the culture plates (25). Consistent with the results from
the MTT assay, DHA or PD98059 alone induced a significant reduction
in electrical resistance after 16 h of incubation (Fig. 6B). With the pretreatment of
PD98059, DHA did not significantly induce additional reduction of
electrical resistance <24 h (P=0.34 at 16 h; P=0.13 at 24 h;
Fig. 6B and C). Therefore, the
anti-proliferative effects of DHA were compromised by PD98059,
suggesting that endothelial cell proliferation is suppressed by DHA
through the inhibition of the ERK signaling pathway.
Discussion
Tumor angiogenesis is closely associated with tumor
aggressiveness, metastasis and patient survival (1). Therefore, disrupting angiogenesis
serves as an important strategy for cancer therapy (26). DHA, a more water-soluble
metabolite of artemisinin derivatives, has exhibited potent
anti-angiogenic activities (11).
Since DHA has been widely used as an anti-malarial drug and proved
to be safe with minimal side effects, it could be used clinically
as a component of cancer chemotherapy (11). In the present study, the molecular
mechanisms underlying the anti-angiogenic activity of DHA were
explored. The results suggested that the ERK signaling pathway
mediates the anti-proliferative effects of DHA on endothelial
cells.
A dose-responsive inhibition was demonstrated on
HUVEC proliferation by DHA. This is similar to the results from
previous studies using other artemisinin derivatives (14,27). A low concentration (20 μM)
of DHA was further defined to significantly inhibit cell
proliferation, but does not induce cell death <24 h. Thus, the
primary effect of DHA at this concentration is restricted to
inhibition of endothelial cell proliferation. In the time course
study, 20 μM DHA induced a significant reduction of ERK1/2
phosphorylation and expression. It has been reported that DHA
prevented phosphorylation of ERK1/2 and other components of MAPK
cascades in specific cell types (18,19). As the activation of the Ras-ERK
cascade leading to endothelial cell proliferation and survival
during angiogenesis (28), DHA
may inhibit this cascade to reduce cell proliferation. In addition,
DHA also decreases total ERK1/2 and suppresses the mRNA expression
of ERK1/2. Although ERK1 and ERK2 proteins share a similar
sequence and function, they are encoded by distinct genes located
at Chr.22q11.2 and Chr.16p11.2, respectively (29–31). The gene expression of ERK1
(MAPK3) and ERK2 (MAPK1) are likely to be
differentially regulated. DHA alters the expression of a variety of
genes in the vascular system. In particular, DHA downregulates the
transcription of key components of the VEGF/VEGFR2 axis, which
promotes angiogenesis (32,33). In addition to inhibiting the
activation of the ERK signaling, the present data suggest that
downregulation of the ERK1/2 gene expression may also contribute to
the anti-angiogenesis effects of DHA.
The proto-oncogenes c-fos and c-myc
are downstream effectors of ERK signaling (22). The induction of the c-fos
and c-myc gene expression is a common response of cells to
stimulation of growth factors, including VEGF and bFGF (34,35). In the present study, DHA decreased
the mRNA and protein expression of c-Fos and c-Myc. In tumor cells
that express high levels of c-myc, DHA induces apoptosis and
permanently reduces the protein level of c-Myc in these cells
(36). In addition, DHA
accelerates degradation of c-Myc protein in tumor cells, and forced
expression of c-Myc sensitizes these cells to DHA-induced apoptosis
(36). The effects of artemisinin
and its derivatives on c-Fos have not been studied previously. The
present data suggest that DHA suppresses responsive gene expression
induced by growth factors, and this suppressive effect may result
from the inhibition of ERK signaling.
The ECIS system monitors transendothelial cell
electronic resistance and provided an accurate method to examine
real-time cell proliferation (25). In this system, application of DHA
to the MEK1/2 inhibitor PD98059 pre-treated HUVECs did not induce
an additional decrease of cell proliferation. Thus, there was no
synergistic or additive effect of DHA and PD98059 in the
suppression of endothelial cell proliferation. Activation of the
ERK signaling pathway increases cell growth, and numerous
extracellular stimuli regulates endothelial cell proliferation
through manipulation of this pathway (9). Pre-treatment with PD98059 reduces
endothelial cell proliferation induced by adiponectin, ghrelin and
adropin (37–39). The present results confirmed the
role of ERK signaling in mediating DHA-induced inhibition of
endothelial cell proliferation.
In conclusion, the present study demonstrated that
DHA suppresses endothelial cell proliferation through inhibition of
the ERK signaling pathway, and these findings provided important
information for understanding the molecular mechanisms of the
anti-angiogenic effects of the artemisinin family of drugs.
Acknowledgments
The present study was supported by grants from the
Science and Technology Development Plan of Shandong Province (no.
2013GSF11805), the Science and Technology Plan of Jinan City (no.
201303026) and the Shandong Taishan Scholarship (J.L. and S.Y.).
The authors are grateful to Dr Thaddeus Allen at the University of
California (San Francisco, USA) for the helpful discussions and
critical reading of the manuscript.
References
|
1
|
Folkman J: Angiogenesis in cancer,
vascular, rheumatoid and other disease. Nat Med. 1:27–31. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ferrara N, Gerber HP and LeCouter J: The
biology of VEGF and its receptors. Nat Med. 9:669–676. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Carmeliet P and Jain RK: Angiogenesis in
cancer and other diseases. Nature. 407:249–257. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Costa C, Soares R and Schmitt F:
Angiogenesis: now and then. APMIS. 112:402–412. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Klagsbrun M and Moses MA: Molecular
angiogenesis. Chem Biol. 6:R217–R224. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Risau W: Mechanisms of angiogenesis.
Nature. 386:671–674. 1997. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Liu J and Kapron CM: Differential
induction of MAP kinase signalling pathways by cadmium in primary
cultures of mouse embryo limb bud cells. Reprod Toxicol.
29:286–291. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Page C and Doubell AF: Mitogen-activated
protein kinase (MAPK) in cardiac tissues. Mol Cell Biochem.
157:49–57. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hoefen RJ and Berk BC: The role of MAP
kinases in endothelial activation. Vascul Pharmacol. 38:271–273.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tu Y: The development of new antimalarial
drugs: qinghaosu and dihydro-qinghaosu. Chin Med J (Engl).
112:976–977. 1999.
|
|
11
|
Crespo-Ortiz MP and Wei MQ: Antitumor
activity of artemisinin and its derivatives: from a well-known
antimalarial agent to a potential anticancer drug. J Biomed
Biotechnol. 2012:2475972012. View Article : Google Scholar
|
|
12
|
Chen HH, Zhou HJ, Wang WQ and Wu GD:
Antimalarial dihydroartemisinin also inhibits angiogenesis. Cancer
Chemother Pharmacol. 53:423–432. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Longo M, Zanoncelli S, Torre PD, et al: In
vivo and in vitro investigations of the effects of the antimalarial
drug dihydroartemisinin (DHA) on rat embryos. Reprod Toxicol.
22:797–810. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chen HH, Zhou HJ and Fang X: Inhibition of
human cancer cell line growth and human umbilical vein endothelial
cell angiogenesis by artemisinin derivatives in vitro. Pharmacol
Res. 48:231–236. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wu GD, Zhou HJ and Wu XH: Apoptosis of
human umbilical vein endothelial cells induced by artesunate.
Vascul Pharmacol. 41:205–212. 2004. View Article : Google Scholar
|
|
16
|
D’Alessandro S, Basilico N, Corbett Y, et
al: Hypoxia modulates the effect of dihydroartemisinin on
endothelial cells. Biochem Pharmacol. 82:476–484. 2011. View Article : Google Scholar
|
|
17
|
Sarina, Yagi Y, Nakano O, et al: Induction
of neurite outgrowth in PC12 cells by artemisinin through
activation of ERK and p38 MAPK signaling pathways. Brain Res.
1490:61–71. 2013. View Article : Google Scholar
|
|
18
|
Wang Y, Huang ZQ, Wang CQ, et al:
Artemisinin inhibits extracellular matrix metalloproteinase inducer
(EMMPRIN) and matrix metalloproteinase-9 expression via a protein
kinase Cδ/p38/extracellular signal-regulated kinase pathway in
phorbol myristate acetate-induced THP-1 macrophages. Clin Exp
Pharmacol Physiol. 38:11–18. 2011. View Article : Google Scholar
|
|
19
|
Lu JJ, Meng LH, Cai YJ, et al:
Dihydroartemisinin induces apoptosis in HL-60 leukemia cells
dependent of iron and p38 mitogen-activated protein kinase
activation but independent of reactive oxygen species. Cancer Biol
Ther. 7:1017–1023. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Guo L, Dong FY, Hou YL, et al:
Dihydroartemisinin inhibits VEGF-induced endothelial cell migration
by a p38 MAPK-independent pathway. Exp Ther Med. 8:1707–1712.
2014.PubMed/NCBI
|
|
21
|
Carmeliet P and Jain RK: Molecular
mechanisms and clinical applications of angiogenesis. Nature.
473:298–307. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Davis RJ: Transcriptional regulation by
MAP kinases. Mol Reprod Dev. 42:459–467. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Monick MM, Geist LJ, Stinski MF and
Hunninghake GW: The immediate early genes of human cytomegalovirus
upregulate expression of the cellular genes myc and fos. Am J
Respir Cell Mol Biol. 7:251–256. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dudley DT, Pang L, Decker SJ, Bridges AJ
and Saltiel AR: A synthetic inhibitor of the mitogen-activated
protein kinase cascade. Proc Natl Acad Sci USA. 92:7686–7689. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hong J, Kandasamy K, Marimuthu M, Choi CS
and Kim S: Electrical cell-substrate impedance sensing as a
non-invasive tool for cancer cell study. Analyst. 136:237–245.
2011. View Article : Google Scholar
|
|
26
|
Ferrara N and Kerbel RS: Angiogenesis as a
therapeutic target. Nature. 438:967–974. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Oh S, Jeong IH, Shin WS and Lee S: Growth
inhibition activity of thioacetal artemisinin derivatives against
human umbilical vein endothelial cells. Bioorg Med Chem Lett.
13:3665–3668. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Feng D, Nagy JA, Pyne K, Hammel I, Dvorak
HF and Dvorak AM: Pathways of macromolecular extravasation across
microvascular endothelium in response to VPF/VEGF and other
vasoactive mediators. Microcirculation. 6:23–44. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Boulton TG and Cobb MH: Identification of
multiple extracellular signal-regulated kinases (ERKs) with
antipeptide antibodies. Cell Regul. 2:357–371. 1991.PubMed/NCBI
|
|
30
|
García F, Zalba G, Páez G, Encío I and de
Miguel C: Molecular cloning and characterization of the human p44
mitogen-activated protein kinase gene. Genomics. 50:69–78. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Owaki H, Makar R, Boulton TG, Cobb MH and
Geppert TD: Extracellular signal-regulated kinases in T cells:
characterization of human ERK1 and ERK2 cDNAs. Biochem Biophys Res
Commun. 182:1416–1422. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
He Y, Fan J, Lin H, et al: The
anti-malaria agent artesunate inhibits expression of vascular
endothelial growth factor and hypoxia-inducible factor-1α in human
rheumatoid arthritis fibroblast-like synoviocyte. Rheumatol Int.
31:53–60. 2011. View Article : Google Scholar
|
|
33
|
Lee J, Zhou HJ and Wu XH:
Dihydroartemisinin downregulates vascular endothelial growth factor
expression and induces apoptosis in chronic myeloid leukemia K562
cells. Cancer Chemother Pharmacol. 57:213–220. 2006. View Article : Google Scholar
|
|
34
|
Yin D, Jia T, Gong W, et al: VEGF blockade
decelerates the growth of a murine experimental osteosarcoma. Int J
Oncol. 33:253–259. 2008.PubMed/NCBI
|
|
35
|
Kosaka C, Sasaguri T, Komiyama Y and
Takahashi H: All-trans retinoic acid inhibits vascular smooth
muscle cell proliferation targeting multiple genes for cyclins and
cyclin-dependent kinases. Hypertens Res. 24:579–588. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lu JJ, Meng LH, Shankavaram UT, et al:
Dihydroartemisinin accelerates c-MYC oncoprotein degradation and
induces apoptosis in c-MYC-overexpressing tumor cells. Biochem
Pharmacol. 80:22–30. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lovren F, Pan Y, Quan A, et al: Adropin is
a novel regulator of endothelial function. Circulation. 122(Suppl
11): S185–S192. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Alvarez G, Visitación Bartolomé M, Miana
M, et al: The effects of adiponectin and leptin on human
endothelial cell proliferation: a live-cell study. J Vasc Res.
49:111–122. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wang L, Chen Q, Li G and Ke D: Ghrelin
stimulates angiogenesis via GHSR1a-dependent MEK/ERK and PI3K/Akt
signal pathways in rat cardiac microvascular endothelial cells.
Peptides. 33:92–100. 2012. View Article : Google Scholar
|