Introduction
Doxorubicin is a chemotherapeutic agent widely used
in the treatment of various types cancers, such as lung, liver,
breast, ovarian and bladder cancer (1–6).
However, cardiotoxicity is a major side-effect of doxorubicin.
Reactive oxygen species (ROS) have been reported as one of the
major factors responsible for cardiotoxicity. Doxorubicin is
reduced by nicotinamide adenine dinucleotide phosphate-oxidase
(NADPH oxidase) to a semi-quinone free radical, which in turn
interacts with oxygen to form superoxide, hydroxyl and
peroxynitrite free radicals (7).
Doxorubicin has been reported to induce oxidative stress and
mitochondria-mediated apoptosis in cardiomyocytes, leading to their
loss and ultimately contributing to progressive heart failure
(8). The administration of
antioxidants has been shown to protect cardiac cells from oxidative
stress induced by doxorubicin (9–11).
Sulforaphane is a naturally occurring isothiocyanate
that is highly abundant in certain cruciferous vegetables (12). L-sulforaphane is the biologically
active isomer, whereas D,L-sulforaphane is a synthetic racemic
analogue of the broccoli constituent, L-sulforaphane (13). Sulforaphane is an antioxidant with
cytoprotective effects and anti-carcinogenic properties (14,15) that has been shown to induce the
activity of phase II enzymes, such as heme oxygenase-1 (HO-1),
quinone reductase, glutathione S-transferase and glutathione
reductase (16). Sulforaphane has
also been reported to protect the kidneys (17) and brain (18) against ischemic injury through the
induction of the activation of transcription factor, NF-E2-related
factor-2 (Nrf2)-dependent phase II enzymes. Under basal conditions,
the cytosolic regulatory protein, Kelch-like ECH-associated protein
1 (Keap1), binds tightly to Nrf2, retaining it in the cytoplasm
(19). Phase II enzyme inducers
can disrupt the Keap1/Nrf2 complex, resulting in the release of
Nrf2 and its subsequent translocation to the nucleus. The induction
of the activation of antioxidant enzymes has been reported to
involve transcriptional activation through antioxidant-responsive
elements (AREs) (19).
Among the phase II enzymes, HO-1 has attracted
significant attention due to its therapeutic effects against
neurodegenerative, cardiovascular and hepatic diseases (20–22). Under conditions of oxidative
stress, the induction of HO-1 accounts for the majority of heme
breakdown, leading to the formation of biliverdin, carbon monoxide
(CO) and ferrous iron. In this way, HO-1 mitigates cellular injury
by producing molecules with antioxidant and anti-apoptotic effects
(20).
The potential cardioprotective effects of
sulforaphane have been confirmed by observing reduced ROS
production, an increased cell viability (23), and the attenuation of ischemic
heart injury through mitochondrial KATP channels and
antioxidant pathways (24).
However, the effects of sulforaphane on the cardiotoxicity induced
by doxorubicin have not been well defined to date. Therefore, the
aim of this study was to determine whether sulforaphane protects
cells against doxorubicin-induced cardiac cell death. Specifically,
we focused on whether the cardioprotective effects of sulforaphane
are related to the activation of the Keap1/Nrf2/ARE pathway and the
subsequent induction of HO-1.
Materials and methods
Reagents
Antibodies against cleaved caspase-3 (#9661) and Bax
(#2772) were purchased from Cell Signaling Technologies (Beverly,
MA, USA). Antibodies against cytochrome c (sc-13156), Bcl-2
(sc-7382), HO-1 (sc-10789), histone H3 (sc-8654), Nrf2 (sc-722),
Keap1 (sc-365626), Hsp60 (sc-13966) and glyceraldehyde 3-phosphate
dehydrogenase (GAPDH) (sc-25778) were purchased from Santa Cruz
Biotechnology, Inc. (Santa Cruz, CA, USA). Dulbecco’s modified
Eagle’s medium (DMEM), fetal bovine serum (FBS), trypsin and other
tissue culture reagents were obtained from Invitrogen Life
Technologies, Inc. (Carlsbad, CA, USA). Hoechst 33258,
5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine
iodide (JC-1), dihydrorhodamine 123 (DHR123) and MitoSOX Red
reagent were purchased from Molecular Probes (Eugene, OR, USA).
Doxorubicin, and L-sulforaphane and D,L-sulforaphane were purchased
from Sigma-Aldrich (St. Louis, MO, USA). All other chemicals were
from Sigma-Aldrich.
Cell culture
The H9c2 rat myoblast cell line (KCLB #21446, Korean
Cell Line Bank, Seoul, Korea) was grown in DMEM supplemented with
10% FBS and antibiotics (100 µg/ml streptomycin/100 U/ml
penicillin mix) in a humidified atmosphere at 37°C with 5%
CO2.
Determination of cell viability
The H9c2 cells were passaged and cultured in 24-well
plates at 5×104 cells/well for 1 day. The cells were
then treated with L-sulforaphane or D,L-sulforaphane for 2 h prior
to the addition of doxorubicin. After 24 h, the cells were assessed
by trypan blue exclusion assay using a light microscope to
determine the percentage of cell death, as previously described
(25). The viable cell ratio was
calculated as follows: viable cell ratio (%) = (unstained cell
number/total cell number) ×100.
Morphological detection of apoptotic
cells
Hoechst 33258, a fluorescent stain used for labeling
DNA, was used to identify the apoptotic H9c2 cells, as described in
a previous study (26). Following
treatment, the cells were washed 3 times with phosphate-buffered
saline (PBS) and then fixed for 5 min with ice-cold 100% methanol.
After fixation, the cells were stained with Hoechst 33258 (10
µg/ml) for 10 min at room temperature in the dark. The cells
were then washed with PBS 3 times and examined under a fluorescence
microscope (Olympus, Tokyo, Japan). The apoptotic cells were
identified by characteristic nuclei condensation, fragmentation and
bright staining, while the nuclei from normal cells demonstrated a
normal uniform chromatin pattern.
Western blot analysis
Western blot analysis was performed as previously
described with some modifications (27). The cells treated for the indicated
periods of time were harvested by washing twice with ice-cold PBS
on ice. For the preparation of whole-cell lysates, the cells were
lysed on ice by the addition of RIPA lysis buffer (25 mM Tris-HCl
pH 7.4, 150 mM NaCl, 1% sodium deoxycholate, 1% Triton X-100, 5 mM
EDTA, 0.1% SDS) plus protease inhibitor cocktail and phosphatase
inhibitor cocktail (Roche Diagnostics, Mannheim, Germany) directly
onto the cells. The cell lysates were incubated for 30 min on ice,
after which they were centrifuged at 10,000 × g for 20 min at 4°C.
The supernatants were then collected and used as protein extracts.
Protein extracts were added to sample buffer, boiled in a water
bath for 5 min and stored at −80°C until use. Protein extracts (30
µg) were run on polyacrylamide gels and transferred onto
PVDF membranes for 40 min at 15 V using a semi-dry transfer system
(Bio-Rad, Hercules, CA, USA). The membranes were blocked with 5%
non-fat dry milk in 0.05% Tween-20/Tris-buffered saline (T-TBS) for
60 min at room temperature. The blots were then probed overnight at
4°C with the relevant primary antibodies, washed and probed again
with species-specific secondary antibodies coupled to horseradish
peroxidase (Santa Cruz Biotechnology, Inc.). Chemiluminescence
reagents (GE Healthcare, Piscataway, NJ, USA) were added for blot
detection. Immunoreactive bands were visualized using a LAS-3000
imaging system (Fujifilm, Tokyo, Japan). Band intensities were
measured and quantified using ImageJ software as described by Luke
Miller (http://lukemiller.org/journal/2007/08/quantifying-western-blots-without.html).
Detection of cytochrome c and Bax by
western blot analysis
To determine the subcellular localization of
cytochrome c and Bax, the cells were fractionated using
digitonin, as previously described (28). Briefly, the cells were suspended
in ice-cold plasma membrane permeabilization buffer (200
µg/ml digitonin, 80 mM KCl in PBS). Following a 5-min
incubation on ice, the cells were centrifuged at 800 × g for 5 min
at 4°C and the soluble extract was taken as the cytosolic fraction.
The insoluble pellet was further resuspended in ice-cold cell lysis
buffer and incubated on ice for 10 min. Following centrifugation at
10,000 × g for 10 min at 4°C, the resulting supernatant was taken
as the membrane-bound organellar fraction enriched with
mitochondria. These two fractions were analyzed by western blot
analysis using antibodies specific for cytochrome c, Bax,
Hsp60 and GAPDH.
Immunostaining for cytochrome c and
Bax
Protocols for the immunofluorescence staining for
cytochrome c and Bax were used as previously described with
some modifications (29). The
H9c2 cells were grown on coverglass-bottom dishes and treated with
the indicated agents. The cells were then fixed with ice-cold
methanol and permeabilized with PBST (PBS containing 0.25% Triton
X-100). Following a 30-min incubation in blocking buffer (1% BSA in
PBST), the cells were incubated with rabbit anti-Bax antibody
(1:300) overnight at 4°C. Subsequently, the cells were washed twice
and stained with FITC-conjugated goat anti-rabbit secondary
antibody (1:300; A24532; Thermo Fisher Scientific, Rockford, IL,
USA) for 1 h. The cells were then incubated with mouse
anti-cytochrome c antibody (1:300) for 1 h and then stained
with TRITC-conjugated goat anti-mouse secondary antibody (1:600;
ab6786; Abcam, Cambridge, UK) for 1 h. Finally, the cells were
mounted using Vectashield mounting medium containing DAPI, and
signals were examined under a fluorescence microscope using FITC,
TRITC and DAPI channels.
JC-1 mitochondrial membrane potential
(ΔΨm) assay
ΔΨm was determined by flow cytometry using the
J-aggregate forming lipophilic cationic probe, JC-1, according to
the manufacturer’s instructions (Molecular Probes). JC-1 stains the
mitochondria in cells with a high ΔΨm by forming red fluorescence
J-aggregates (30), whereas in
cells with depolarized mitochondria, JC-1 is present as a green
fluorescent monomer. In this way, mitochondrial depolarization can
be determined by a decreased ratio of red-to-green fluorescence
intensity. The cells were grown in glass-bottom dishes (SPL Life
Sciences Co., Ltd., Pochoen, Korea). Following treatment, JC-1 was
dissolved in dimethyl sulfoxide (1 mg/ml), diluted to a final
concentration of 1 µg/ml in serum-free medium and then added
to the cells followed by incubation for 10 min at 37°C; the cells
were then washed twice with PBS. Subsequently, the cells were
incubated in 1 ml of culture medium and analyzed under a
fluorescence microscope (Olympus).
DHR123
DHR123 is a cell-permeable fluorogenic probe and an
indicator of peroxynitrite levels (31,32). Specifically, DHR123 is oxidized by
peroxynitrite to cationic rhodamine 123, which localizes in the
mitochondria and exhibits green fluorescence (33). Neither nitric oxide, superoxide,
nor hydrogen peroxide alone appear to oxidize DHR (34). In order to determine the level of
mitochondrial peroxynitrite by fluorescence microscopy, the cells
were grown in glass-bottom dishes (SPL Life Sciences Co., Ltd.).
Following treatment, DHR123 was dissolved in dimethyl sulfoxide (5
mM) and diluted to 1.25 µM in serum-free medium. The cells
were treated with DHR123 for 30 min at 37°C and then washed twice
with PBS. Subsequently, the cells were incubated in 1 ml of culture
medium and analyzed under a fluorescence microscope (Olympus).
MitoSOX
MitoSOX Red is a novel fluorogenic dye that serves
as an indicator of mitochondrial superoxide levels in live cells
(35,36). MitoSOX Red reagent is live-cell
permeant and is rapidly and selectively targeted to the
mitochondria. Once in the mitochondria, MitoSOX Red reagent is
oxidized by superoxide and emits red fluorescence. In order to
determine the levels of mitochondrial superoxide by fluorescence
microscopy, the cells were grown in glass-bottom dishes (SPL Life
Sciences Co., Ltd.). Following treatment, MitoSOX reagent was
dissolved in dimethyl sulfoxide (5 mM), diluted to 5 µM in
serum-free medium, and was then added to the cells followed by
incubation for 10 min at 37°C; the cells were then washed twice
with PBS. Subsequently, the cells were incubated in 1 ml of culture
medium and analyzed under a fluorescence microscope (Olympus).
Detection of Nrf2 and Keap1 by western
blot analysis
Nuclear and cytoplasmic extracts of H9c2 cells were
prepared using NE-PER nuclear and cytoplasmic extraction reagent
(Pierce Biotechnology, Rockford, IL, USA) as recommended by the
manufacturer. The two fractions were analyzed by western blot
analysis with antibodies specific to Nrf2 and Keap1.
Immunostaining for Nrf2 and Keap1
The H9c2 cells were grown on coverglass-bottom
dishes and treated with the indicated agents. The cells were then
fixed with ice-cold methanol and permeabilized with PBST (PBS
containing 0.25% Triton X-100). Following incubation for 30 min in
blocking buffer (1% BSA in PBST), the cells were incubated with
rabbit anti-Nrf2 antibody (1:400) overnight at 4°C. The cells were
then washed twice and stained with FITC-conjugated goat anti-rabbit
secondary antibody (1:400) for 1 h. Subsequently, the cells were
incubated with mouse anti-Keap1 antibody (1:100) for 1 h and then
stained with TRITC-conjugated goat anti-mouse secondary antibody
(1:200) for 1 h. Finally, the cells were mounted using Vectashield
mounting medium with DAPI. Signals were examined by fluorescence
microscopy using the FITC, TRITC and DAPI channels.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from the H9c2 cells using
TRIzol reagent (Invitrogen Life Technologies). The
ethanol-precipitated RNA fraction (500 ng) was reverse transcribed
using the PrimeScript™ RT reagent kit (RR037A; Takara, Shiga,
Japan) according to the manufacturer’s protocol. The primers used
for PCR were as follows: 5′-AGAGTTTCCGCCTCCAACCA-3′ and
5′-CGGGACTGGGCTAGTTCAGG-3′ for rat HO-1 and
5′-CAGTCAAGGCTGAGAATGG-3′ and 5′-CGACATACT CAGCACCAGC-3′ for rat
GAPDH, as described in a previous study (37). Relative gene expression was
determined by quantitative (real-time) PCR (qPCR) using an Applied
Biosystems 7900HT Fast Real-Time PCR system (Applied Biosystems,
Foster City, CA, USA) according to the instructions provided by the
manufacturer.
ARE luciferase activity assay
The cells were transfected with an ARE promoter
luciferase reporter plasmid for 24 h and then treated with
doxorubicin in the absence or presence of L-sulforaphane or
D,L-sulforaphane for various periods of time. Cell lysates were
prepared and assayed for luciferase activity using the Luciferase
Assay System (Promega, Madison, WI, USA) according to the
manufacturer’s instructions. Changes in luciferase activity with
respect to the controls (untreated cells) were then calculated.
Statistical analysis
All data are presented as the means ± SEM. A
two-tailed Student’s t-test was applied to examine the statistical
significance of differences between groups. Origin 8.0 software
(OriginLab Corp., Northampton, MA, USA) was used for statistical
calculations. Values of P<0.05 were considered to indicate
statistically significant differences.
Results
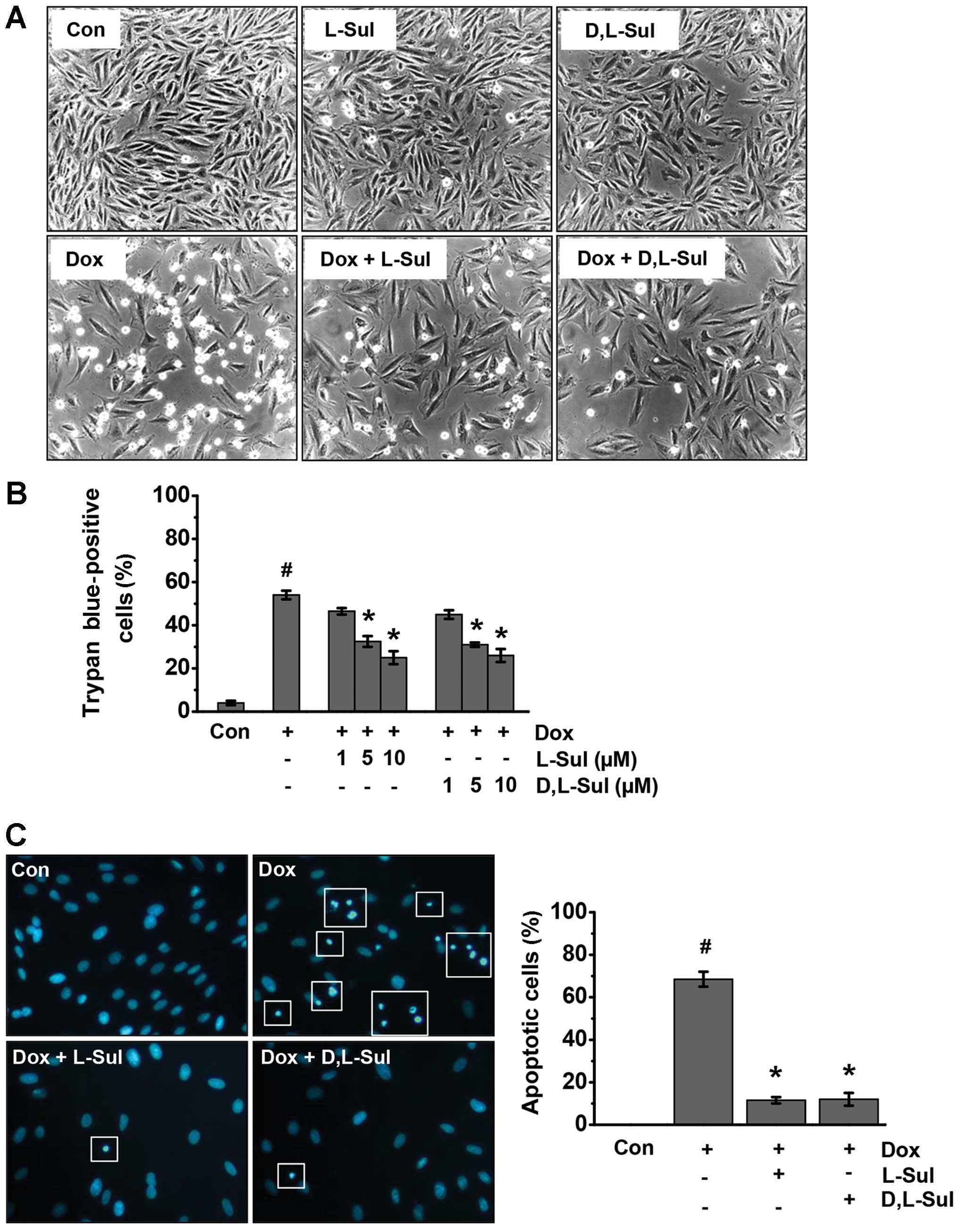
L-sulforaphane and D,L-sulforaphane
protect H9c2 myoblasts against doxorubicin-induced cell death
The H9c2 cells were exposed to doxorubicin and cell
viability was examined after 24 h under a light microscope and by
trypan blue exclusion assay. Based on the results obtained by light
microscopy, treatment of the H9c2 cells with doxorubicin induced
morphological changes, including rounding up and detachment.
Treatment with L-sulforaphane and D,L-sulforaphane clearly
protected the H9c2 cells against doxorubicin-induced cell death
(Fig. 1A). In addition, treatment
with either L-sulforaphane or D,L-sulforaphane alone was not toxic
to the H9c2 cells. Analysis of trypan blue dye uptake also revealed
that both L-sulforaphane and D,L-sulforaphane increased cell
viability in a dose-dependent manner (Fig. 1B). The results of the analysis of
apoptotic cells using Hoechst 33258 staining are shown on the left
panel of Fig. 1C. Following
treatment with doxorubicin, the H9c2 cells exhibited numerous
brightly condensed and broken fluorescent nuclei. Conversely, the
number of apoptotic cells treated with doxorubicin was
significantly decreased in the cells pre-treated with
L-sulforaphane or D,L-sulforaphane. The results of the
quantification of apoptotic cells are shown on the right panel of
Fig. 1C.
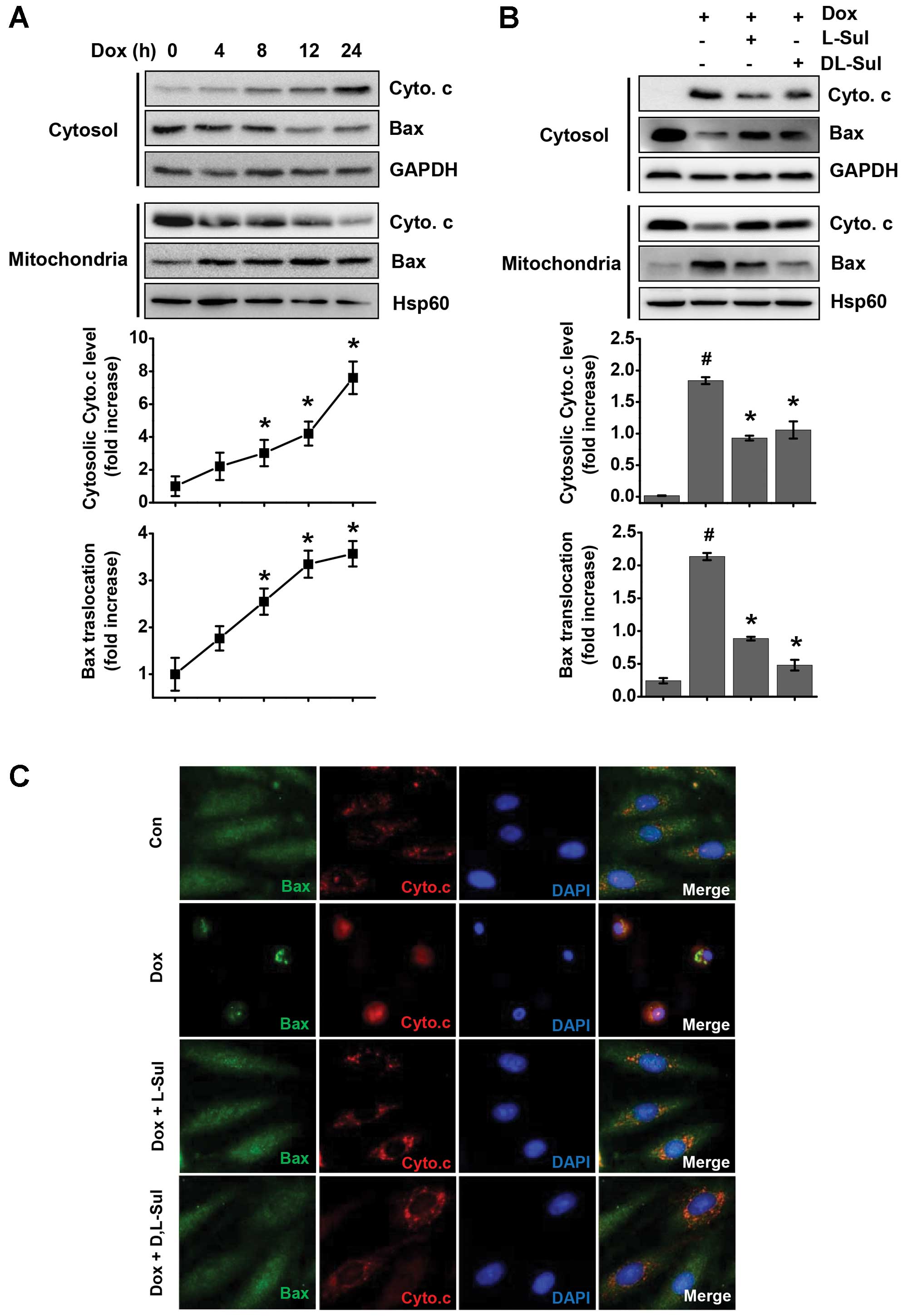
L-sulforaphane and D,L-sulforaphane
protect H9c2 myoblasts against the doxorubicin-induced
translocation of Bax to the mitochondria and the release of
cytochrome c
We then evaluated the effects of L-sulforaphane and
D,L-sulforaphane on translocation of Bax to the mitochondria and
the subsequent release of cytochrome c following treatment
with doxorubicin using cellular fractionation and western blot
analysis. Kinetic analysis of the appearance of the main signs of
apoptosis in the doxorubicin-treated cells revealed the rapid
release of mitochondrial cytochrome c into the cytosol of
H9c2 cells within 4 h of treatment (Fig. 2A). The presence of L-sulforaphane
and D,L-sulforaphane prevented the release of cytochrome c
into the cytosol in comparison to the group treated with
doxorubicin alone (Fig. 2B).
Similarly, in the cells treated with doxorubicin alone, we observed
a time-dependent increase in the translocation of Bax to the
mitochondria and a concomitant decrease in cytosolic Bax levels
(Fig. 2A). Pre-treatment with
L-sulforaphane and D,L-sulforaphane prevented the translocation of
Bax into the cytosol compared to the cells treated with doxorubicin
alone (Fig. 2B). We also
investigated the subcellular distribution of Bax and cytochrome
c in the H9c2 cells by dual immunofluorescence staining of
Bax and cytochrome c. The control cells displayed a
cytosolic distribution pattern of Bax and a punctate pattern of
cytochrome c immunostaining (Fig. 2C). During apoptosis induced by
doxorubicin, Bax translocated to the mitochondria and displayed a
punctate pattern. The Bax-positive cells displayed a diffuse
cytosolic pattern of cytochrome c staining, as well as a
condensed and shrunken nucleus as assessed by Hoechst 33258
staining (Fig. 1C). Consistent
with the results from western blot analysis (Fig. 2B), pre-treatment with
L-sulforaphane and D,L-sulforaphane prevented the translocation of
Bax to the mitochondria and the release of cytochrome c
(Fig. 2C).
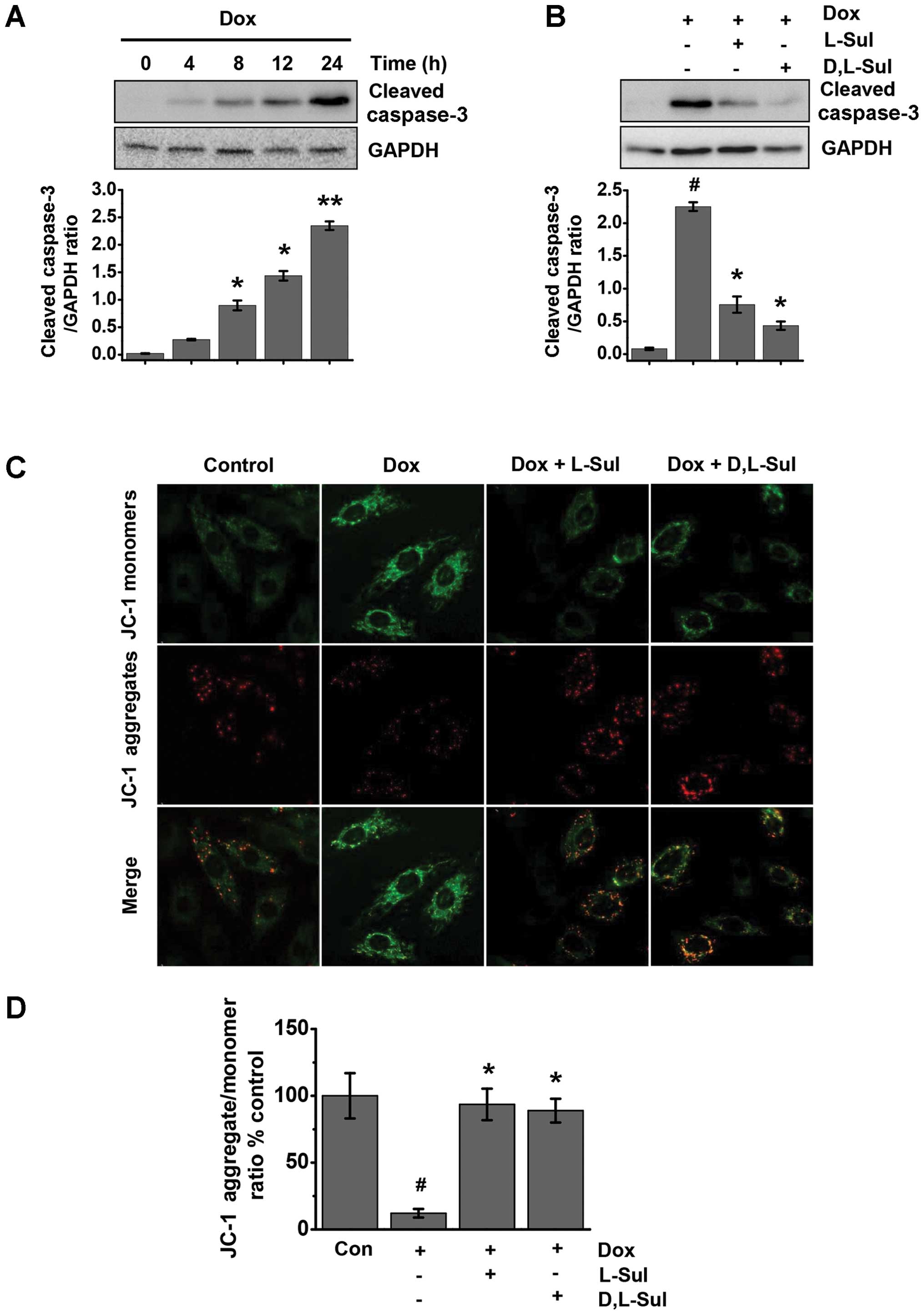
L-sulforaphane and D,L-sulforaphane
prevent the doxorubicin-induced activation of caspase-3 and protect
cells against doxorubicin-induced changes in ΔΨm
Doxorubicin was found to induce time-dependent cell
apoptosis in the H9c2 cells. Treatment with doxorubicin
significantly increased the levels of cleaved caspase-3 at 8 h;
these levels increased further in a time-dependent manner (Fig. 3A). As shown in Fig. 3B, both L-sulforaphane and
D,L-sulforaphane attenuated the doxorubicin-induced increase in the
levels of cleaved caspase-3. ΔΨm is one of the key events during
apoptosis (38). Mitochondrial
permeability transition has been implicated in the collapse of ΔΨm.
To monitor ΔΨm, we used the JC-1 dye and measured the emission
ratio at 590 to 527 nm. As shown in Fig. 3C, the control cells exhibited
mostly brightly stained mitochondria emitting red fluorescence,
whereas the H9c2 cells treated with doxorubicin produced green
fluorescence indicative of mitochondrial depolarization and the
collapse of ΔΨm. We observed an approximately 80% loss in ΔΨm in
the cells treated with doxorubicin alone compared with the control
cells (Fig. 3D). Conversely, the
cells pre-treated with either L-sulforaphane or D,L-sulforaphane
exhibited a significant preservation of red fluorescence compared
with the cells treated with doxorubicin alone (Fig. 3C and D).
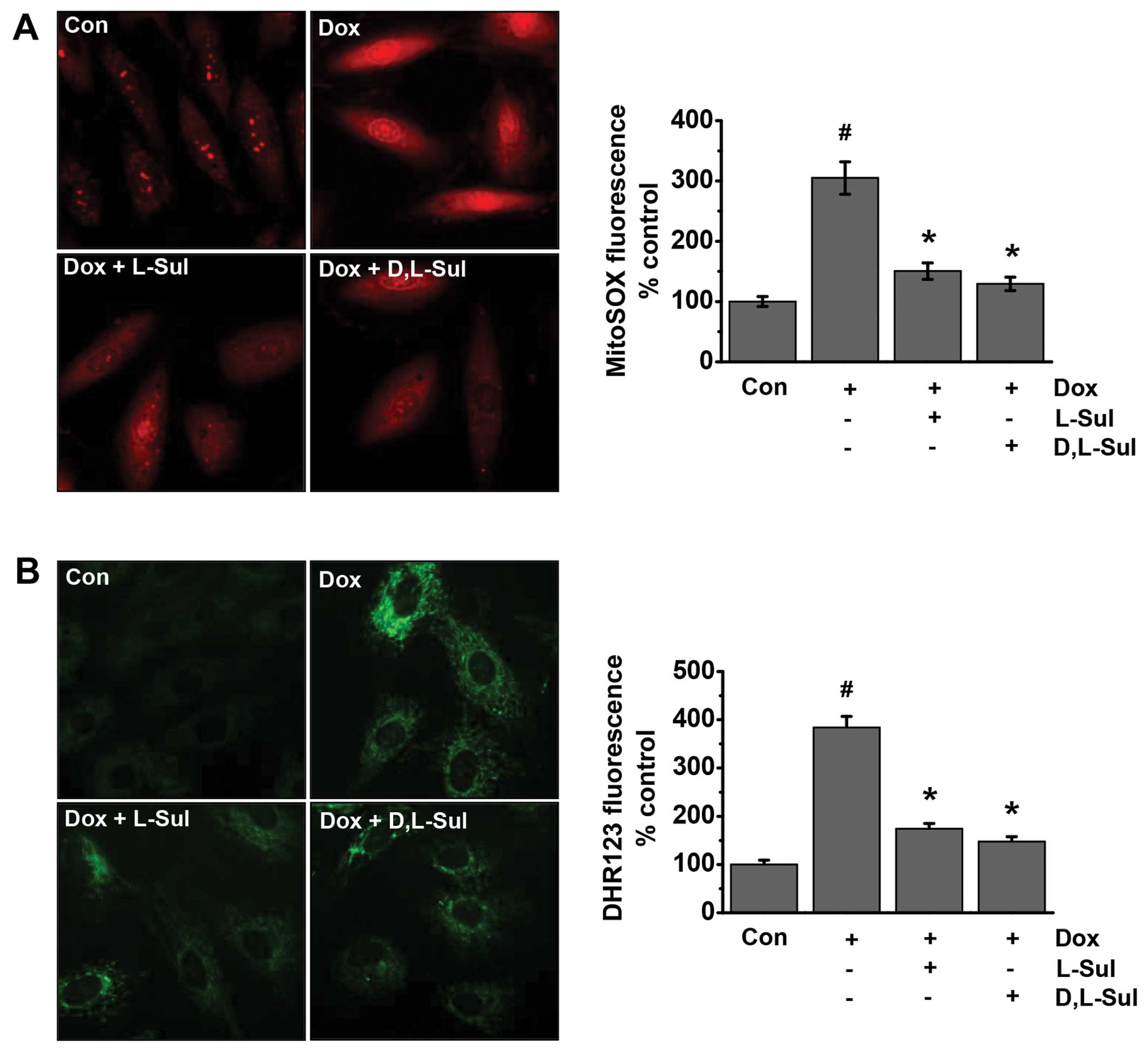
L-sulforaphane and D,L-sulforaphane
reduce the doxorubicin-induced generation of mitochondrial ROS in
H9c2 cells
ROS are involved in doxorubicin-induced cell
apoptosis (39–41). Previous studies have suggested
that cardiomyocyte mitochondria are important intracellular targets
of ROS during doxorubicin-induced cardiotoxicity. Superoxide and
peroxynitrite are the major ROS induced by doxorubicin (42,43). Thus, in the present study, we
evaluated whether L-sulforaphane and D,L-sulforaphane influence the
generation of doxorubicin-induced ROS. The cells were analyzed for
mitochondrial superoxide generation by fluorescence microscopy
using the mitochondria-targeted dye, dihydroethidium (MitoSOX Red),
as a probe (Fig. 4A). Our results
indicated that doxorubicin increased mitochondrial superoxide
generation compared to the untreated control cells. Conversely, the
doxorubicin-induced MitoSOX fluorescence intensity was attenuated
by pre-treatment with L-sulforaphane and D,L-sulforaphane (Fig. 4A). We also investigated the
generation of mitochondrial peroxynitrite using the dye, DHR123, as
a probe. As shown in Fig. 4B, the
doxorubicin-induced DHR123 fluorescence was attenuated by
pre-treatment wtih L-sulforaphane and D,L-sulforaphane. Taken
together, these results suggest that both L-sulforaphane and
D,L-sulforaphane protect H9c2 cells against doxorubicin-induced
apoptosis by preventing doxorubicin-induced mitochondrial ROS
generation.
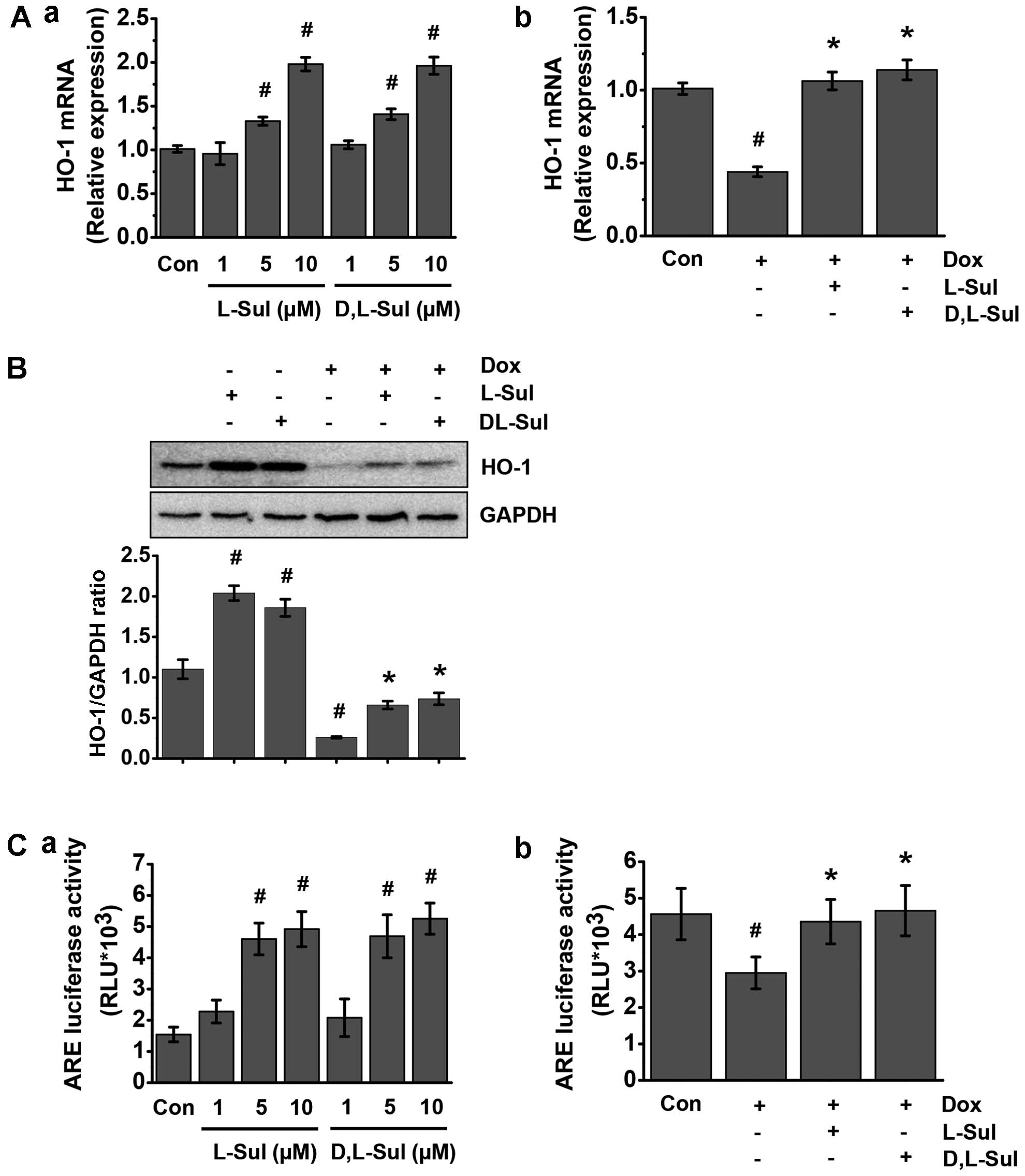
L-sulforaphane and D,L-sulforaphane
activate HO-1 through AREs in H9c2 cells
L-sulforaphane and D,L-sulforaphane protected the
H9c2 cells from doxorubicin-induced oxidative insults (Fig. 4). This protective action of
sulforaphane may be related to the induction of HO-1, which, along
with other phase II enzymes, serves as a defense system against
oxidative stress (44,45). Using RT-qPCR, we found that
pre-treatment with L-sulforaphane and D,L-sulforaphane induced HO-1
mRNA expression in a dose-dependent manner (Fig. 5A panel a). In addition,
pre-treatment with L-sulforaphane and D,L-sulforaphane reversed the
decrease in HO-1 mRNA expression observed in the cells treated with
doxorubicin alone (Fig. 5A panel
b). We then measured HO-1 protein expression levels by western blot
analysis. Consistent with our mRNA data, pre-treatment with
L-sulforaphane and D,L-sulforaphane induced a significant increase
in HO-1 protein expression (Fig.
5B). Furthermore, HO-1 protein expression was higher in the
cells pre-treated with L-sulforaphane and D,L-sulforaphane before
the addition of doxorubicin compared with cells treated only with
doxorubicin. We also found that L-sulforaphane and D,L-sulforaphane
stimulated ARE-dependent transcriptional activity in a
dose-dependent manner (Fig. 5C
panel a). Similarly, pretreatment with L-sulforaphane and
D,L-sulforaphane increased ARE-dependent transcriptional activity
compared to the cells treated with doxorubicin alone (Fig. 5C panel b). Taken together, these
results demonstrate that sulforaphane induces HO-1 mRNA and protein
expression by activating AREs.
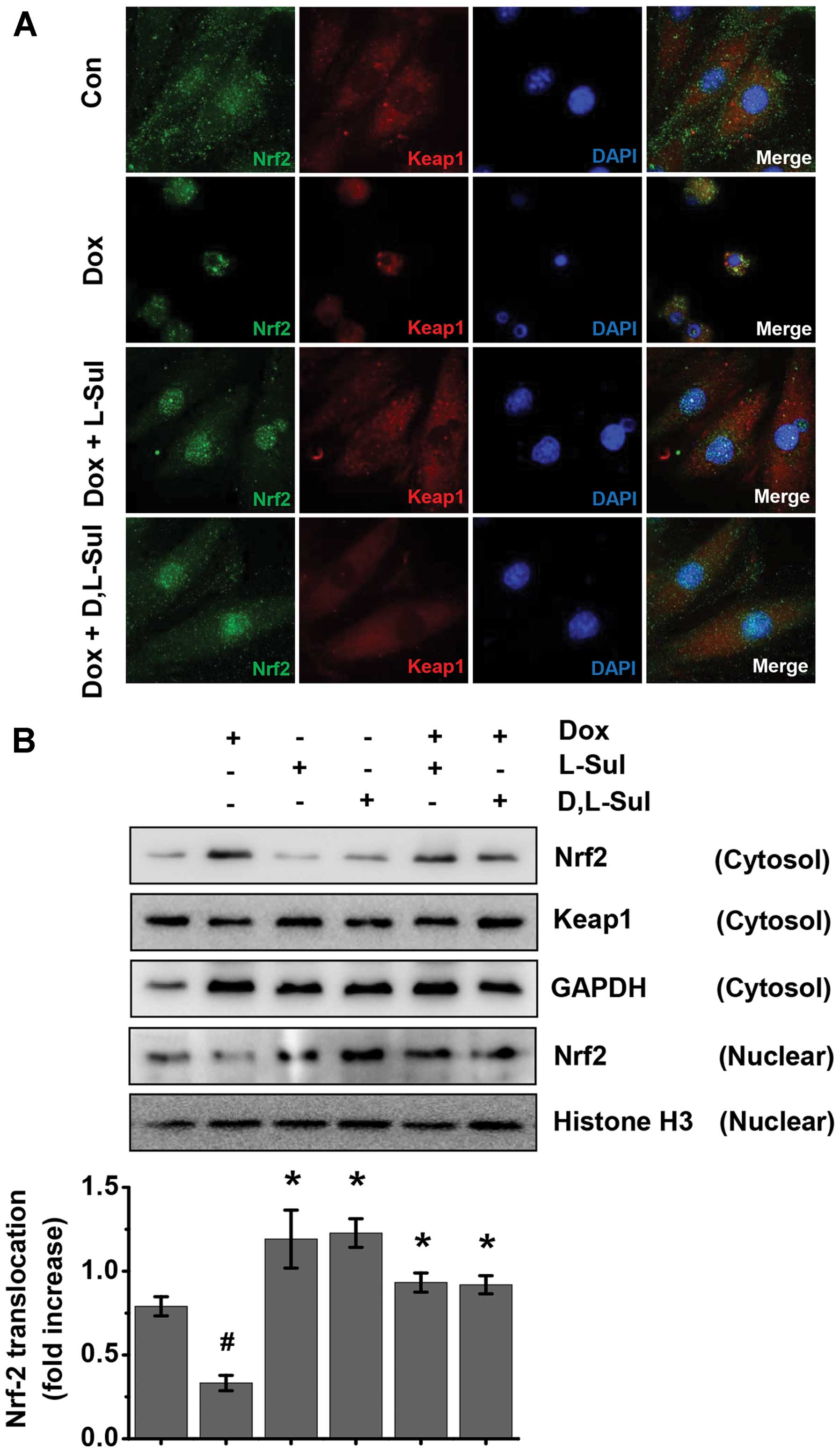
Activation of the Keap1/Nrf2 pathway by
L-sulforaphane and D,L-sulforaphane in H9c2 cells
We then investigated the activation status of Nrf2
in the H9c2 cells by assessing the nuclear translocation of Nrf2 by
western blot analysis of the cytosolic and nuclear fractions and by
immunofluorescence staining of Nrf2 and Keap1 (Fig. 6). Immunofluorescence staining and
confocal microscopy revealed that Nrf2 and Keap1 were predominantly
localized in the cytoplasm under basal conditions. In the
doxorubicin-treated positive cells, Nrf2 was almost completely
absent in the nuclear fraction. Conversely, the nuclear Nrf2
content was increased in the presence of L-sulforaphane and
D,L-sulforaphane, whereas Keap1 remained localized in the cytoplasm
(Fig. 6A). Similarly, western
blot analysis revealed that Nrf2 expression induced by
L-sulforaphane and D,L-sulforaphane was present at much higher
levels in the nucleus than in the cytoplasmic fraction (Fig. 6B). Taken together, these results
demonstrate that L-sulforaphane and D,L-sulforaphane activate the
Keap1/Nrf2 pathway in H9c2 cells.
Discussion
Recent evidence suggests a critical role of ROS in
doxorubicin-induced cardiotoxicity, leading to left ventricular
pathological hypertrophy and ultimately, heart failure (46). Therefore, the induction of the
activation endogenous antioxidants and phase II enzymes by dietary
means may be a promising cardio-protective strategy. In this study,
we found that sulforaphane protected H9c2 cells against cell death
induced by doxorubicin. Sulforaphane induced Nrf2-activated HO-1
expression, which consequently reduced ROS levels induced by
doxorubicin. In this study, we used H9c2 cells as a model for in
vitro studies of doxorubicin-induced cardiotoxicity. The H9c2
cell line was originally derived from embryonic rat ventricular
tissue (47), which is important
to note, as cardiac hypertrophy resulting from hypertension occurs
primarily in the ventricular muscle of the heart. Although H9c2
cells have lost their ability to spontaneously contract, they still
show many similarities to primary cardiomyocytes (48,49). Indeed, previous studies, using a
cell culture approach have investigated doxorubicin-induced cardiac
hypertrophy with rat H9c2 ventricular myocardial cells as a model
(50,51).
The data from the present study demonstrated that
doxorubicin activated apoptotic signaling as indicated by a
significant increase in the number of apoptotic cells, the
upregulation of pro-apoptotic proteins (Bax, caspase-3 and
cytochrome c) and an increase in ΔΨm. Excessive oxidative
stress by products in the mitochondria likely serves as an upstream
trigger of the apoptosis cascade (52). Importantly, oxidative
stress-induced apoptosis is a final common pathway for progressive
heart failure (53). In this
study, pre-treatment of the cells with sulforaphane decreased the
number of apoptotic cells, down-regulated the levels of
pro-apoptotic proteins (Bax, caspase-3 and cytochrome c) and
decreased ΔΨm.
Since ROS act as key mediators in doxorubicin
cardiotoxicity models, antioxidant defense mechanisms are necessary
to maintain normal cellular function. The Nrf2-phase II enzyme
system functions as one of the most important antioxidant defense
mechanisms by upregulating antioxidant response element-related
detoxification (17). In the
present study, we focused on one important Nrf2-target protein,
HO-1, which was increased by pre-treatment with sulforaphane during
doxorubicin-induced oxidative stress. Specifically, doxorubicin
significantly increased mitochondrial ROS levels, whereas
pre-treatment with sulforaphane reversed this increase. The
reversal effects of sulforaphane towards the mitochondrial ROS
levels confirmed its role as an antioxidant in cardiac injury.
Although sulforaphane is not a direct antioxidant, it activates the
transcription of phase II genes, the products of which provide
chemically versatile, often catalytic and prolonged ‘indirect’
antioxidant protection (54).
Although several natural or synthetic compounds may be used to
prevent doxorubicin-induced cardiotoxicity (11,55–59), one of the major advantages of
sulforaphane is that it is already a component of the human diet
and is therefore likely to be relatively safe for chronic
administration (54). In
addition, we observed that L-sulforaphane and D,L-sulforaphane had
similar effects. Sulforaphane is one of the promising
chemo-preventive phytochemicals (16,60). Sulforaphane is able to increase
the efficacy of doxorubicin and induce apoptosis in
doxorubicin-resistant p53 mutant cells (60). Sulforaphane has been shown to
significantly enhance doxorubicin cytoxicity particularly in A549
lung cancer cells, but not in other cancer cells (61). Additional studies are required to
further elucidate the mechanisms involved.
In conclusion, the findings of the present study
demonstrated that sulforaphane effectively reduced ROS production
and apoptosis induced by doxorubicin in H9c2 cells, and that the
protective mechanisms of action of sulforaphane were mediated by
preconditioning through the activation of Nrf2 and the subsequent
induction of HO-1. Additional studies are warranted to obtain
further insight into the clinical modalities through which
sulforaphane exerts its beneficial effects on patients who are at
risk of heart injury after receiving doxorubicin chemotherapy by
improving heart function. Nevertheless, the dietary consumption of
sulforaphane-containing cruciferous vegetables, such as broccoli
may prove to be useful in patients to prevent doxorubicin-induced
cardiotoxicity.
Acknowledgments
This study was supported by grants from the National
Research Foundation of Korea (2008-0062279, 2015R1A2A1A13001849 and
2012R1A2A2A01045214).
References
|
1
|
Vilaseca J, Guardia J, Bacardi R and Monné
J: Doxorubicin for liver cancer. Lancet. 1:13671978. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Arcamone F, Cassinelli G, Fantini G, Grein
A, Orezzi P, Pol C and Spalla C: Adriamycin, 14-hydroxydaunomycin,
a new antitumor antibiotic from S. peucetius var. caesius.
Biotechnol Bioeng. 11:1101–1110. 1969. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hortobágyi GN: Anthracyclines in the
treatment of cancer. An overview. Drugs. 54(Suppl 4): S1–S7.
1997.
|
|
4
|
Gewirtz DA: A critical evaluation of the
mechanisms of action proposed for the antitumor effects of the
anthracycline antibiotics adriamycin and daunorubicin. Biochem
Pharmacol. 57:727–741. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Muggia FM: Clinical efficacy and prospects
for use of pegylated liposomal doxorubicin in the treatment of
ovarian and breast cancers. Drugs. 54(Suppl 4): S22–S29. 1997.
View Article : Google Scholar
|
|
6
|
Naito S, Kotoh S, Omoto T, Osada Y,
Sagiyama K, Iguchi A, Ariyoshi A, Hiratsuka Y and Kumazawa J; The
Kyushu University Urological Oncology Group: Prophylactic
intravesical instillation chemotherapy against recurrence after a
transurethral resection of superficial bladder cancer: A randomized
controlled trial of doxorubicin plus verapamil versus doxorubicin
alone. Cancer Chemother Pharmacol. 42:367–372. 1998. View Article : Google Scholar
|
|
7
|
Mukhopadhyay P, Rajesh M, Bátkai S,
Kashiwaya Y, Haskó G, Liaudet L, Szabó C and Pacher P: Role of
superoxide, nitric oxide, and peroxynitrite in doxorubicin-induced
cell death in vivo and in vitro. Am J Physiol Heart Circ Physiol.
296:H1466–H1483. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Childs AC, Phaneuf SL, Dirks AJ, Phillips
T and Leeuwenburgh C: Doxorubicin treatment in vivo causes
cytochrome c release and cardiomyocyte apoptosis, as well as
increased mitochondrial efficiency, superoxide dismutase activity,
and Bcl-2:Bax ratio. Cancer Res. 62:4592–4598. 2002.PubMed/NCBI
|
|
9
|
Tatlidede E, Sehirli O, Velioğlu-Oğünc A,
Cetinel S, Yeğen BC, Yarat A, Süleymanoğlu S and Sener G:
Resveratrol treatment protects against doxorubicin-induced
cardiotoxicity by alleviating oxidative damage. Free Radic Res.
43:195–205. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang XL, Wang X, Xiong LL, Zhu Y, Chen HL,
Chen JX, Wang XX, Li RL, Guo ZY, Li P and Jiang W: Salidroside
improves doxorubicin-induced cardiac dysfunction by suppression of
excessive oxidative stress and cardiomyocyte apoptosis. J
Cardiovasc Pharmacol. 62:512–523. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kim DS, Woo ER, Chae SW, Ha KC, Lee GH,
Hong ST, Kwon DY, Kim MS, Jung YK, Kim HM, et al: Plantainoside D
protects adriamycin-induced apoptosis in H9c2 cardiac muscle cells
via the inhibition of ROS generation and NF-kappaB activation. Life
Sci. 80:314–323. 2007. View Article : Google Scholar
|
|
12
|
Zhang Y, Talalay P, Cho CG and Posner GH:
A major inducer of anticarcinogenic protective enzymes from
broccoli: Isolation and elucidation of structure. Proc Natl Acad
Sci USA. 89:2399–2403. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sakao K and Singh SV:
D,L-sulforaphane-induced apoptosis in human breast cancer cells is
regulated by the adapter protein p66Shc. J Cell Biochem.
113:599–610. 2012. View Article : Google Scholar :
|
|
14
|
Nestle M: Broccoli sprouts as inducers of
carcinogen-detoxifying enzyme systems: Clinical, dietary, and
policy implications. Proc Natl Acad Sci USA. 94:11149–11151. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tanito M, Masutani H, Kim YC, Nishikawa M,
Ohira A and Yodoi J: Sulforaphane induces thioredoxin through the
antioxidant-responsive element and attenuates retinal light damage
in mice. Invest Ophthalmol Vis Sci. 46:979–987. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Surh YJ: Cancer chemoprevention with
dietary phytochemicals. Nat Rev Cancer. 3:768–780. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yoon HY, Kang NI, Lee HK, Jang KY, Park JW
and Park BH: Sulforaphane protects kidneys against
ischemia-reperfusion injury through induction of the Nrf2-dependent
phase 2 enzyme. Biochem Pharmacol. 75:2214–2223. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhao J, Kobori N, Aronowski J and Dash PK:
Sulforaphane reduces infarct volume following focal cerebral
ischemia in rodents. Neurosci Lett. 393:108–112. 2006. View Article : Google Scholar
|
|
19
|
Satoh T, Okamoto SI, Cui J, Watanabe Y,
Furuta K, Suzuki M, Tohyama K and Lipton SA: Activation of the
Keap1/Nrf2 pathway for neuroprotection by electrophilic [correction
of electrophillic] phase II inducers. Proc Natl Acad Sci USA.
103:768–773. 2006. View Article : Google Scholar
|
|
20
|
Barbagallo I, Galvano F, Frigiola A,
Cappello F, Riccioni G, Murabito P, D’Orazio N, Torella M, Gazzolo
D and Li Volti G: Potential therapeutic effects of natural heme
oxygenase-1 inducers in cardiovascular diseases. Antioxid Redox
Signal. 18:507–521. 2013. View Article : Google Scholar
|
|
21
|
Jazwa A and Cuadrado A: Targeting heme
oxygenase-1 for neuroprotection and neuroinflammation in
neurodegenerative diseases. Curr Drug Targets. 11:1517–1531. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang RQ, Nan YM, Wu WJ, Kong LB, Han F,
Zhao SX, Kong L and Yu J: Induction of heme oxygenase-1 protects
against nutritional fibrosing steatohepatitis in mice. Lipids
Health Dis. 10:312011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Angeloni C, Leoncini E, Malaguti M,
Angelini S, Hrelia P and Hrelia S: Modulation of phase II enzymes
by sulforaphane: Implications for its cardioprotective potential. J
Agric Food Chem. 57:5615–5622. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Piao CS, Gao S, Lee GH, Kim S, Park BH,
Chae SW, Chae HJ and Kim SH: Sulforaphane protects ischemic injury
of hearts through antioxidant pathway and mitochondrial K(ATP)
channels. Pharmacol Res. 61:342–348. 2010. View Article : Google Scholar
|
|
25
|
Strober W: Trypan blue exclusion test of
cell viability. Curr Protoc Immunol Appendix 3. Appendix 3B. 2001.
View Article : Google Scholar
|
|
26
|
Latt SA and Wohlleb JC: Optical studies of
the interaction of 33258 Hoechst with DNA, chromatin, and metaphase
chromosomes. Chromosoma. 52:297–316. 1975. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chae HJ, Kim HR, Xu C, Bailly-Maitre B,
Krajewska M, Krajewski S, Banares S, Cui J, Digicaylioglu M, Ke N,
et al: BI-1 regulates an apoptosis pathway linked to endoplasmic
reticulum stress. Mol Cell. 15:355–366. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ludke AR, Sharma AK, Akolkar G, Bajpai G
and Singal PK: Downregulation of vitamin C transporter SVCT-2 in
doxorubicin-induced cardiomyocyte injury. Am J Physiol Cell
Physiol. 303:C645–C653. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jiang M, Wang CY, Huang S, Yang T and Dong
Z: Cisplatin-induced apoptosis in p53-deficient renal cells via the
intrinsic mitochondrial pathway. Am J Physiol Renal Physiol.
296:F983–F993. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Salvioli S, Ardizzoni A, Franceschi C and
Cossarizza A: JC-1, but not DiOC6(3) or rhodamine 123, is a
reliable fluorescent probe to assess delta psi changes in intact
cells: Implications for studies on mitochondrial functionality
during apoptosis. FEBS Lett. 411:77–82. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kooy NW, Royall JA, Ischiropoulos H and
Beckman JS: Peroxynitrite-mediated oxidation of dihydrorhodamine
123. Free Radic Biol Med. 16:149–156. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Tarpey MM and Fridovich I: Methods of
detection of vascular reactive species: Nitric oxide, superoxide,
hydrogen peroxide, and peroxynitrite. Circ Res. 89:224–236. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sobreira C, Davidson M, King MP and
Miranda AF: Dihydrorhodamine 123 identifies impaired mitochondrial
respiratory chain function in cultured cells harboring
mitochondrial DNA mutations. J Histochem Cytochem. 44:571–579.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Crow JP: Dichlorodihydrofluorescein and
dihydrorhodamine 123 are sensitive indicators of peroxynitrite in
vitro: Implications for intracellular measurement of reactive
nitrogen and oxygen species. Nitric Oxide. 1:145–157. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Robinson KM, Janes MS and Beckman JS: The
selective detection of mitochondrial superoxide by live cell
imaging. Nat Protoc. 3:941–947. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Mukhopadhyay P, Rajesh M, Yoshihiro K,
Haskó G and Pacher P: Simple quantitative detection of
mitochondrial superoxide production in live cells. Biochem Biophys
Res Commun. 358:203–208. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ito T, Okumura H, Tsukue N, Kobayashi T,
Honda K and Sekizawa K: Effect of diesel exhaust particles on mRNA
expression of viral and bacterial receptors in rat lung epithelial
L2 cells. Toxicol Lett. 165:66–70. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ly JD, Grubb DR and Lawen A: The
mitochondrial membrane potential (deltapsi(m)) in apoptosis; an
update. Apoptosis. 8:115–128. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Tsang WP, Chau SP, Kong SK, Fung KP and
Kwok TT: Reactive oxygen species mediate doxorubicin induced
p53-independent apoptosis. Life Sci. 73:2047–2058. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Sawyer DB, Fukazawa R, Arstall MA and
Kelly RA: Daunorubicin-induced apoptosis in rat cardiac myocytes is
inhibited by dexrazoxane. Circ Res. 84:257–265. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kotamraju S, Konorev EA, Joseph J and
Kalyanaraman B: Doxorubicin-induced apoptosis in endothelial cells
and cardiomyocytes is ameliorated by nitrone spin traps and
ebselen. Role of reactive oxygen and nitrogen species. J Biol Chem.
275:33585–33592. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Mihm MJ, Yu F, Weinstein DM, Reiser PJ and
Bauer JA: Intracellular distribution of peroxynitrite during
doxorubicin cardiomyopathy: Evidence for selective impairment of
myofibrillar creatine kinase. Br J Pharmacol. 135:581–588. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Luanpitpong S, Chanvorachote P, Nimmannit
U, Leonard SS, Stehlik C, Wang L and Rojanasakul Y: Mitochondrial
superoxide mediates doxorubicin-induced keratinocyte apoptosis
through oxidative modification of ERK and Bcl-2 ubiquitination.
Biochem Pharmacol. 83:1643–1654. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Lee YJ, Jeong HY, Kim YB, Lee YJ, Won SY,
Shim JH, Cho MK, Nam HS and Lee SH: Reactive oxygen species and
PI3K/Akt signaling play key roles in the induction of Nrf2-driven
heme oxygenase-1 expression in sulforaphane-treated human
mesothelioma MSTO-211H cells. Food Chem Toxicol. 50:116–123. 2012.
View Article : Google Scholar
|
|
45
|
Surh YJ, Na HK and Lee SS: Transcription
factors and mitogen-activated protein kinases as molecular targets
for chemoprevention with anti-inflammatory phytochemicals.
Biofactors. 21:103–108. 2004. View Article : Google Scholar
|
|
46
|
Scott JM, Khakoo A, Mackey JR, Haykowsky
MJ, Douglas PS and Jones LW: Modulation of anthracycline-induced
cardiotoxicity by aerobic exercise in breast cancer: Current
evidence and underlying mechanisms. Circulation. 124:642–650. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Kimes BW and Brandt BL: Properties of a
clonal muscle cell line from rat heart. Exp Cell Res. 98:367–381.
1976. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Watkins SJ, Borthwick GM and Arthur HM:
The H9C2 cell line and primary neonatal cardiomyocyte cells show
similar hypertrophic responses in vitro. In Vitro Cell Dev Biol
Anim. 47:125–131. 2011. View Article : Google Scholar
|
|
49
|
Prathapan A, Vineetha VP, Abhilash PA and
Raghu KG: Boerhaavia diffusa L. attenuates angiotensin II-induced
hypertrophy in H9c2 cardiac myoblast cells via modulating oxidative
stress and down-regulating NF-κB and transforming growth factor β1.
Br J Nutr. 110:1201–1210. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Karagiannis TC, Lin AJ, Ververis K, Chang
L, Tang MM, Okabe J and El-Osta A: Trichostatin A accentuates
doxorubicin-induced hypertrophy in cardiac myocytes. Aging (Albany
NY). 2:659–668. 2010.
|
|
51
|
Merten KE, Jiang Y, Feng W and Kang YJ:
Calcineurin activation is not necessary for Doxorubicin-induced
hypertrophy in H9c2 embryonic rat cardiac cells: Involvement of the
phosphoinositide 3-kinase-Akt pathway. J Pharmacol Exp Ther.
319:934–940. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Wang GW, Klein JB and Kang YJ:
Metallothionein inhibits doxorubicin-induced mitochondrial
cytochrome c release and caspase-3 activation in cardiomyocytes. J
Pharmacol Exp Ther. 298:461–468. 2001.PubMed/NCBI
|
|
53
|
Hare JM: Oxidative stress and apoptosis in
heart failure progression. Circ Res. 89:198–200. 2001.PubMed/NCBI
|
|
54
|
Gao X and Talalay P: Induction of phase 2
genes by sulforaphane protects retinal pigment epithelial cells
against photooxidative damage. Proc Natl Acad Sci USA.
101:10446–10451. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Al-Abd AM, Al-Abbasi FA, Asaad GF and
Abdel-Naim AB: Didox potentiates the cytotoxic profile of
doxorubicin and protects from its cardiotoxicity. Eur J Pharmacol.
718:361–369. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Jay SM, Murthy AC, Hawkins JF, Wortzel JR,
Steinhauser ML, Alvarez LM, Gannon J, Macrae CA, Griffith LG and
Lee RT: An engineered bivalent neuregulin protects against
doxorubicin-induced cardiotoxicity with reduced proneoplastic
potential. Circulation. 128:152–161. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Osman AM, Al-Harthi SE, AlArabi OM, Elshal
MF, Ramadan WS, Alaama MN, Al-Kreathy HM, Damanhouri ZA and Osman
OH: Chemosensetizing and cardioprotective effects of resveratrol in
doxorubicin-treated animals. Cancer Cell Int. 13:522013. View Article : Google Scholar
|
|
58
|
Ahmed LA and El-Maraghy SA: Nicorandil
ameliorates mitochondrial dysfunction in doxorubicin-induced heart
failure in rats: Possible mechanism of cardioprotection. Biochem
Pharmacol. 86:1301–1310. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Choi HJ, Seon MR, Lim SS, Kim JS, Chun HS
and Park JH: Hexane/ethanol extract of Glycyrrhiza uralensis
licorice suppresses doxorubicin-induced apoptosis in H9c2 rat
cardiac myoblasts. Exp Biol Med (Maywood). 233:1554–1560. 2008.
View Article : Google Scholar
|
|
60
|
Fimognari C, Nüsse M, Lenzi M, Sciuscio D,
Cantelli-Forti G and Hrelia P: Sulforaphane increases the efficacy
of doxorubicin in mouse fibroblasts characterized by p53 mutations.
Mutat Res. 601:92–101. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Hu L, Miao W, Loignon M, Kandouz M and
Batist G: Putative chemopreventive molecules can increase
Nrf2-regulated cell defense in some human cancer cell lines,
resulting in resistance to common cytotoxic therapies. Cancer
Chemother Pharmacol. 66:467–474. 2010. View Article : Google Scholar
|