Introduction
Generalized arterial calcification of infancy (GACI)
is an autosomal recessive disorder of spontaneous infantile
arterial and periarticular calcification (1–3).
This life-threatening disease is caused by loss-of-function
mutations in the ectonucleotide pyrophosphatase/phosphodiesterase 1
(ENPP1) gene, a key regulator of biomineralisation and
vascular calcification (4–8).
ENPP1 is a cell-surface glycoprotein enzyme that
functions in synergy with the anklyosis protein (ANK) to
respectively form and intracellularly channel inorganic
pyrophosphate (PPi), an inhibitor of hydroxyapatite
formation, from nucleoside triphosphates (9–12).
The extracellular concentration of PPi is further
influenced by tissue non-specific alkaline phosphatase (TNSALP),
another cell-surface enzyme located on the cell membrane of
osteoblasts and chondrocytes, as well as on the membranes of their
matrix vesicles (MVs) (13).
TNSALP exerts its effects by hydrolysing PPi reducing
the concentration of this mineralisation inhibitor and establishing
a phosphate (Pi)/PPi ratio permissive for the formation
of hydroxyapatite crystals (14–17). Phosphatase, orphan 1 (PHOSPHO1) is
another essential phosphatase, located within osteoblast- and
chondrocyte-derived MVs with high phosphohydrolase activity toward
phosphoethanolamine and phosphocholine (18–22), which contributes Pi for the
initiation of skeletal mineralisation. Together, ENPP1, ANK, TNSALP
and PHOSPHO1 control the Pi/PPi ratio conducive to physiological
skeletal mineralisation. Thus, ENPP1 in GACI reduces extracellular
PPi levels and predisposes to ectopic calcification.
This was further exemplified in a previous study of ours, in which
we determined that vascular smooth muscle cells from mice deficient
in Enpp1 have increased TNSALP levels (23).
In naturally occurring mouse models, the link
between defective Enpp1 expression and altered
mineralisation was initially demonstrated in ‘tiptoe walking’
(ttw/ttw) mice (24–28). These animals are homozygous for a
G→T substitution resulting in the introduction of a stop codon in
the NPP1 coding sequence. The subsequent truncated protein leads to
the loss of a vital calcium binding domain and two putative
glycosylation sites (25). The
phenotype of this mouse includes the postnatal development of
progressive ankylosing intervertebral and peripheral joint
hyperostosis, as well as spontaneous arterial and articular
cartilage calcification and increased vertebral cortical bone
formation (24–28). Transgenic mice that are homozygous
for a disruption in exon 9 of the Enpp1 gene
(Enpp1−/− mice) exhibit abnormalities that are
almost identical to those present in ttw/ttw mice (29). These include decreased levels of
extracellular PPi, with phenotypic characteristics,
including significant alterations in bone mineralisation in long
bones and calvariae, and pathological, severe peri-spinal soft
tissue and arterial calcification (30–32).
Effective treatment for infants and young children
with GACI is critical as without it, 85% of patients succumb to the
disease within 6 months of age. First used off-label in the
treatment of Fibrodysplasia ossificans progressiva, the
‘first generation’ bisphosphonate, etidronate (EHDP;
ethane-1-hydroxy-1,1-diphosphonic acid, also known as
1-hydroxyethylidene-bisphosphonate) is an analogue of
PPi and has also been used in the treatment of GACI.
Bisphosphonates are potent inhibitors of osteoclast activity, and
are widely used in clinical practice to prevent the bone loss
associated with conditions, such as Paget’s disease, metastatic
bone disease and osteoporosis (33). The inhibitory effects of
bisphosphonates on osteoblast function have also been demonstrated
(34–37).
In 2008, a retrospective observational analysis of
55 patients with GACI revealed survival beyond infancy with
etidronate therapy (38)
corroborated by a recent study highlighting that 15 out of 22 GACI
survivors received etidronate (39). However, studies on uremic rats
have suggested that the administration of etidronate may not be
able to prevent arterial calcification without inhibiting bone
formation (40). Furthermore, a
recent case report has highlighted the profound inhibition of
skeletal mineralisation with paradoxical joint calcifications
following protracted etidronate therapy in a 7-year-old boy with
GACI (41). Taken together, these
findings have led us to herein assess the effects of etidronate on
bone architecture and arterial calcification in the
Enpp1−/− mouse model of GACI.
Materials and methods
Animals. Enpp1−/− and wild-type
(WT) mice were generated and maintained as previously described
(1,5,29,42). Male mice were administered
etidronate at 100 µg/kg, intraperitoneally twice a week from
11 to 22 weeks of age. The dosage of etidronate used in this study
was based on the dose reported in a previously study (43). Animals were administered saline as
a placebo (vehicle treatment). All animals were weighed once a
week. The animals were sacrificed at 22 weeks of age and the
tissues were dissected for further analysis. All animal experiments
were approved by The Roslin Institute’s Animal Users Committee and
the animals were maintained in accordance with the UK Home Office
guidelines for the care and use of laboratory animals (PIL number
DD 60/3828).
Preparation of tissue
The aortae and tibiae were dissected as previously
described (5). The aortae were
fixed in 10% neutral buffered formalin (NBF) for 48 h before being
transferred to 70% ethanol. The tibiae were immediately frozen in
distilled water pending analysis.
Micro-computed tomography (µCT) of the
aortae
Prior to scanning, the aortae were immersed for a
minimum period of time (10 min) in a macro-molecular
iopamidol-based contrast agent (Niopam 300; Brako UK Ltd., High
Wycombe, Buckinghamshire, UK) diluted 1:4 in water as previously
described (44). To allow tissue
differentiation, aortic luminae were filled with corn oil and the
aortae were submersed in oil for the duration of the scan. Tissues
were imaged using a Skyscan 1172 X-Ray Microtomograph (Bruker
Daltonics, Brussels, Belgium). Sequential high-resolution scans
were acquired using a rotation step of 0.3° with the averaging of 3
frames at each step, applying a 0.5-mm aluminium filter, with an
X-ray source set at 60 kV and 167 µA, and with an isotropic
voxel size of 7 µm. The scans were reconstructed using
NRecon (Bruker Daltonics). Noise in the reconstructed images was
reduced by applying a median filter (radius = 1). The region of
interest was selected to be the aortic arch, 200 lices (1.4 mm)
under the subclavian artery. Soft and calcified tissue was
identified by thresholding using CTAn software (Bruker
Daltonics).
µCT of the tibiae
High-resolution scans with an isotropic voxel size
of 5 µm were acquired with a µCT system (60 kV, 0.5
mm aluminium filter, 0.6° rotation, Skyscan 1172; Bruker
Daltonics). Scans were reconstructed using NRecon software (Bruker
Daltonics). A 1,000-µm section of the metaphysis 250
µm off the reference plate was taken for analysis of the
trabecular bone. The base of the growth plate was used as a
standard reference point. A 250-µm metaphysis section of the
mid-diaphysis, 1,500 µm below the reference plate, was
scanned for the analysis of cortical structure. Data were analysed
with CTAn software (Bruker Daltonics). The following parameters
were analysed using CTAn software (Bruker Daltonics): percentage
bone volume/trabecular bone volume (%BV/TV), trabecular number
(Tb.N;/mm), trabecular patten factor (Tb.Pf), bone mineral density
(BMD; g/cm3), trabecular thickness (Tb.Th; mm),
trabecular separation (Tb. Sp) and the structure model index (SMI)
were evaluated. In the cortical bone, %BV/TV, BMD
(g/cm3), cortical thickness, cross-sectional area
(mm2), the percentage of closed pores and polar moment
of inertia (mm4) were evaluated.
Mechanical testing
Mechanical testing of the cortical bone was carried
out using a Zwick materials testing machine (Zwick Armaturen GmbH,
Ennepetal, Germany) and data were analysed as previously described
(45). The span was fixed at 6.0
mm. The cross-head was lowered at 1 mm/min and data were recorded
after every 0.1 mm change in deflection. Each bone was tested to
fracture. Failure and fracture points were identified from the
load-extension curve as the point of maximum load and where the
load rapidly decreased to zero, respectively. The maximum stiffness
was defined as the maximum gradient of the rising portion of this
curve, and the yield point, the point at which the gradient reduced
to 95% of this value. Both values were calculated from a polynomial
curve fitted to the rising region of the load-extension curve.
Serum marker analysis
To determine differences in bone formation and
resorption, plasma serum was collected from the mice at 22 weeks of
age. A sandwich ELISA P1NP (IDS Ltd., Boldons, UK) and a C-terminal
telopoptide of type I collagen (CTx) ELISA kit (RatLaps™; IDS) were
used respectively, and analyses were performed according to the
manufacturer’s instructions.
Statistical analysis
General linear model analysis, the Student’s t-test,
the Mann-Whitney non-parametric test and Pearson’s correlation
anlaysis were used to assess the data where appropriate. All data
are expressed as the means ± SEM. Statistical analysis was
performed using SPSS (IBM Software, New York, NY, USA). A value of
P<0.05 was considered to indicate a statistically significant
difference.
Results
Enpp1−/− mouse growth
phenotype
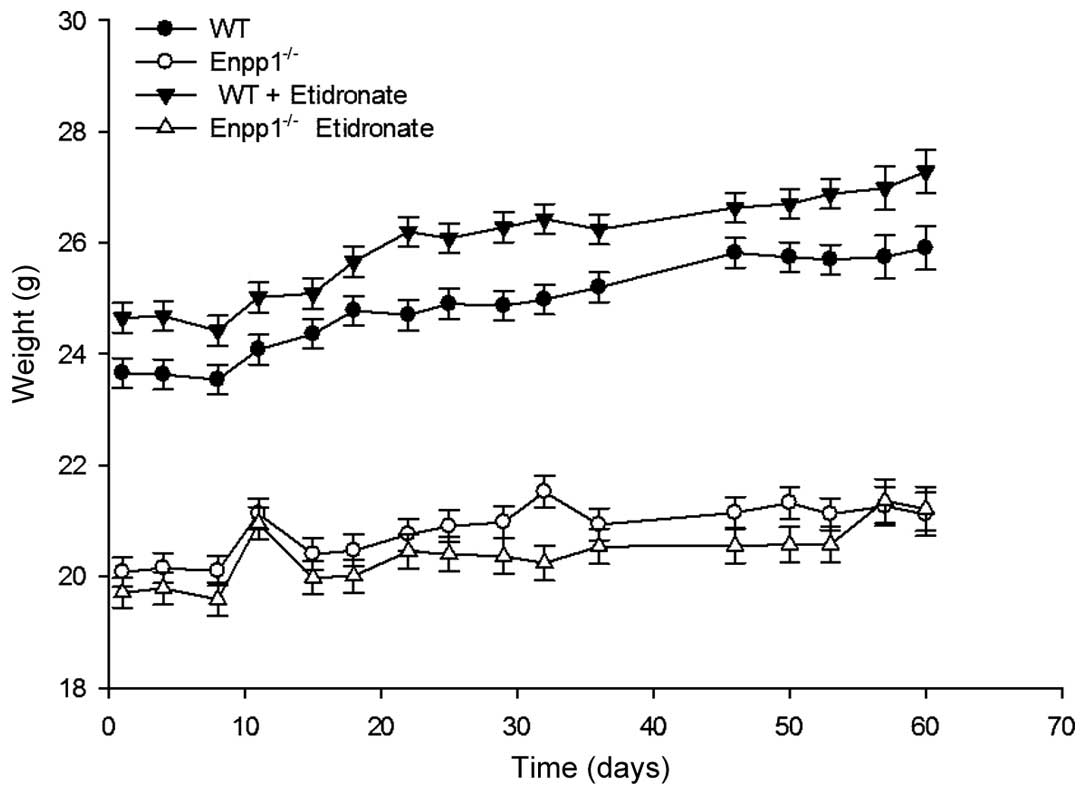
In initial experiments, we examined whether the
treatment of Enpp1−/− and WT mice with 100
µg/kg etidronate affects their growth. In accordance with
our previous study, the Enpp1−/− mice exhibited a
reduced growth in comparison to the WT mice (18.4% smaller than the
age-matched WT controls; P<0.05) (5). Intraperitoneal injections of
etidronate had no effect on the total body weight of the WT mice,
nor the Enpp1−/− mice in comparison to the
respective vehicle-treated mice (Fig.
1). Notably, the Enpp1−/− mice appeared to
lose weight from approximately 12 weeks of age, which may be a
consequence of their limited movement due to excessive joint
calcification (Fig. 1) (1,5).
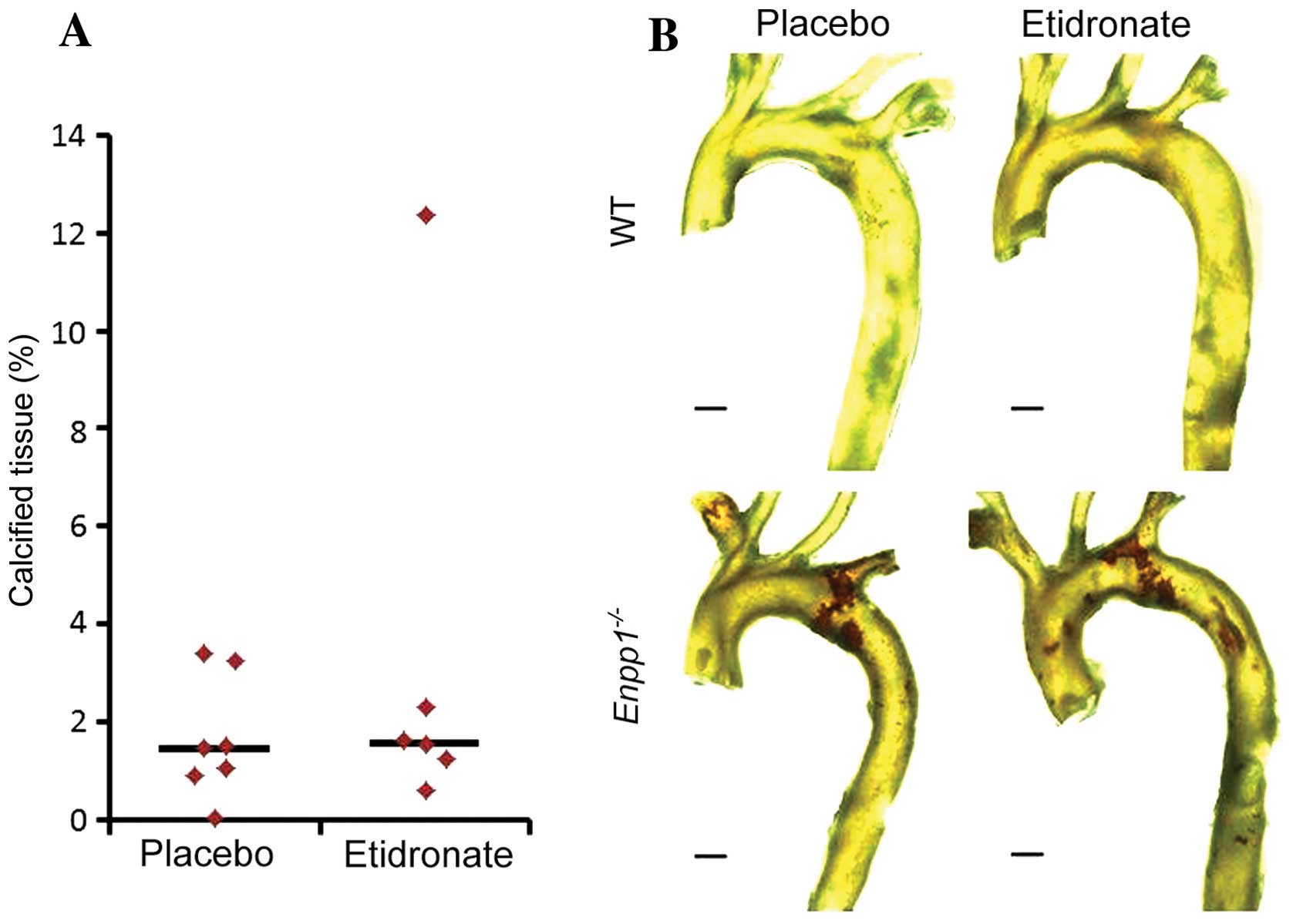
Aortic calcification
We have previously demonstrated that
Enpp1−/− mice exhibit arterial calcification from
11 weeks of age (31). In this
study, we employed our recently developed three-dimensional (3D)
µCT protocol (44) for the
quantification of aortic calcification to examine the effects of
treatment with etidronate on mice lacking Enpp1. As
expected, the Enpp1−/− mice exhibited extensive
aortic calcification in comparison to the WT mice at 22 weeks of
age (Fig. 2B). However, treatment
with etidronate did not prevent de novo calcification, and
did not arrest the progression of established calcification of the
aorta in these mice (Fig. 2).
µCT analysis of bone
microarchitecture
Enpp1−/− mice have previously been
reported to display reduced mineral content in bone, with a
reduction in bone volume fraction and trabecular thickness
(5). The present study extended
these observations by fully examining the effects of the
administration of etidronate on the bone phenotype of
Enpp1−/− mice. µCT analysis of the tibiae
from Enpp1−/− mice in comparison to those from WT
mice (both vehicle-treated) at 22 weeks of age revealed a
significant decrease in trabecular bone mass, as reflected by a
decrease in %BV/TV, trabecular thickness and trabecular number
(P<0.05; Table I). Moreover,
we observed a significant decrease in cortical parameters in the
tibiae of the 22-week-old Enpp1−/− mice in
comparison to the age-matched WT mice, except for cortical porosity
(P<0.05; Table II). Treatment
with etidronate had no significant effect on cortical or trabecular
bone parameters in the WT mice (Tables I and II). In the Enpp1−/−
mice, treatment with etidronate resulted in an increase in
trabecular number and %BV/TV, as reflected by the significant
decrease in trabecular separation (P<0.05, in comparison to the
vehicle-treated Enpp1−/− mice) (Table I). The Enpp1−/−
mice treated with etidronate did show a significant decrease in SMI
[quantification of the plate- or rod-like geometry of trabecular
structures, as previously described (46)] compared to the vehicle-treated
Enpp1−/− mice (P<0.05; Table I).
 | Table IµCT analysis of trabecular
bone in male placebo (vehicle)- and etidronate-treated
Enpp1−/− and WT mice. |
Table I
µCT analysis of trabecular
bone in male placebo (vehicle)- and etidronate-treated
Enpp1−/− and WT mice.
| WT
|
Enpp1−/−
|
|---|
| Placebo | Etidronate | Placebo | Etidronate |
|---|
| BV/TV (%) | 8.79±1.59 | 8.75±1.79 | 3.39±1.06a,e | 4.63±1.57a,e |
| BMD
(g/cm3) | 0.139±0.021 | 0.13±0.024 | 0.042±0.020a,e | 0.062±0.025a,e |
| Tb.Th
(µm) | 60.62±4.31 | 57.34±8.34 | 48.35±3.08a,e | 47.99±3.54a,d |
| Tb.Sp
(µm) | 298.24±12.68 | 290.47±26.56 |
363.66±48.07a,d |
319.66±48.44b,c |
| Tb.N |
0.00146±0.00023 | 0.0015±0.00029 |
0.00070±0.00024a,e |
0.00097±0.00034a,e |
| Tb.Pf | 0.027±0.0026 | 0.026±0.0033 |
0.042±0.0036a,e |
0.0392±0.0053a,e |
| SMI | 2.48±0.11 | 2.33±0.23 | 2.78±0.093a,d | 2.69±0.22a,e |
| DA | 2.30±0.19 | 2.08±0.15 | 2.42±0.28 | 2.39±0.31a,c |
| Table IIµCT analysis of cortical bone
in male placebo (vehicle)- and etidronate-treated
Enpp1−/− and WT mice. |
Table II
µCT analysis of cortical bone
in male placebo (vehicle)- and etidronate-treated
Enpp1−/− and WT mice.
| WT
|
Enpp1−/−
|
|---|
| Placebo | Etidronate | Placebo | Etidronate |
|---|
| Co.BMD
(g/cm3) | 1.18±0.018 | 1.18±0.020 | 1.20±0.024 | 1.20±0.011a |
| Co.Po (%) | 53.27±7.26 | 54.79±5.64 | 61.75±3.71a,c | 63.11±4.85a,c |
| Co.Th
(µm) | 197.90±1.91 | 195.64±3.84 | 175.98±5.96a,d |
169.86±15.92a,d |
| Co.Area
(µm2) |
5.14E+06±5.57E+05 |
5.09E+06±3.72E+05 |
4.47E+06±4.2E+05a,e |
4.17E+06±4.3E+05a,e |
Mechanical testing
The changes in bone geometry observed as a result of
treatment with etidronate in the Enpp1−/− mice
are likely to alter the biomechanical properties of long bones. In
order to examine this hypothesis, we carried out 3-point bending
analysis of the tibiae. Mechanical testing revealed a significant
decrease in all mechanical parameters examined (stiffness, load at
failure, work to failure, load at fracture, work to fracture, and
yield) except work post-failure, in the Enpp1−/−
mice compared to the WT mice at 22 weeks of age (Table III; P<0.05), reflecting
reduced bone strength and stiffness as we have previously reported
(5). In the WT mice, treatment
with 100 µg/kg etidronate significantly improved work to
fracture and increased work post-failure (Table III; P<0.05, in comparison to
the vehicle-treated WT mice); this suggests that more energy is
required to fracture these etidronate-treated bones in comparison
to the vehicle-treated bones. This significant increase, however,
was not observed in the Enpp1−/−mice treated with
etidronate (Table III).
| Table IIIMeasurements of tibia mechanical
properties in male placebo (vehicle)- and etidronate-treated
Enpp1−/− and WT mice. |
Table III
Measurements of tibia mechanical
properties in male placebo (vehicle)- and etidronate-treated
Enpp1−/− and WT mice.
| WT
|
Enpp1−/−
|
|---|
| Placebo | Etidronate | Placebo | Etidronate |
|---|
| Stiffness
(N/mm) | 44.56±7.08 | 48.82±23.43 | 22.42±7.19a,d | 22.52±6.64a,e |
| Load at failure
(N) | 17.22±2.90 | 15.34±4.13 | 9.21±1.38a,e | 8.89±1.84a,e |
| Work to failure
(J) | 0.0066±0.00066 | 0.0064±0.0019 |
0.0033±0.00081a,e |
0.0035±0.00092a,e |
| Load at fracture
(N) | 1.72±0.29 | 1.53±0.41 | 0.92±0.14a,e | 0.89±0.18a,e |
| Work to fracture
(J) | 0.0069±0.00087 |
0.0086±0.0019b,c |
0.0038±0.0011a,e |
0.0044±0.00082a,e |
| Work post-failure
(J) |
0.00037±0.00041 |
0.0023±0.0014b,d |
0.00055±0.00082 |
0.00088±0.0012a,c |
| Yield (N) | 14.41±3.95 | 12.97±4.42 | 6.96±1.45a,e | 7.11±1.79a,d |
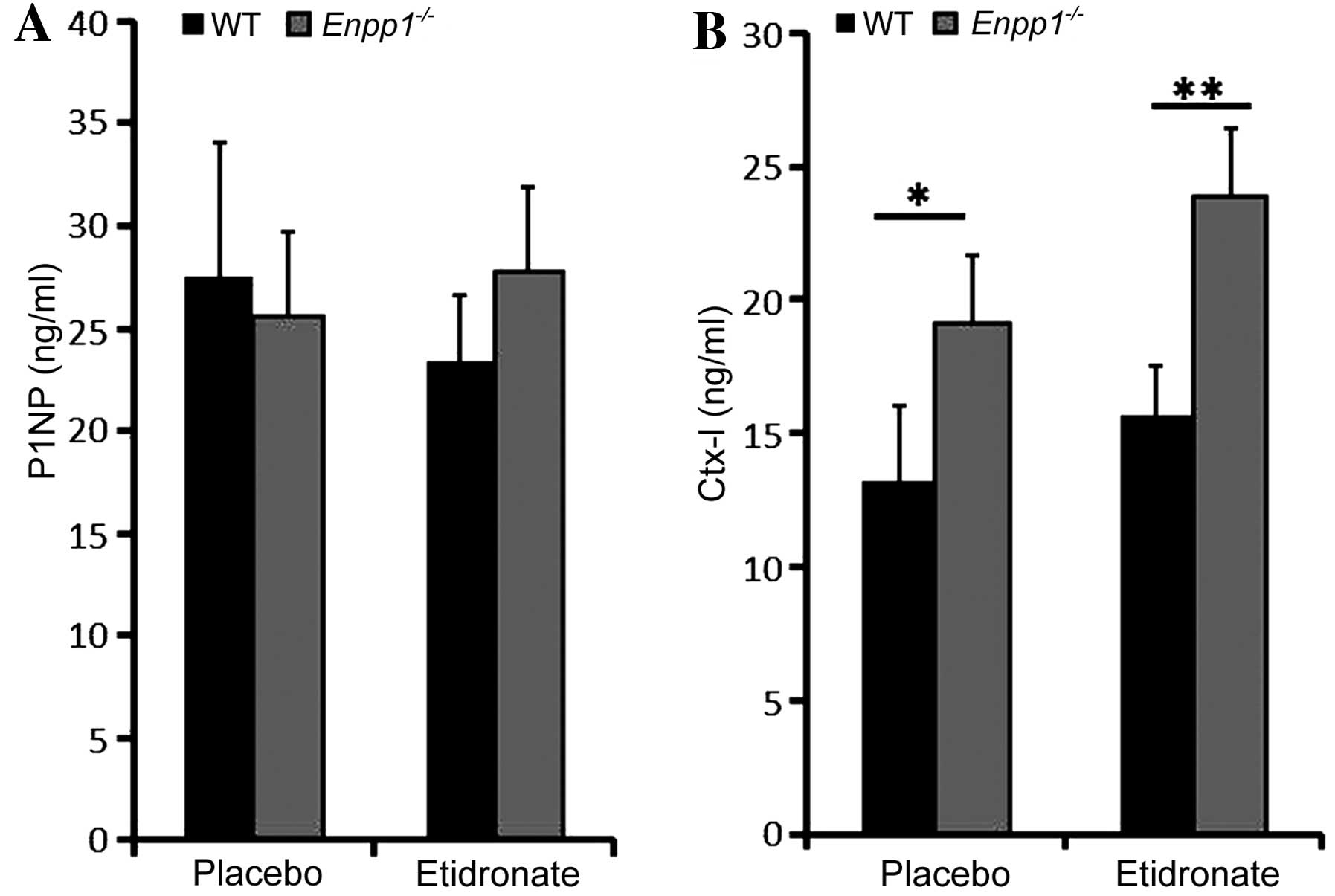
Plasma biochemical markers
The level of osteoblast and osteoclast activity was
assessed by ELISA of serum taken from the etidronate-treated and
vehicle-treated 22-week-old male Enpp1−/− and WT
mice. The plasma concentrations of P1NP, a marker of bone
formation, were unaltered in the Enpp1−/− and WT
mice at 22 weeks of age (Fig.
3A). Moreover, no significant differences in bone formation
were observed upon the administration of etidronate in the WT or
Enpp1−/− mice (Fig.
3A). The plasma concentrations of CTx, a marker of bone
resorption, were increased in the Enpp1−/− mice
in comparison to the WT mice in both the etidronate- and
vehicle-treated mice (P<0.05; Fig.
3B). This is in concordance with our previous observation of
this marker in Enpp1−/− mice (5). However, there were no significant
differences observed between the etidronate- and vehicle-treated
mice in either parameter (Fig.
3).
Discussion
Studies have associated treatment with
bisphosphonates, chiefly etidronate, with improved survival in
patients with GACI, an autosomal recessive disorder of spontaneous
infantile arterial and periarticular calcification which is
attributed to mutations in the ENPP1 gene (38). Animal models have proven to be key
to the understanding of pathological ectopic mineralisation
(4,47). In particular, the
Enpp1−/− mouse is of particular importance in
advancing our understanding of GACI. Thus, the present study was
undertaken to determine the effects of etidronate on
Enpp1−/− mice.
Our data confirm and extend those of our previous
study (5), demonstrating that
tibiae from Enpp1−/− mice have a reduced
trabecular bone mass and cortical thickness in comparison to WT
mice, which explains the altered bone mechanical properties noted
in the present study. This, therefore, is consistent with the
depletion of NPP1 activity reducing extracellular PPi to
abnormally low levels, resulting in insufficient PPi a
substrate for TNAP to generate Pi for normal mineral
formation. In the present study, we also used our novel 3D
µCT protocol to provide evidence of the severe
hypermineralisation of the arteries in Enpp1−/−
mice, consistent with reduced extracellular PPi levels
predisposing the vascular system to ectopic calcification.
Bisphosphonates are typically prescribed for the
treatment of osteoporosis and to reduce fracture risk, preventing
bone loss primarily by the inhibition of osteoclast function
(33). However, there is evidence
to suggest that bisphosphonates impair the anabolic response of
bone to parathyroid hormone (48), inhibiting osteoblast function and
suppressing bone formation (34–37). Furthermore, the
hydrolysis-resistant P-C-P motif of bisphosphonates resembles the
TNSALP susceptible core of PPi, (49) permitting bisphosphonates to impair
calcium phosphate crystallisation (50). First used off-label in the
treatment of Fibrodysplasia ossificans progressiva, the
‘first generation’ bisphosphonate, etidronate, is an analogue of
PPi, and whilst it is currently being investigated as a
treatment for GACI, a number of studies have highlighted a number
of detrimental effects with this approach, including the
development of rickets or osteomalacia during the protracted
administration of etidro-nate (40,41,51–56).
In this study, to clarify the effects of
bisphosphonates on GACI, we treated WT and
Enpp1−/− mice with 100 µg/kg etidro-nate.
In the WT mice, treatment with etidronate had no effect on cortical
or trabecular parameters as determined by µCT analysis.
Despite this, treatment with etidronate significantly improved work
to fracture and increased work post-failure, thus suggesting that
more energy is required to fracture these bones in comparison to
the vehicle-treated bones. The assessment of bone architecture in
Enpp1−/− mice treated with etidronate revealed a
significant decrease in SMI, a method for the determination of the
plate- or rod-like geometry of trabecular structures. This change
in bone geometry did not, however, affect the bone mechanical
properties. Consistent with this, the assessment of plasma markers
of bone formation and resorption revealed that this dosage of
etidronate did not significantly affect the bone remodelling
process in the Enpp1−/− mice, nor in WT mice.
These findings add further support to those of a recent in
vivo study using mice, which also reported no effects of the
administration of etidronate on bone resorption (57). Furthermore, the anti-resorptive
effects of alendronate, risedro-nate and minodronate were revealed
to be 1,000-, 3,300- and 10,000-fold greater than those of
etidronate, respectively (57).
Surprisingly, and in contrast to previous data on
rats with experimental renal failure (40), treatment with etidronate did not
prevent de novo calcification, and did not arrest the
progression of established calcification of the aorta in our
Enpp1−/− mice. Taken together, these data suggest
that the skeleton here is displaying an expected response to
etidronate treatment, but this is not yet toxic to aortic
calcification.
The mild effects of etidronate observed in this
study may be explained by the dosage used, the frequency of
administration and/or species differences. Future studies may aim
to investigate a higher dosage and/or daily administration of
etidronate, as previously it has been shown that high doses of
etidronate significantly reduce mineralisation (58). Furthermore, in the present study,
treatment of the mice commenced when the mice were 11 weeks of age,
the point at which calcification is observed in this model
(5). However, this initiation of
treatment may have been too late to resolve underlying,
pre-existing calcifications and additional investigation into this
would allow further conclusions to be drawn.
In conclusion, despite the changes in bone
microarchitecture, we did not observe an inhibition of aortic
calcification or bone formation in the Enpp1−/−
mice treated with etidronate. Additional studies are, therefore,
required to fully determine whether etidronate is the most
appropriate therapy for the treatment of GACI.
Acknowledgments
The present study was supported by an Institute
Strategic Programme Grant and Institute Career Path Fellowship
funding from the Biotechnology and Biological Sciences Research
Council (BBSRC) (BB/F023928/1).
References
|
1
|
Rutsch F, Ruf N, Vaingankar S, Toliat MR,
Suk A, Höhne W, Schauer G, Lehmann M, Roscioli T, Schnabel D, et
al: Mutations in ENPP1 are associated with ‘idiopathic’ infantile
arterial calcification. Nat Genet. 34:379–381. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rutsch F, Vaingankar S, Johnson K,
Goldfine I, Maddux B, Schauerte P, Kalhoff H, Sano K, Boisvert WA,
Superti-Furga A, et al: PC-1 nucleoside triphosphate
pyrophosphohydrolase defi-ciency in idiopathic infantile arterial
calcification. Am J Pathol. 158:543–554. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Nitschke Y and Rutsch F: Modulators of
networks: Molecular targets of arterial calcification identified in
man and mice. Curr Pharm Des. 20:5839–5852. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mackenzie NC, Huesa C, Rutsch F and MacRae
VE: New insights into NPP1 function: Lessons from clinical and
animal studies. Bone. 51:961–968. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mackenzie NC, Zhu D, Milne EM, van’t Hof
R, Martin A, Darryl Quarles L, Millán JL, Farquharson C and MacRae
VE: Altered bone development and an increase in FGF-23 expression
in Enpp1−/−mice. PLoS One. 7:e321772012. View Article : Google Scholar
|
|
6
|
Apschner A, Huitema LF, Ponsioen B,
Peterson-Maduro J and Schulte-Merker S: Zebrafish enpp1 mutants
exhibit pathological mineralization, mimicking features of
generalized arterial calcification of infancy (GACI) and
pseudoxanthoma elasticum (PXE). Dis Model Mech. 7:811–822. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hajjawi MO, MacRae VE, Huesa C, Boyde A,
Millán JL, Arnett TR and Orriss IR: Mineralisation of collagen rich
soft tissues and osteocyte lacunae in Enpp1−/−mice. Bone.
69:139–147. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Li Q, Guo H, Chou DW, Berndt A, Sundberg
JP and Uitto J: Mutant Enpp1asj mice as a model for generalized
arterial calcification of infancy. Dis Model Mech. 6:1227–1235.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hakim FT, Cranley R, Brown KS, Eanes ED,
Harne L and Oppenheim JJ: Hereditary joint disorder in progressive
ankylosis (ank/ank) mice. I. Association of calcium hydroxyapatite
deposition with inflammatory arthropathy. Arthritis Rheum.
27:1411–1420. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Terkeltaub R, Rosenbach M, Fong F and
Goding J: Causal link between nucleotide pyrophosphohydrolase
overactivity and increased intracellular inorganic pyrophosphate
generation demonstrated by transfection of cultured fibroblasts and
osteoblasts with plasma cell membrane glycoprotein-1. Relevance to
calcium pyrophosphate dihydrate deposition disease. Arthritis
Rheum. 37:934–941. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Addison WN, Azari F, Sørensen ES,
Kaartinen MT and McKee MD: Pyrophosphate inhibits mineralization of
osteoblast cultures by binding to mineral, up-regulating
osteopontin, and inhibiting alkaline phosphatase activity. J Biol
Chem. 282:15872–15883. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Staines KA, MacRae VE and Farquharson C:
The importance of the SIBLING family of proteins on skeletal
mineralisation and bone remodelling. J Endocrinol. 214:241–255.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Anderson HC: Molecular biology of matrix
vesicles. Clin Orthop Relat Res. (314): 266–280. 1995.PubMed/NCBI
|
|
14
|
Moss DW, Eaton RH, Smith JK and Whitby LG:
Association of inorganic-pyrophosphatase activity with human
alkaline-phosphatase preparations. Biochem J. 102:53–57.
1967.PubMed/NCBI
|
|
15
|
Majeska RJ and Wuthier RE: Studies on
matrix vesicles isolated from chick epiphyseal cartilage.
Association of pyrophosphatase and ATPase activities with alkaline
phosphatase. Biochim Biophys Acta. 391:51–60. 1975. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hessle L, Johnson KA, Anderson HC,
Narisawa S, Sali A, Goding JW, Terkeltaub R and Millan JL:
Tissue-nonspecific alkaline phosphatase and plasma cell membrane
glycoprotein-1 are central antagonistic regulators of bone
mineralization. Proc Natl Acad Sci USA. 99:9445–9449. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Murshed M, Schinke T, McKee MD and
Karsenty G: Extracellular matrix mineralization is regulated
locally; different roles of two glacontaining proteins. J Cell
Biol. 165:625–630. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Macrae VE, Davey MG, McTeir L, Narisawa S,
Yadav MC, Millan JL and Farquharson C: Inhibition of PHOSPHO1
activity results in impaired skeletal mineralization during limb
development of the chick. Bone. 46:1146–1155. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Roberts S, Narisawa S, Harmey D, Millán JL
and Farquharson C: Functional involvement of PHOSPHO1 in matrix
vesicle-mediated skeletal mineralization. J Bone Miner Res.
22:617–627. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Roberts SJ, Owen HC and Farquharson C:
Identification of a novel splice variant of the haloacid
dehalogenase: PHOSPHO1. Biochem Biophys Res Commun. 371:872–876.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Stewart AJ, Roberts SJ, Seawright E, Davey
MG, Fleming RH and Farquharson C: The presence of PHOSPHO1 in
matrix vesicles and its developmental expression prior to skeletal
mineralization. Bone. 39:1000–1007. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yadav MC, Simão AM, Narisawa S, Huesa C,
McKee MD, Farquharson C and Millán JL: Loss of skeletal
mineralization by the simultaneous ablation of PHOSPHO1 and
alkaline phosphatase function: A unified model of the mechanisms of
initiation of skeletal calcification. J Bone Miner Res. 26:286–297.
2011. View Article : Google Scholar
|
|
23
|
Narisawa S, Harmey D, Yadav MC, O’Neill
WC, Hoylaerts MF and Millán JL: Novel inhibitors of alkaline
phosphatase suppress vascular smooth muscle cell calcification. J
Bone Miner Res. 22:1700–1710. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sakamoto M, Hosoda Y, Kojimahara K,
Yamazaki T and Yoshimura Y: Arthritis and ankylosis in twy mice
with hereditary multiple osteochondral lesions: With special
reference to calcium deposition. Pathol Int. 44:420–427. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Okawa A, Goto S and Moriya H: Calcitonin
simultaneously regulates both periosteal hyperostosis and
trabecular osteopenia in the spinal hyperostotic mouse (twy/twy) in
vivo. Calcif Tissue Int. 64:239–247. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Okawa A, Nakamura I, Goto S, Moriya H,
Nakamura Y and Ikegawa S: Mutation in Npps in a mouse model of
ossification of the posterior longitudinal ligament of the spine.
Nat Genet. 19:271–273. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Baba H, Furusawa N, Fukuda M, Maezawa Y,
Imura S, Kawahara N, Nakahashi K and Tomita K: Potential role of
streptozotocin in enhancing ossification of the posterior
longitudinal ligament of the cervical spine in the hereditary
spinal hyperostotic mouse (twy/twy). Eur J Histochem. 41:191–202.
1997.PubMed/NCBI
|
|
28
|
Furusawa N, Baba H, Imura S and Fukuda M:
Characteristics and mechanism of the ossification of posterior
longitudinal ligament in the tip-toe walking Yoshimura (twy) mouse.
Eur J Histochem. 40:199–210. 1996.PubMed/NCBI
|
|
29
|
Sali A, Favaloro J, Terkeltaub R and
Goding J: Germline deletion of the nucleoside triphosphate
pyrophosphohydrolase (NTPPPH) plasma cell membrane glycoprotein-1
(PC-1) produces abnormal calcification of periarticular tissues.
Ecto-ATPases and Related Ectoenzymes. Vanduffel L and Lemmems R:
Shaker Publishing BV; Maastricht, The Netherlands: pp. 267–282.
1999
|
|
30
|
Harmey D, Hessle L, Narisawa S, Johnson
KA, Terkeltaub R and Millán JL: Concerted regulation of inorganic
pyrophosphate and osteopontin by akp2, enpp1, and ank: An
integrated model of the pathogenesis of mineralization disorders.
Am J Pathol. 164:1199–1209. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Anderson HC, Harmey D, Camacho NP,
Garimella R, Sipe JB, Tague S, Bi X, Johnson K, Terkeltaub R and
Millán JL: Sustained osteomalacia of long bones despite major
improvement in other hypophosphatasia-related mineral deficits in
tissue nonspecific alkaline phosphatase/nucleotide pyrophosphatase
phosphodi-esterase 1 double-deficient mice. Am J Pathol.
166:1711–1720. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Johnson K, Goding J, Van Etten D, Sali A,
Hu SI, Farley D, Krug H, Hessle L, Millán JL and Terkeltaub R:
Linked deficiencies in extracellular PPi and osteopontin
mediate pathologic calcification associated with defective PC-1 and
ANK expression. J Bone Miner Res. 18:994–1004. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Russell RG: Bisphosphonates: From bench to
bedside. Ann NY Acad Sci. 1068:367–401. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Orriss IR, Key ML, Colston KW and Arnett
TR: Inhibition of osteoblast function in vitro by
aminobisphosphonates. J Cell Biochem. 106:109–118. 2009. View Article : Google Scholar
|
|
35
|
Idris AI, Rojas J, Greig IR, Van’t Hof RJ
and Ralston SH: Aminobisphosphonates cause osteoblast apoptosis and
inhibit bone nodule formation in vitro. Calcif Tissue Int.
82:191–201. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Iwata K, Li J, Follet H, Phipps RJ and
Burr DB: Bisphosphonates suppress periosteal osteoblast activity
independently of resorption in rat femur and tibia. Bone.
39:1053–1058. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Tobias JH, Chow JW and Chambers TJ:
3-Amino-1-hydroxypropylidine-1-bisphosphonate (AHPrBP) suppresses
not only the induction of new, but also the persistence of existing
bone-forming surfaces in rat cancellous bone. Bone. 14:619–623.
1993. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Rutsch F, Böyer P, Nitschke Y, Ruf N,
Lorenz-Depierieux B, Wittkampf T, Weissen-Plenz G, Fischer RJ,
Mughal Z, Gregory JW, et al: GACI Study Group: Hypophosphatemia,
hyperphosphaturia, and bisphosphonate treatment are associated with
survival beyond infancy in generalized arterial calcification of
infancy. Circ Cardiovasc Genet. 1:133–140. 2008. View Article : Google Scholar
|
|
39
|
Chong CR and Hutchins GM: Idiopathic
infantile arterial calcification: The spectrum of clinical
presentations. Pediatr Dev Pathol. 11:405–415. 2008. View Article : Google Scholar
|
|
40
|
Lomashvili KA, Monier-Faugere MC, Wang X,
Malluche HH and O’Neill WC: Effect of bisphosphonates on vascular
calcification and bone metabolism in experimental renal failure.
Kidney Int. 75:617–625. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Otero JE, Gottesman GS, McAlister WH, Mumm
S, Madson KL, Kiffer-Moreira T, Sheen C, Millán JL, Ericson KL and
Whyte MP: Severe skeletal toxicity from protracted etidronate
therapy for generalized arterial calcification of infancy. J Bone
Miner Res. 28:419–430. 2013. View Article : Google Scholar
|
|
42
|
Huesa C, Zhu D, Glover JD, Ferron M,
Karsenty G, Milne EM, Millan JL, Ahmed SF, Farquharson C, Morton
NM, et al: Deficiency of the bone mineralization inhibitor NPP1
protects mice against obesity and diabetes. Dis Model Mech.
7:1341–1350. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Sugiyama T, Meakin LB, Galea GL, Jackson
BF, Lanyon LE, Ebetino FH, Russell RG and Price JS: Risedronate
does not reduce mechanical loading-related increases in cortical
and trabecular bone mass in mice. Bone. 49:133–139. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Huesa C, Millán JL, van’t Hof RJ and
MacRae VE: A new method for the quantification of aortic
calcification by three-dimensional micro-computed tomography. Int J
Mol Med. 32:1047–1050. 2013.PubMed/NCBI
|
|
45
|
Huesa C, Yadav MC, Finnilä MA, Goodyear
SR, Robins SP, Tanner KE, Aspden RM, Millán JL and Farquharson C:
PHOSPHO1 is essential for mechanically competent mineralization and
the avoidance of spontaneous fractures. Bone. 48:1066–1074. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Hildebrand T and Ruegsegger P: A new
method for the model-independent assessment of thickness in
three-dimensional images. J Microscopy (Oxford). 185:67–75. 1997.
View Article : Google Scholar
|
|
47
|
Li Q and Uitto J:
Mineralization/anti-mineralization networks in the skin and
vascular connective tissues. Am J Pathol. 183:10–18. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Black DM, Greenspan SL, Ensrud KE, Palermo
L, McGowan JA, Lang TF, Garnero P, Bouxsein ML, Bilezikian JP and
Rosen CJ: PaTH Study Investigators: The effects of parathyroid
hormone and alendronate alone or in combination in postmenopausal
osteoporosis. N Engl J Med. 349:1207–1215. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Fleisch H, Russell RG and Straumann F:
Effect of pyrophosphate on hydroxyapatite and its implications in
calcium homeostasis. Nature. 212:901–903. 1966. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Felix R, Herrmann W and Fleisch H:
Stimulation of precipitation of calcium phosphate by matrix
vesicles. Biochem J. 170:681–691. 1978.PubMed/NCBI
|
|
51
|
Thiaville A, Smets A, Clercx A and
Perlmutter N: Idiopathic infantile arterial calcification: A
surviving patient with renal artery stenosis. Pediatr Radiol.
24:506–508. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Van Dyck M, Proesmans W, Van Hollebeke E,
Marchal G and Moerman P: Idiopathic infantile arterial
calcification with cardiac, renal and central nervous system
involvement. Eur J Pediatr. 148:374–377. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Thomas T, Lafage MH and Alexandre C:
Atypical osteomalacia after 2 year etidronate intermittent cyclic
administration in osteoporosis. J Rheumatol. 22:2183–2185.
1995.PubMed/NCBI
|
|
54
|
Silverman SL, Hurvitz EA, Nelson VS and
Chiodo A: Rachitic syndrome after disodium etidronate therapy in an
adolescent. Arch Phys Med Rehabil. 75:118–120. 1994.PubMed/NCBI
|
|
55
|
Russell RG, Smith R, Preston C, Walton RJ
and Woods CG: Diphosphonates in Paget’s disease. Lancet. 1:894–898.
1974.PubMed/NCBI
|
|
56
|
Smith R, Russell RG and Woods CG: Myositis
ossificans progressiva. Clinical features of eight patients and
their response to treatment. J Bone Joint Surg Br. 58:48–57.
1976.PubMed/NCBI
|
|
57
|
Kim S, Seiryu M, Okada S, Kuroishi T,
Takano-Yamamoto T, Sugawara S and Endo Y: Analgesic effects of the
non-nitrogen-containing bisphosphonates etidronate and clodronate,
independent of anti-resorptive effects on bone. Eur J Pharmacol.
699:14–22. 2013. View Article : Google Scholar
|
|
58
|
Li Q, Sundberg JP, Levine MA, Terry SF and
Uitto J: The effects of bisphosphonates on ectopic soft tissue
mineralization caused by mutations in the ABCC6gene. Cell Cycle.
14:1082–1089. 2015. View Article : Google Scholar
|