Introduction
Pulmonary arterial hypertension (PAH) ranks as a
fatal disease with high morbidity and mortality, and is clinically
identified by detecting the mean pulmonary artery pressure (PAP) at
>25 mmHg at rest or >30 mmHg with exercise. PAH is now widely
accepted as a progressive disorder, which is characterized by a
marked increase in pulmonary arterial pressure, vascular remodeling
and right ventricular hypertrophy, leading to shortness of breath,
dizziness, fainting, leg swelling and other symptoms (1–3).
In spite of the number of studies available, the underlying
mechanisms of the pathological progression of PAH have largely
remained elusive.
Pulmonary hypertension often occurs either as a
primary or secondary disease following cardiac or pulmonary
diseases. Accumulating evidence has shown that the histological
lung biopsies in PAH patients display critical vascular remodeling,
including smooth muscle cell proliferation with medial hypertrophy
and endothelial cell proliferation with subsequent intimal
thickening (4,5). Endothelial cell proliferation and
smooth muscle cell (SMC) migration lead to the formation of
so-called plexiform lesions, which are characteristics of primary
and secondary pulmonary hypertension (1,6).
The hyperplasia of pulmonary artery smooth muscle cells (PASMCs)
has been demonstrated in monocrotaline (MCT)-induced PAH rats, in
which pre-conditioning with fluoxetine significantly inhibited
PASMC proliferation and subsequent PH development (7). In addition, abnormal endothelial
cell growth in the lungs of PH patients and hypoxia-induced PAH
models has been observed, which is therefore a characteristic of
PAH (6,8).
Autophagy is an evolutionarily conserved lysosomal
degradation pathway and is involved in various vascular diseases,
including atherosclerosis, heart failure and ischemia/re-perfusion
injury (9–11). Recently, the pivotal roles of
autophagy in pulmonary diseases, including acute lung injury,
pulmonary hypertension and chronic obstructive pulmonary disease,
have drawn increasing interest (12). It has been reported that apelin
overexpression significantly inhibited PASMC proliferation and
migration by suppressing the autophagic pathway, indicating a
potential role of autophagy in PAH (13). Furthermore, impaired angiogenesis
has been corroborated in fetal lambs with persistent PH; however,
blocking of autophagy enhanced angiogenesis, implying a possible
beneficial function of autophagy in PAH progression (14). Numerous studies have demonstrated
a critical role of mammalian target of rapamycin (mTOR) in
regulating the autophagic pathway (15,16). mTOR is known as a well-conserved
serine/threonine kinase that exerts a pivotal function in the
signaling network controlling cell metabolism, growth,
proliferation and survival in response to various environmental
factors. Recently, mTOR pathways were proved to be involved in the
apelin-induced inhibitory effect of autophagy on PASMC
proliferation (13). Even though
numerous studies have validated the negative effect of mTOR on the
autophagic pathway, its effect on PAH has remained elusive.
The present study examined the expression of mTOR in
lung tissues from PAH patients. Furthermore, the function of mTOR
in PAH triggered by chronic hypoxia was explored, and the
underlying mechanism was investigated.
Materials and methods
Reagents
Rabbit anti-human mTOR monoclonal antibody (#2972)
was obtained from Cell Signaling Technology, Inc., (Danvers, MA,
USA). Rabbit anti-mouse mTOR polyclonal antibody (ab2732) was
purchased from Abcam (Cambridge, MA, USA). The polyclonal antibody
against mouse light chain (LC)3 (NB100-2220) was from NOVUS
(Littleton, CO, USA). Rabbit polyclonal anti-human LC3 (ab128025),
p62 (ab91526) and -autophagy-related 5 (ATG5; ab78073) were from
Abcam. Rabbit anti-human p62 antibodies (sc-25730) were from Santa
Cruz Biotechnology (Dallas, TX, USA).
Specimen collections
The present study comprised 13 patients (aged 28–60
years) with PAH and six control subjects (aged 27–62 years). All
human lung tissues were from volunteers with PAH and were processed
according to the recommendations of the Second Affiliated Hospital
of Medical College, Xi’an Jiaotong University (Xi’an, China)
(17). Normal lungs were obtained
from 6 patients who died from traumatic injury unrelated to the
lung. The study was conducted in compliance with the Helsinki
Declaration and all patients gave written informed consent. All
specimens were preserved in liquid nitrogen for subsequent
experiments.
Construction of the recombinant
adenoviral vector
The mouse mTOR cDNA fragments were subcloned into
the p adenovirus (Ad) Track-cytomegalovirus (CMV) vector expressing
green fluorescent protein (GFP) to construct the recombinant vector
pAdTrack-CMV-mTOR-GFP.
The recombinant shuttle plasmids pAdTrack-CMV and
pAdEasy-1 were then homogeneously re-combined in the Escherichia
coli strain BJ5183 (Stratagene, La Jolla, CA, USA). The
obtained recombinant plasmids were then transfected into 293 cells
(ATCC, Rockville, MD, USA) to produce the recombinant adenovirus.
Following amplification and purification, the p24 ELISA kit (Cell
Biolabs, Inc., San Diego, CA, USA) was used to determine the virus
titers and vectors were then stored at −80°C until use.
Animal model of pulmonary
hypertension
For experiments requiring animal use, thirty
wild-type C57BL/6 mice (nine weeks old; male; weighing 20–25 g)
were used. All animal experiments were performed with the approval
from the Institutional Animal Care and Use Committee of the Second
Affiliated Hospital of Medical College, Xi’an Jiaotong University
(Xi’an, China). All animals were housed under a controlled 12-h
light/dark cycle and temperature conditions, with free access to
water and chow. The mice were injected with the virus via their
tail veins. For each injection, ~0.2-ml viral suspension with a
titer of 1×107 IU/ml was administered every other day.
Mice were then exposed to hypoxia (10% O2) or normoxia
in a chamber for three weeks. The mice were then anaesthetized with
sodium pentobarbital (50 mg/kg) (Beyotime Biotech, Shanghai, China)
for lung tissue harvesting. Western blot analysis was performed to
determine mTOR levels and autophagy.
Hemodynamic studies and right ventricular
hypertrophy
Three weeks following injection with the vector, all
animals were anesthetized with sodium pentobarbital (60 mg/kg) for
hemodynamic assessment. The body weight (BW) of mice was recorded,
and a 23G needle was then inserted into the right ventricle for
measuring the right ventricle (RV) pressure. For RV hypertrophy
analysis, the heart was removed. The ratio of right ventricle
weight to body weight (RVW/BW) or by the ratio of RVW to combined
left ventricle and septum weight [RV/(LV + Sep)] was detected to
assess the degree of RV hypertrophy. In addition, the wall
thickness in all groups of animals was also measured and calculated
as: Wall thickness (%) = (areaext -
areaint)/areaext ×100. The areaext
represents the external diameter and the areaint
signifies the internal diameter of each vessel. All parameters were
analyzed using PVAN 3.5 software (Millar Instruments, Houston, TX,
USA).
Cell culture
Human pulmonary artery endothelial cells (PAECs)
were purchased from Lonza (Basel, Switzerland). Passages from PAEC
5–8 were grown to ~80% confluence in endothelial cell growth
medium-2 (EGM-2; Lonza Walkersville Inc., Walkersville, MD, USA)
containing EGM-2 SingleQuots™ (Lonza) and 2% fetal bovine serum
(FBS; (Lonza). Cells were then transfected with 2 nmol/l Ad-GFP or
Ad-mTOR vector using the DharmaFECT transfection reagent
(Dharmacon, Lafayette, CO, USA), followed by exposure to hypoxia
(10% O2) or normoxia for 72 h. All cells were maintained
at 37°C.
Small interfering (si)RNA
transfection
For specifically silencing ATG5 expression, the
targeted siRNA fragments of ATG5 were designed and obtained from
Qiagen (Hilden, Germany) (NM_004849). The scrambled siRNA (NC) was
obtained form Santa Cruz Biotechnology. For transfection, cells
were seeded into 24-well micro-plates to reach 40–50% confluence
with a density of 1×105 cells/well. Subsequently, 2
µg/ml ATG5 siRNA or scrambled siRNA were transfected into
cells together with 1 ml Lipofectamine™ RNAi-MAX (Invitrogen,
Carlsbad, CA, USA) using the GeneSilencer® siRNA
transfection reagent (Gene Therapy Systems, Inc., San Diego, CA,
USA). Twenty-four hours later, the transfection efficiency was
assessed by western blot analysis.
Reverse transcription quantitative
polymerase chain reaction (RT-PCR)
Total RNA from the above tissue samples was
extracted with TRIzol reagent (Sigma-Aldrich, St Louis, MO, USA).
Subsequently, RT was performed using ImProm II
reverse-transcriptase (Promega, Madison, WI, USA) with oligo-dT
priming to obtain the cDNA. To evaluate the mRNA levels of mTOR in
tissues, the obtained cDNA was used as a template to perform PCR
amplification with the SYBR Premix Ex Taq™ II kit (Takara, Shiga,
Japan) using an ABI PRISM Sequence Detector System 7500 (Applied
Biosystems, Foster City, CA, USA). The specific primers for mTOR
were 5′-ATTTGATCAGGTGTGCCAGT-3′ (forward sequence) and
5′-GCTTAGGACATGGTTCATGG-3′ (reverse sequence). Each 20-µl
reaction system was comprised of 2 µl cDNA, 10 µl
SYBR® Premix EX Taq™ II and 10 µmol/l of forward
and reverse primers. The PCR program was 95°C for 10 min, followed
by 38 PCR cycles (95°C for 10 sec, 56°C for 40 sec, 72°C for 30
sec) and a final extension for 5 min at 72°C, followed by a
standard melting curve analysis. Amplification reactions were
performed in triplicate for each sample and the results were
normalized to β-actin (11). All
primers were obtained from GenePharma (Shanghai, China). The
2−ΔΔCt method was used to quantify results (11).
Western blot analysis
Following lysis with radioimmunoprecipitation assay
lysis buffer (100 mM NaCl, 50 mM Tris-HCl pH 7.5, 1% Triton X-100,
1 mM EDTA, 10 mM β-glycerophosphate, 2 mM sodium vanadate and
protease inhibitor; Beyotime Biotech), the protein concentration in
the above samples was measured using the Pierce™ BCA Protein Assay
Kit (23227; Pierce, Rockford, IL, USA). Approximately 100 µl
protein (25 µg/lane) was subjected to 12% SDS-PAGE, followed
by the transfer onto a polyvinylidene difluoride membrane
(Millipore, Bedford, MA, USA) in a semi-dry trans-blot apparatus.
Then, 5% non-fat dry milk in phosphate-buffered saline was added to
block the non-specific binding at 4°C overnight. The membranes were
then incubated with primary antibodies against mTOR (1:1,000),
light chain LC3 I, LC3 II (1:500), p62 (1:200) and ATG5 (1:500) for
1 h at room temperature. Following three washes with Tris-buffered
saline containing Tween 20, horseradish peroxidase (HRP)-conjugated
secondary antibodies were introduced for 1 h. LumiGLo reagent
(Pierce) was added to detect the bound antibodies. The images were
obtained on Kodak film and quantified using ImageJ software version
1.46 (National Institutes of Health, Bethesda, MD, USA).
Cell proliferation assay
To assess cell proliferation, an MTT assay was
performed. Briefly, cells were seeded into 96-well plates at a
density of 1×105 cells/well. Following pre-conditioning
with Ad-mTOR transfection, the culture medium was replaced with
fresh medium containing 500 µg/ml MTT reagent. Following 5
hours of incubation at 37°C, 200 µl isopropanol was added to
dissolve the formazan product. Cell viability was then analyzed by
detecting the absorbance of MTT at 590 nm with a micro-ELISA reader
(3550; Bio-Rad Laboratories, Inc., Hercules, CA, USA). All samples
were performed in triplicate.
Statistical analysis
All assays were performed in triplicate and values
are expressed as the mean ± standard deviation. SPSS 13.0 (SPSS,
Inc., Chicago, IL, USA) was used for statistical analysis.
Student’s t-test was performed using GraphPad InStat Statistics
software (version 1.12; GraphPad, Inc., La Jolla, CA, USA) to
determine statistical significance. P<0.05 was considered to
indicate a statistically significant difference between values.
Results
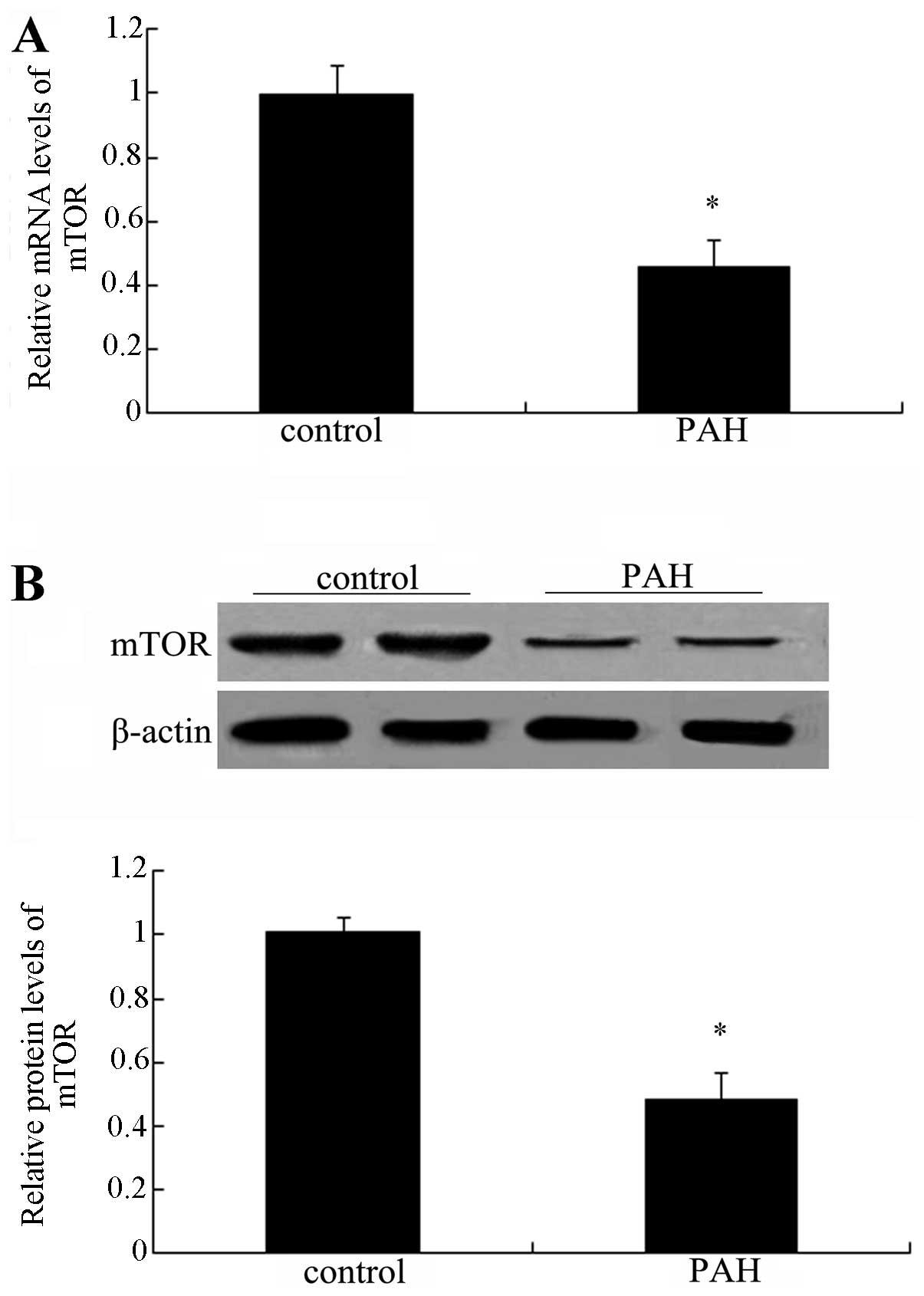
mTOR is downregulated in patients with
PAH
To clarify the association between mTOR and PAH,
lung tissues from normal patients and patients with PAH were
collected and subjected to PCR and western blot analysis of mTOR.
As shown in Fig. 1A, a
significant downregulation of mTOR mRNA levels was observed
compared with those in the normal groups (P<0.05).
Simultaneously, the protein levels of mTOR were also markedly
decreased in patients with PAH (Fig.
1B). Collectively, these results suggested a marked
downregulation of mTOR in patients with PAH, indicating an
association between mTOR and the development of PAH.
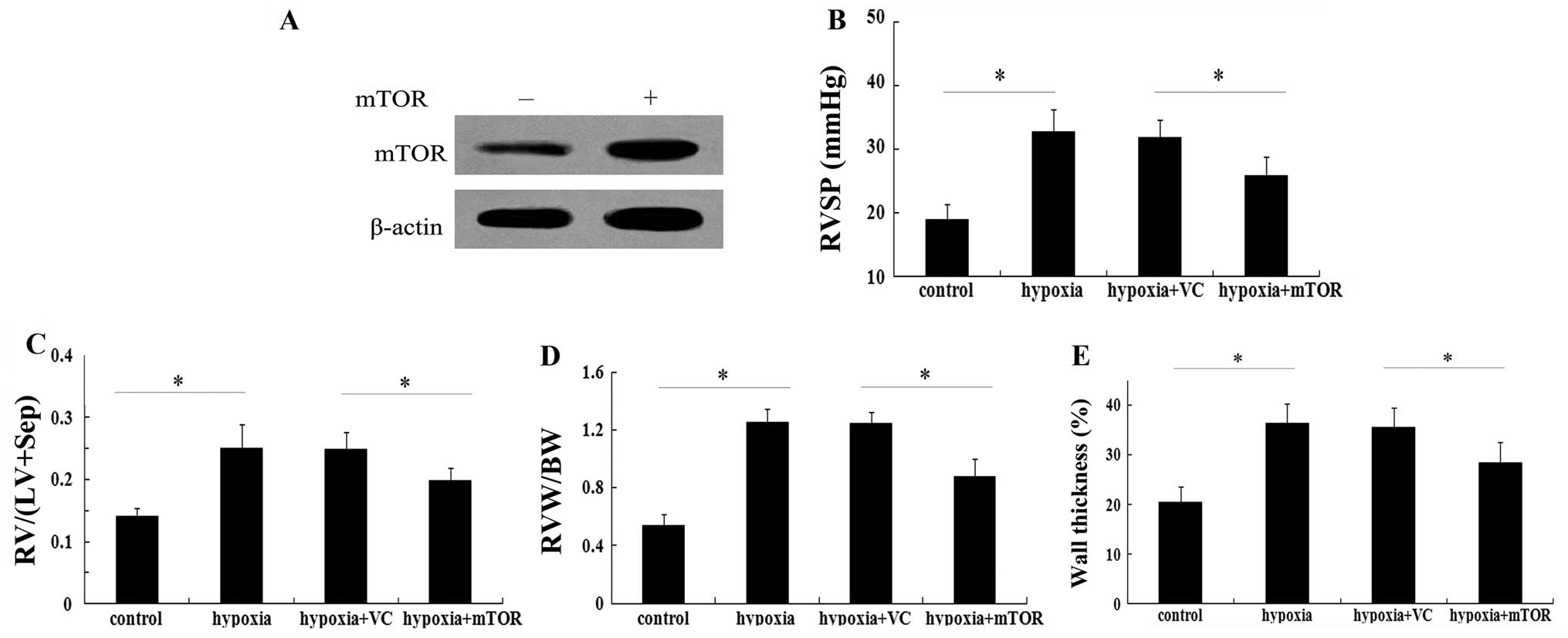
Elevated mTOR expression attenuates
chronic hypoxia-induced pulmonary artery hypertension
Based on the above results, the present study
evaluated the possible function of mTOR in the pathological
progression of PAH in vivo. Following injection of
recombinant Ad-mTOR, elevated mTOR protein levels were detected in
C57BL/6 mice with chronic hypoxia-induced pulmonary artery
hypertension (Fig. 2A). Of note,
mTOR overexpression vector administered for three weeks markedly
inhibited the elevation of right ventricular systolic pressure
(RVSP) in mice exposed to hypoxia (P<0.05; Fig. 2B). In terms of right ventricular
hypertrophy, hypoxia increased the ratio of RV/(LV+Sep) by
~1.8-fold relative to the baseline value, which was markedly
attenuated in the group injected with Ad-mTOR (P<0.05; Fig. 2C). Consistent with the above
results, the increase in the ratio of RVW/BW induced by hypoxia was
significantly reduced in the mTOR overexpression group (P<0.05;
Fig. 2D). Of note, exposure to
hypoxia exaggerated vascular remodeling with an obvious increase in
wall thickness of pulmonary arterioles (36.25±4.10%, compared with
20.51±3.11% in the control group; P<0.05) (Fig. 2E). When mTOR was overexpressed,
this upregulation in wall thickness of pulmonary arterioles was
markedly mitigated (P<0.05). These results suggested a potential
protective effect of mTOR against PAH.
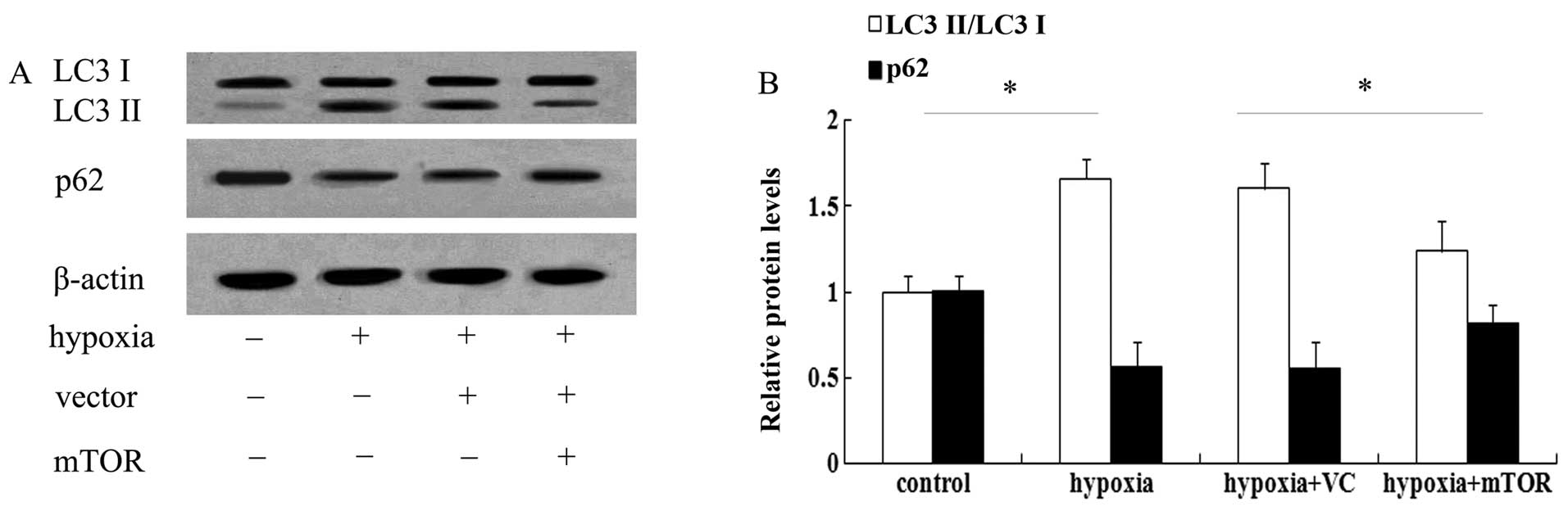
mTOR overexpression abrogates increased
autophagy in mice with PAH
It is widely accepted that mTOR is associated with
autophagy. To test whether the underlying mechanism of the
beneficial effect of mTOR in PAH involves autophagic pathways, LC3
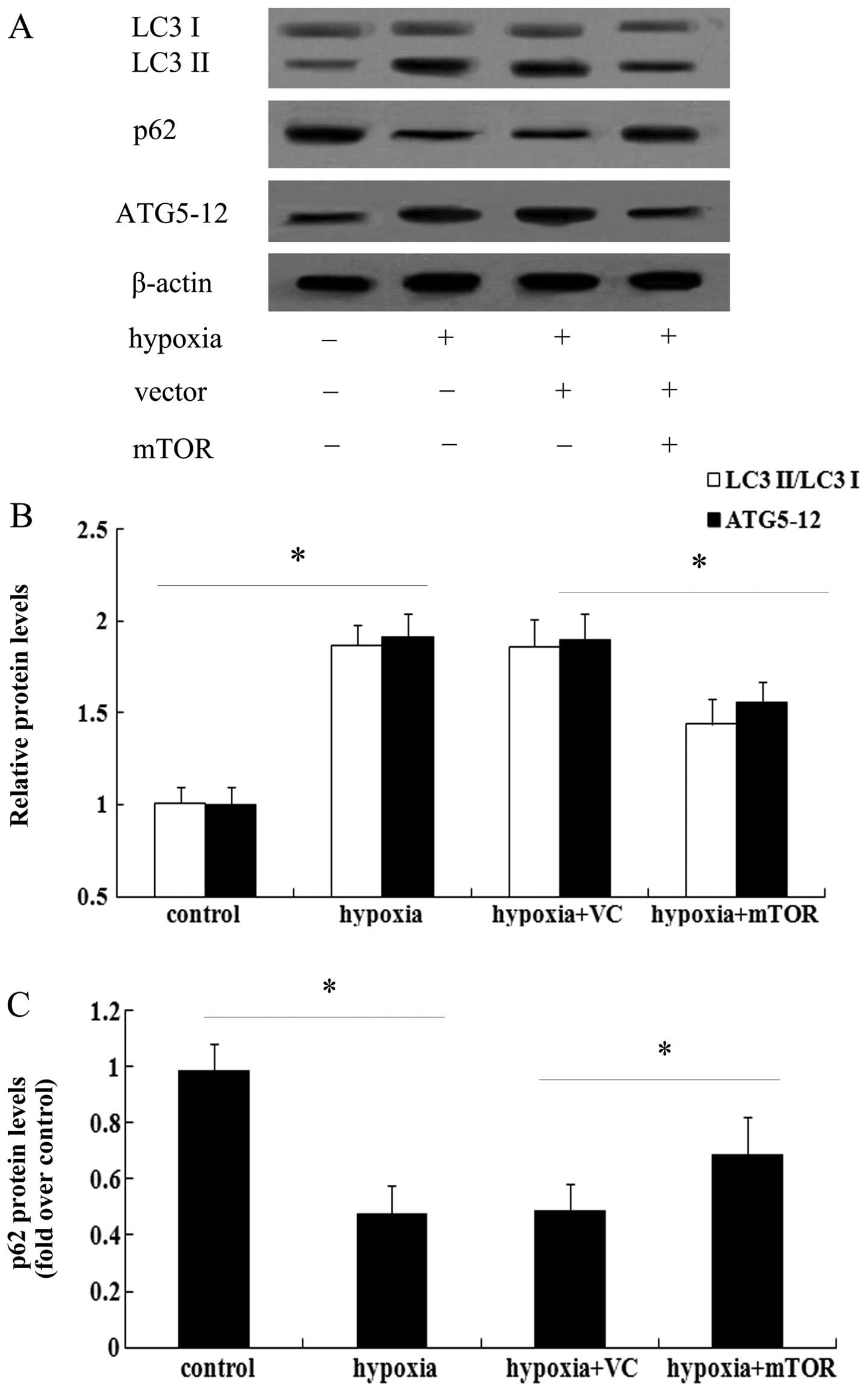
was assessed, which is a known marker for autophagy (17). As shown in Fig. 3A, exposure to hypoxia for three
weeks enhanced the expression of LC3 II and induced a 1.56-fold
increase in the ratio of LC3 II/LC3 I in the lungs of mice with PAH
(Fig. 3B). However, mTOR
overexpression significantly attenuated the increase in LC3 levels
(P<0.05). As a common autophagic substrate, p62 serves as a
useful marker for measuring autophagic flux (11,14). Following exposure to hypoxia, the
expression levels of p62 were obviously abrogated in mice with PAH
(Fig. 3), which was restored in
the mTOR-overexpressing group. The above results suggested that
mTOR may exert a protective effect against PAH by inhibiting the
autophagic pathway.
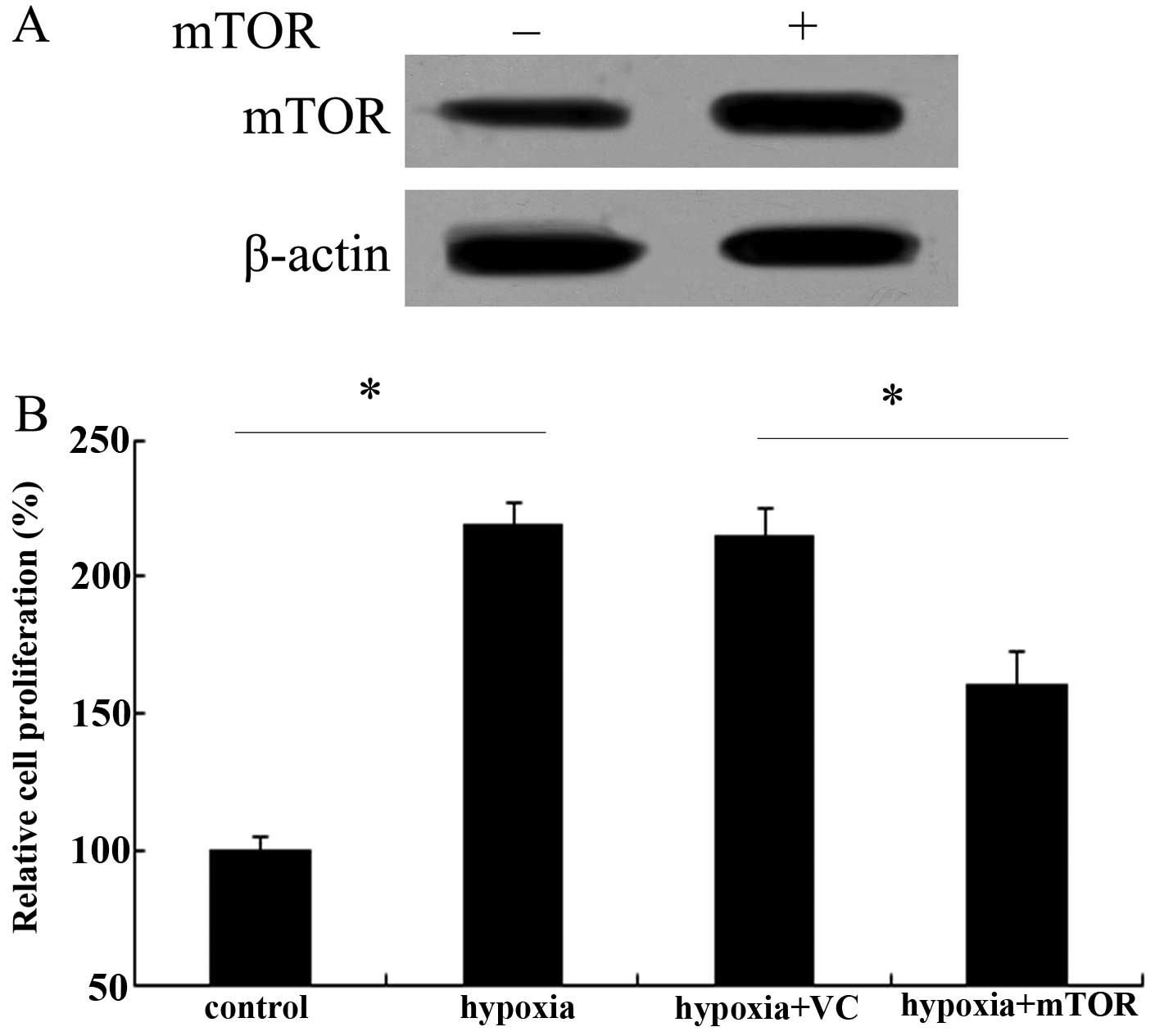
mTOR overexpression impedes hypoxic
proliferation of PAECs
It is well known that abnormal endothelial cell
growth occurs in pulmonary arteries and lungs of patients with PAH
(8). Therefore, the effect of
mTOR on PAEC proliferation was assessed. After transfected with
Ad-mTOR, the expression levels of mTOR protein were markedly
upregulated in PAECs (Fig. 4A).
Following exposure to chronic hypoxia, an ~2.17-fold increase in
cell proliferation ability was demonstrated (Fig. 4B). However, mTOR overexpression
obviously attenuated this increase in cell proliferation triggered
by hypoxia (P<0.05), indicating a critical role of mTOR in the
hypoxic proliferation of PAECs.
mTOR inhibits hypoxia-induced cell
autophagy in PAECs
As the effect of mTOR on hypoxia-induced PAEC
proliferation had been confirmed, it was next sought to elucidate
whether mTOR inhibited autophagy in PAECs. Western blot analysis
showed that hypoxia induced an obvious increase in the LC3 II/LC3 I
ratio, concomitant with the marked upregulation of ATG5-12 levels
(Fig. 5A). Quantification of the
blots revealed that hypoxia triggered ~1.87- and 1.91-fold
increases in LC3 and ATG5-12 levels, respectively (P<0.05),
which were abrogated by mTOR overexpression (P<0.05) (Fig. 5B). Furthermore, an obvious
0.48-fold downregulation of p62 levels was noted in PAECs exposed
to hypoxia (P<0.05), which was partly restored by mTOR
overexpression (P<0.05) (Fig.
5C).
Blocking of autophagy antagonizes cell
proliferation of PAECs
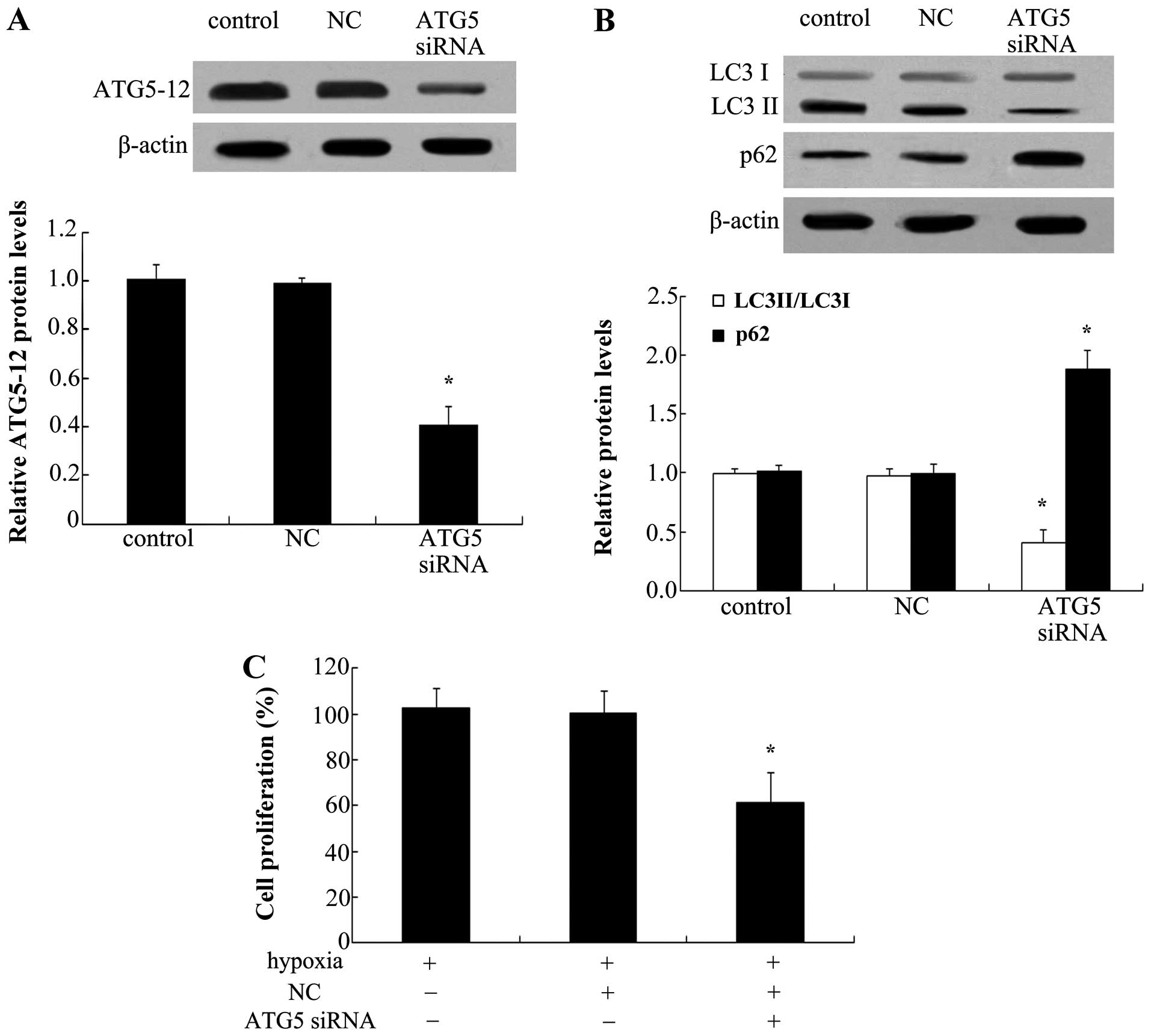
To explore the effect of autophagy on the
proliferation of PAECs, ATG5 siRNA was used to silence the
expression of ATG5-12. As shown in Fig. 6A, ATG5 siRNA transfection markedly
abrogated the protein expression of ATG5-12 in PAECs. Of note, ATG5
silencing reduced hypoxia-induced increases in LC3 II levels
(Fig. 6B). Consistently, a marked
upregulation of p62 protein levels was also observed when blocking
ATG5 levels, indicating that ATG5 knockdown inhibited the cell
autophagy pathway. Furthermore, silencing of ATG5 significantly
reduced hypoxia-induced cell proliferation (P<0.05) (Fig. 6C).
Discussion
Pulmonary hypertension (PH) is a progressive
pulmonary vascular disorder comprising increased proliferation
ability of PASMCs and PAECs, resulting in high morbidity and
mortality (8,18,19). As a well-conserved
serine/threonine kinase, mTOR controls various major cellular
processes and is increasingly being associated with pathological
conditions (20). Recently, mTOR
has attracted broad interest of scientists and clinicians due to
being a potential therapeutic target against a number of diseases
associated with proliferative and metabolic abnormalities,
including atherosclerosis, cancer and neurodegeneration (21,22). However, to date, its function in
PAH has remained elusive. The present study reported an obvious
downregulation of mTOR in lung tissues from patients with PAH,
indicating a critical role of mTOR in the development of PAH.
Pulmonary hypertension has been proven to contribute
to the morbidity and mortality of adult and pediatric patients with
various lung and heart diseases, which are mainly associated with
persistent or intermittent hypoxia (23). The fact that hypoxia can induce
PAH and significant structural remodeling of pulmonary arteries
(PAs) in experimental models has been widely confirmed and will
contribute to disease development (24,25). To investigate the role of mTOR in
PAH, a mouse model of PAH was established in the present study by
exposing the animals to chronic hypoxia. Through simultaneous
injection with a specific adenovector, mTOR-overexpressing mice
with PAH were successfully generated. Following prolonged exposure
to hypoxia, a significant enhancement of the RVSP was observed,
which was obviously attenuated in C57BL/6 mice injected with
Ad-mTOR. Furthermore, mTOR overexpression markedly abrogated right
ventricular hypertrophy as decreased hypoxia-induced increases in
the RV/(LV+Sep) and RVW/BW ratios. Of note, the wall thickness of
pulmonary arterioles was markedly mitigated by overexpression of
mTOR. Therefore, the above results confirmed the critical
protective role of mTOR in the progression of PAH.
Autophagy is a lysosomal pathway which can degrade
intracellular organelles and proteins to maintain cellular
homeostasis (26). The
microtubule-associated protein LC3 is frequently used as a specific
marker to monitor macroautophagy in vitro and in
vivo, and increases in LC3 II indicate the induction of
autophagosome formation (27).
The present study confirmed that autophagy was elevated in PAH,
which was previously observed in monocrotaline-triggered PAH in
rats (28). A recent study
confirmed that inhibition of autophagy was able to prevent
pulmonary hypertension in rats (29). The fact that mTOR is known as a
negative regulator of autophagy has been widely observed (30). To further clarify the underlying
mechanism of the protective effect of mTOR against PAH, the effect
of mTOR on the autophagic pathway was investigated. As expected,
overexpression of mTOR abrogated the expression levels of LC3 II
induced by chronic hypoxia. As a common autophagic substrate, p62
is recognized as a useful marker for measuring autophagic flux
(11,14). In the present study, mTOR
overexpression markedly ameliorated the inhibitory effect of
hypoxia on p62 expression levels. These results indicated that
autophagic pathways are involved in the mTOR-mediated attenuation
of PAH induced by chronic hypoxia.
It has been demonstrated that hypoxia can contribute
to the development and progression of PAH, mainly by de-regulating
endothelial cell functions, including excessive growth of
endothelial cells (31). Abnormal
endothelial cell growth in the lungs and pulmonary arteries of PH
patients has been reported previously (8,32).
In the present study, hypoxia treatment markedly enhanced the
proliferation of endothelial cells. Although a previous study has
suggested that hypoxia stimulation failed to induce pulmonary
artery endothelial cell proliferation (33), other studies have reported results
similar to those of the present study (34,35). The present study manifested that
overexpression of mTOR significantly reduced hypoxia-induced PAEC
proliferation. Of note, stimulation with hypoxia also enhanced the
autophagic pathway as evidenced by the upregulation of LC3 II and
ATG5-12, as well as the decease of p62 expression. ATG5 is an E3
ubiquitin ligase and is required for autophagy due to its role in
autophagosome elongation (36).
Following ATG5 knockdown with its specific siRNA, the autophagic
pathway was obviously blocked. Of note, ATG5 silencing markedly
inhibited hypoxia-induced PAEC proliferation, implying a critical
role of mTOR in hypoxia-induced PAEC proliferation by regulating
the autophagic pathway.
In conclusion, the present study reported an obvious
downregulation of mTOR in PAH induced by chronic hypoxia. In the
present study, high expression levels of mTOR were able to abrogate
the development of hypoxia-triggered PAH by suppressing PAEC
proliferation and through blocking the autophagic pathway.
Accordingly, the present study illustrated the potential protective
effect of mTOR against the development and progression of PAH and
suggested mTOR as a promising therapeutic agent for the future
development of anti-PAH therapies.
Acknowledgments
The present study was supported by the Key Project
of the National Natural Science Foundation of China (no. 81330002)
and the Social Development Technology Project of Shaanxi Province
(no. 2015S099).
Abbreviations:
|
PAH
|
pulmonary arterial hypertension
|
|
mTOR
|
mammalian target of rapamycin
|
|
RVSP
|
right ventricular systolic
pressure
|
|
PAECs
|
pulmonary artery endothelial cell
|
|
PASMCs
|
pulmonary artery smooth muscle
cells
|
|
GFP
|
green fluorescent protein
|
|
PVDF
|
polyvinylidene difluoride
|
|
BW
|
body weight
|
|
RVW
|
right ventricle weight
|
|
LV
|
left ventricle
|
References
|
1
|
Tuder RM, Abman SH, Braun T, et al:
Development and pathology of pulmonary hypertension. J Am Coll
Cardiol. 54:S3–S9. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
McLaughlin VV, Davis M and Cornwell W:
Pulmonary arterial hypertension. Curr Probl Cardiol. 36:461–517.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Voelkel NF, Gomez-Arroyo J, Abbate A,
Bogaard HJ and Nicolls MR: Pathobiology of pulmonary arterial
hypertension and right ventricular failure. Eur Respir J.
40:1555–1565. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tuder RM, Marecki JC, Richter A,
Fijalkowska I and Flores S: Pathology of pulmonary hypertension.
Clin Chest Med. 28:23–42. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rabinovitch M: Molecular pathogenesis of
pulmonary arterial hypertension. J Clin Invest. 118:2372–2379.
2008. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Xu W and Erzurum SC: Endothelial cell
energy metabolism, proliferation, and apoptosis in pulmonary
hypertension. Compr Physiol. 1:357–372. 2011.PubMed/NCBI
|
|
7
|
Guignabert C, Raffestin B, Benferhat R, et
al: Serotonin transporter inhibition prevents and reverses
monocrotaline-induced pulmonary hypertension in rats. Circulation.
111:2812–2819. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Masri FA, Xu W, Comhair SA, et al:
Hyperproliferative apoptosis-resistant endothelial cells in
idiopathic pulmonary arterial hypertension. Am J Physiol Lung Cell
Mol Physiol. 293:L548–L554. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Choi AM, Ryter SW and Levine B: Autophagy
in human health and disease. N Engl J Med. 368:651–662. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Slomovitz BM and Coleman RL: The
PI3K/AKT/mTOR pathway as a therapeutic target in endometrial
cancer. Clin Cancer Res. 18:5856–5864. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang X, Li L, Niu X, et al: mTOR enhances
foam cell formation by suppressing the autophagy pathway. DNA Cell
Biol. 33:198–204. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ryter SW, Nakahira K, Haspel JA and Choi
AM: Autophagy in pulmonary diseases. Annu Rev Physiol. 74:377–401.
2012. View Article : Google Scholar
|
|
13
|
Zhang H, Gong Y, Wang Z, et al: Apelin
inhibits the proliferation and migration of rat PASMCs via the
activation of PI3K/Akt/mTOR signal and the inhibition of autophagy
under hypoxia. J Cell Mol Med. 18:542–553. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mizumura K, Cloonan SM, Haspel JA and Choi
AM: The emerging importance of autophagy in pulmonary diseases.
Chest. 142:1289–1299. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nyfeler B, Bergman P, Wilson CJ and Murphy
LO: Quantitative visualization of autophagy induction by mTOR
inhibitors. Methods Mol Biol. 821:239–250. 2012. View Article : Google Scholar
|
|
16
|
Jung CH, Ro SH, Cao J, Otto NM and Kim DH:
mTOR regulation of autophagy. FEBS Lett. 584:1287–1295. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang X, Li W, Hou Y, Niu Z, Zhong Y,
Zhang Y and Yang S: Comparative membrane proteomic analysis between
lung adenocarcinoma and normal tissue by iTRAQ labeling mass
spectrometry. Am J Transl Res. 6:267–280. 2014.PubMed/NCBI
|
|
18
|
Veyssier-Belot C and Cacoub P: Role of
endothelial and smooth muscle cells in the physiopathology and
treatment management of pulmonary hypertension. Cardiovasc Res.
44:274–282. 1999. View Article : Google Scholar
|
|
19
|
Perros F, Dorfmüller P, Souza R, et al:
Fractalkine-induced smooth muscle cell proliferation in pulmonary
hypertension. Eur Respir J. 29:937–943. 2007. View Article : Google Scholar
|
|
20
|
Laplante M and Sabatini DM: mTOR signaling
in growth control and disease. Cell. 149:274–293. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang X, Li L, Li M, et al: Knockdown of
mTOR by lentivirus-mediated RNA interference suppresses
atherosclerosis and stabilizes plaques via a decrease of
macrophages by autophagy in apolipoprotein E-deficient mice. Int J
Mol Med. 32:1215–1221. 2013.PubMed/NCBI
|
|
22
|
Zoncu R, Efeyan A and Sabatini DM: mTOR:
from growth signal integration to cancer, diabetes and ageing. Nat
Rev Mol Cell Biol. 12:21–35. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Stenmark KR, Fagan KA and Frid MG:
Hypoxia-induced pulmonary vascular remodeling cellular and
molecular mechanisms. Circ Res. 99:675–691. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Orr R, Smith LJ and Cuttica MJ: Pulmonary
hypertension in advanced chronic obstructive pulmonary disease.
Curr Opin Pulm Med. 18:138–143. 2012. View Article : Google Scholar
|
|
25
|
Hoshikawa Y, Ono S, Suzuki S, et al:
Generation of oxidative stress contributes to the development of
pulmonary hypertension induced by hypoxia. J Appl Physiol.
90:1299–1306. 2001.PubMed/NCBI
|
|
26
|
Mizushima N: Autophagy: process and
function. Genes Dev. 21:2861–2873. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kuma A, Matsui M and Mizushima N: LC3, an
autophagosome marker, can be incorporated into protein aggregates
independent of autophagy. Autophagy. 3:323–328. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gomez-Arroyo JG, Farkas L, Alhussaini AA,
et al: The monocrotaline model of pulmonary hypertension in
perspective. Am J Physiol Lung Cell Mol Physiol. 302:L363–L369.
2012. View Article : Google Scholar
|
|
29
|
Long L, Yang X, Southwood M, et al:
Chloroquine prevents progression of experimental pulmonary
hypertension via inhibition of autophagy and lysosomal bone
morphogenetic protein type II receptor degradation. Circ Res.
112:1159–1170. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ravikumar B, Vacher C, Berger Z, et al:
Inhibition of mTOR induces autophagy and reduces toxicity of
polyglutamine expansions in fly and mouse models of Huntington
disease. Nat Genet. 36:585–595. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Schaefer CA, Kuhlmann CRW, Weiterer S, et
al: Statins inhibit hypoxia-induced endothelial proliferation by
preventing calcium-induced ROS formation. Atherosclerosis.
185:290–296. 2006. View Article : Google Scholar
|
|
32
|
Tuder RM, Groves B, Badesch DB and Voelkel
NF: Exuberant endothelial cell growth and elements of inflammation
are present in plexiform lesions of pulmonary hypertension. Am J
Pathol. 144:275–285. 1994.PubMed/NCBI
|
|
33
|
Yu L and Hales CA: Hypoxia does neither
stimulate pulmonary artery endothelial cell proliferation in mice
and rats with pulmonary hypertension and vascular remodeling nor in
human pulmonary artery endothelial cells. J Vasc Res. 48:465–475.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Porter KM, Kang BY, Adesina SE, Murphy TC,
Hart CM and Sutliff RL: Chronic hypoxia promotes pulmonary artery
endothelial cell proliferation through
H2O2-induced 5-lipoxygenase. PloS One.
9:e985322014. View Article : Google Scholar
|
|
35
|
Kang BY, Kleinhenz JM, Murphy TC and Hart
CM: The PPARγ ligand rosiglitazone attenuates hypoxia-induced
endothelin signaling in vitro and in vivo. Am J Physiol Lung Cell
Mol Physiol. 301:L881–L891. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Poon A, Eidelman D, Laprise C and Hamid Q:
ATG5, autophagy and lung function in asthma. Autophagy. 8:694–695.
2012. View Article : Google Scholar : PubMed/NCBI
|