Introduction
Ischemic heart disease (IHD) is the leading cause of
mortality worldwide, particularly in developed countries (1). Mesenchymal stem cells (MSCs) have
been widely applied in regenerative medicine, with significant
beneficial effects on post-infarction heart failure (2,3).
However, the therapeutic potential of MSCs is impeded by their poor
survival rate following transplantation into the harsh
microenvironment of the infarcted myocardium. As shown in previous
studies, the survival rates of human MSCs were <0.44% on day 4
following transplantation in immunodeficient mice (4) and <1% following autologous cell
transplantation in humans (5).
Thus, in order to improve the MSC-mediated beneficial effects on
post-infarction heart failure, research should focus on finding
strategies to increase the survival ability of donor MSCs. In
vivo, the loss of nutritional factors and limited blood supply
to the ischemic region are thought to be the key factors
responsible for the high rate of MSC attrition (6). Correspondingly, hypoxia and serum
deprivation (H/SD) have been designed to mimic the hostile ischemic
microenvironment in vitro (6). Therefore, strategies that enhance
survival under conditions of H/SD are pivotal for improving the
efficacy of MSCs.
Adenosine triphosphate (ATP)-sensitive potassium
(KATP) channels are unique proteins that directly
provide a link associating cellular energetics with electrical
activity (7). KATP
channels include the plasma membrane (sKATP) and the
inner mitochondrial membrane (mitoKATP). Previous
studies have suggested that mitoKATP, rather than
sKATP channels, are possible effectors of
cardioprotection against ischemic injury (8,9).
In addition, the mitoKATP channel has been shown to
exert profound beneficial effects, including lowering of blood
pressure, rectifying hypoglycemia, mimicking ischemic
preconditioning, modifying arrhythmia like J-wave syndrome, atrial
fibrillation and heart failure (10–12).
Nicorandil, a drug with both nitrate-like and
KATP channel-activating properties, is the only
mitoKATP channel opener used in clinical practice for
the treatment of ischemic heart disease (13). Numerous experimental and clinical
studies have reported the protective effects of nicorandil against
myocardial ischemia (14,15), mainly in terms of limiting the
size of the infarct area (16),
reducing the no-reflow phenomenon (17), having anti-atherogenic properties
(18) and inhibiting inflammation
(19). Recently, nicorandil was
reported to reduce the activation of the inflammasome and the
subsequent release of caspase-1, interleukin (IL)-1β and IL-18
(20). However, the pro-survival
effects of nicorandil on MSCs for regeneration purposes have not
been examined thus far. In the present study, we hypothesized that
nicorandil would prevent the apoptosis induced by exposure to H/SD
and would thus improve the survival of MSCs. To confirm this
theory, we examined the effects of nicorandil on the H/SD-induced
apoptosis of MSCs and the related signaling pathways.
Materials and methods
Culture of MSCs
MSCs were isolated from the bone marrow of
Sprague-Dawley (SD) rats (weighing 60–80 g) using a previously
published method with minor modifications (21,22). All the SD rats were obtained from
the Laboratory Animal Science Department, the Second Affiliated
Hospital of Harbin Medical University, Harbin, China. The
axperimental animal procedures were approved by the Local Ethics
Committee for the Care and Use of Laboratory antimals of Harbin
Medical University. Briefly, the femurs and tibias were removed
from the SD rats, and the bone marrow was flushed out using 10 ml
Iscove’s Modified Dulbecco’s medium (IMEM; Gibco, Grand Island, NY,
USA) with 1% penicillin/streptomycin (Beyotime Institute of
Biotechnology, Nantong, China). The cells were centrifuged at 300 x
g for 5 min. The resulting cell pellets were resuspended in 6 ml
IMEM supplemented with 10% fetal bovine serum (Gibco) and 1%
penicillin/streptomycin and plated in a 25 cm2 plastic
flask at 37°C in a humidified atmosphere containing 5%
CO2 to allow the adherence of the MSCs. After 3 days,
the medium was changed, and the non-adherent cells were removed.
The medium was replaced every 3 days. Approximately 8–10 days after
seeding, the cells became 80–90% confluent. The adherent cells were
released from the dishes using 0.25% trypsin (Beyotime Institute of
Biotechnology) and expanded at a 1:2 or 1:3 dilution. All
subsequent experiments were performed using MSCs of passages
3–5.
For the identification of the MSC phenotype, the
cells were harvested, washed with phosphate-buffered saline (PBS)
and labeled with the following conjugated antibodies: FITC-labeled
anti-CD44 (BD Pharmingen-550974; BD Biosciences, Franklin Lakes,
NJ, USA), anti-CD45 (eBioscience-11-0461; eBioscience, San Diego,
CA, USA), anti-CD29 (BD Pharmingen-555005; BD Biosciences),
anti-CD34 (sc-7324; Santa Cruz Biotechnology, Inc., Dallas, TX,
USA) and phycoerythrin-labeled anti-CD90 (BD Pharmingen-551401; BD
Biosciences). The labeled cells were analyzed by flow cytometry and
FACSDiva Pro Software (Becton-Dickinson, San Jose, CA, USA).
Treatment of MSCs
The induction of apoptosis in vitro by H/SD,
designed to mimic the in vivo conditions of the ischemic
myocardium, was initiated as previously described by Zhu et
al (6). In terms of the
experimental design, the MSCs were washed with PBS, exposed to
various concentrations (0, 10, 100, 500 and 1,000 μM) of
nicorandil (Sigma, St. Louis, MO, USA) in serum-free medium and
incubated in a glove box (855-AC; Plas-Labs Inc., Lansing, MI, USA)
to scavenge free oxygen at 37°C. Cells cultured in complete medium
alone were used as the non-ischemic controls.
To investigate the mechanism of MSC apoptosis
further, the phosphoinositide 3-kinase (PI3K) inhibitor, LY294002
(25 μM; Cell Signaling Technology, Danvers, MA, USA), or the
reactive oxygen species (ROS) scavenger, N-acetyl-L-cysteine (NAC)
(500 μM; Sigma-Aldrich, St. Louis, MO, USA), were added 1 h
prior to treatment with nicorandil.
Measurement of apoptosis and ROS
levels
Cell death was assessed using the Annexin
V-FITC/propidium iodide (PI) Apoptosis Detection kit (BD
Biosciences). In accordance with the instructions provided by the
manufacturer, the cells were harvested, washed with binding buffer
(BD Biosciences) and resuspended in 200 μl binding buffer.
Annexin V solution (5 μl; BD Biosciences) was added to the
cell suspension followed by incubation for 20 min in the dark at
4°C. Subsequently, 5 μl PI were added for 5 min, and the
cell suspension was immediately analyzed by bivariate flow
cytometry using BD FACSCanto II equipped with BD FACSDiva Software
(Becton-Dickinson). Approximately 1×105 cells were
analyzed per sample. Annexin V−/PI− staining
represented surviving cells, Annexin V+/PI−
cells signified early apoptosis and PI+ cells indicated
necrotic or apoptotic cells at the terminal stage.
Cellular ROS levels were determined using a Reactive
Oxygen Species assay kit (Beyotime Institute of Biotechnology).
Briefly, the cells were incubated with the diluted fluoroprobe,
2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA; Beyotime
Institute of Biotechnology), for 20 min at 37°C with slight shaking
every 5 min. After washing with serum-free culture medium, the
cells were collected and examined by flow cytometry (FACSCanto II)
at excitation and emission wavelengths of 488 and 525 nm,
respectively, or examined under a fluorescence microscope
(DMI4000B; Leica, Wetzlar, Germany).
Detection of mitochondrial membrane
potential (MMP or ΔΨm)
The loss of ΔΨm was determined using the JC-1
Mitochondrial Membrane Potential assay kit (Beyotime Institute of
Biotechnology). Briefly, the MSCs were seeded in 6-well plates.
Following treatment, the cells were washed with PBS and 5 μM
JC-1 was added, followed by incubation at 37°C for 20 min.
Subsequently, the cells were washed twice with cold JC-1 staining
buffer and visualized under a fluorescence microscope.
Western blot analysis
At the end of the treatment period, the MSCs were
harvested and lysed with ice-cold RIPA lysis buffer, and the
homogenate was centrifuged at 12,000 × g for 10 min at 4°C. Total
protein in the supernatant was quantified using a BCA Protein assay
kit, and an aliquot (30–50 μg) from each sample was
separated by 12% sodium dodecyl sulfate-polyacrylamide gel
electrophoresis (SDS-PAGE). The protein band was transferred onto
polyvinylidene difluoride (PVDF) membranes blocked with 8% fat-free
milk in Tris-buffered saline (TBS) with 0.5% Tween-20 for 60 min at
37°C, followed by treatment with the following primary antibodies
at 4°C overnight: rabbit monoclonal against Akt (cst-4691s),
phosphorylated Akt [p-Akt (Ser473); cst-4060s], caspase-3
(cst-9662s), Bax (cst-2772s), Bcl-2 (cst-2876s) and cytochrome
c (cst-4272s) (all from Cell Signaling Technology) and mouse
polyclonal antibody against β-actin (TA-09; Zhongshan Golden Bridge
Biotechnology, Beijing, China). After washing in TBS with Tween-20
(TBS-T) buffer, the membranes were further incubated with
horseradish peroxidase-conjugated anti-mouse (ZB-2305; Zhongshan
Goldenbridge Biotechnology) and anti-rabbit (sc-2357) secondary
antibodies (Santa Cruz Biotechnology, Inc.) for 60 min at 37°C.
Subsequently, the membranes were washed in TBS-T solution 3 times,
followed by the addition of TBS solution, and visualization using
the ECL chemiluminescence detection system with BeyoECL Plus
(Beyotime Institute of Biotechnology). Densitometric analysis of
the protein bands was carried out using Quantity One software
(Bio-Rad, Hercules, CA, USA).
Cell viability and proliferation
assay
Cell viability was assessed using the cell counting
kit-8 (CCK-8). Briefly, the MSCs were cultured in 96-well plates at
a density of 1,000 cells/well. Following an ~70% fusion of the
cells, the indicated treatments were carried out. Subsequently, 10
μl CCK-8 solution were added to each well and the plates
incubated for 2 h. The absorbance at 450 nm was measured using a
microplate reader (Tecan Infinite M200 microplate reader; LabX,
Midland, ON, Canada). The mean optical density (OD) of 4 wells in
each group was used to calculate the percentage of cell viability.
The experiments were carried out in triplicate.
In order to determine the cell proliferative
ability, the MSCs were cultured in 96-well plates for 24 h,
followed by exposure to various concentrations of nicorandil for
the indicated periods of time. An aliquot of
5-ethynyl-2′-deoxyuridine (EdU; 50 μM; Ribobio, Guangzhou,
China) was added to the culture medium for 2 h. The cells were
washed with PBS, fixed with 4% paraformaldehyde, and dyed with
Apollo staining reaction liquid (Ribobio). Hoechst 33342 (Ribobio)
was used for the labeling of the nuclei. Images were acquired using
a fluorescence microscope. Five fields were randomly selected from
each dish, and at least 3 dishes were counted per
concentration.
Statistical analysis
Data were analyzed using SPSS 19.0 software (SPSS,
Inc., Chicago, IL, USA). All values are expressed as the means ±
SD. Differences among groups were examined by one-way ANOVA. The
Student’s t-test was used to compare differences between 2 groups.
Differences were considered statistically significant at
P<0.05.
Results
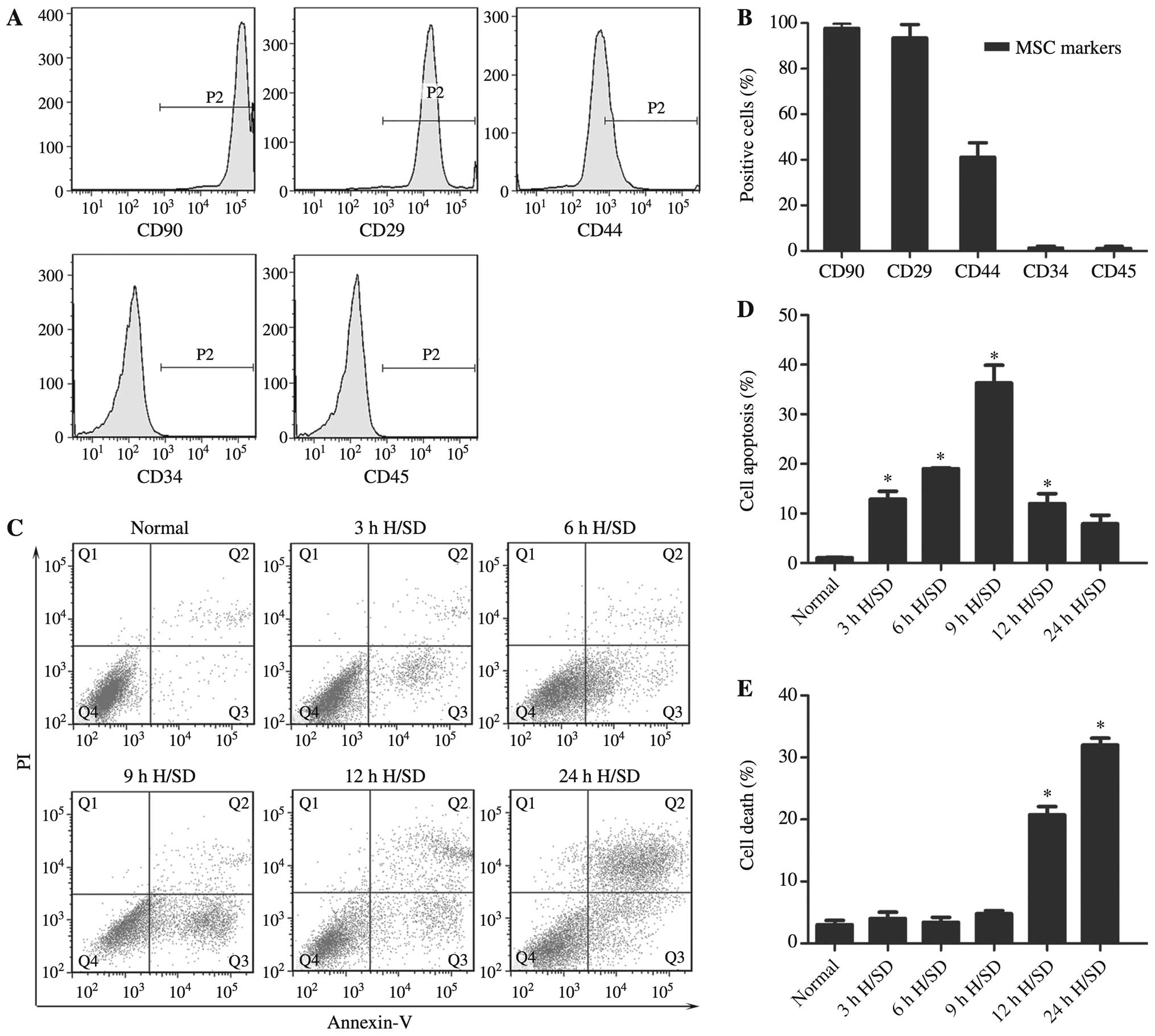
Characterization of the rat MSCs
The MSCs obtained from the bone marrow of the SD
rats exhibited a fibroblast-like appearance (data not shown). The
results of flow cytometry revealed that the majority of the
adherent MSCs from passage 3 expressed the common markers, CD90
(99.70±2.01%), CD29 (93.46±5.89%) and CD44 (41.16±6.27%), but were
negative for CD34 (1.32±0.82%) and CD45 (1.15±0.88%) (Fig. 1A and B). Therefore, cells at
passages 3–5 were used for the subsequent experiments.
H/SD conditions induce the apoptosis of
MSCs
The apoptosis of MSCs induced by H/SD (3–24 h) was
examined (Fig. 1C). The early
apoptotic rate was observed with a peak at 9 h in the MSCs exposed
to H/SD (cells exposed to H/SD, 39.20±5.11% vs. normal cells,
1.07±0.11%; Fig. 1D). Following
longer periods of treatment, the populations of PI+
cells representing necrotic or apoptotic cells at the terminal
stage were significantly increased (Fig. 1E).
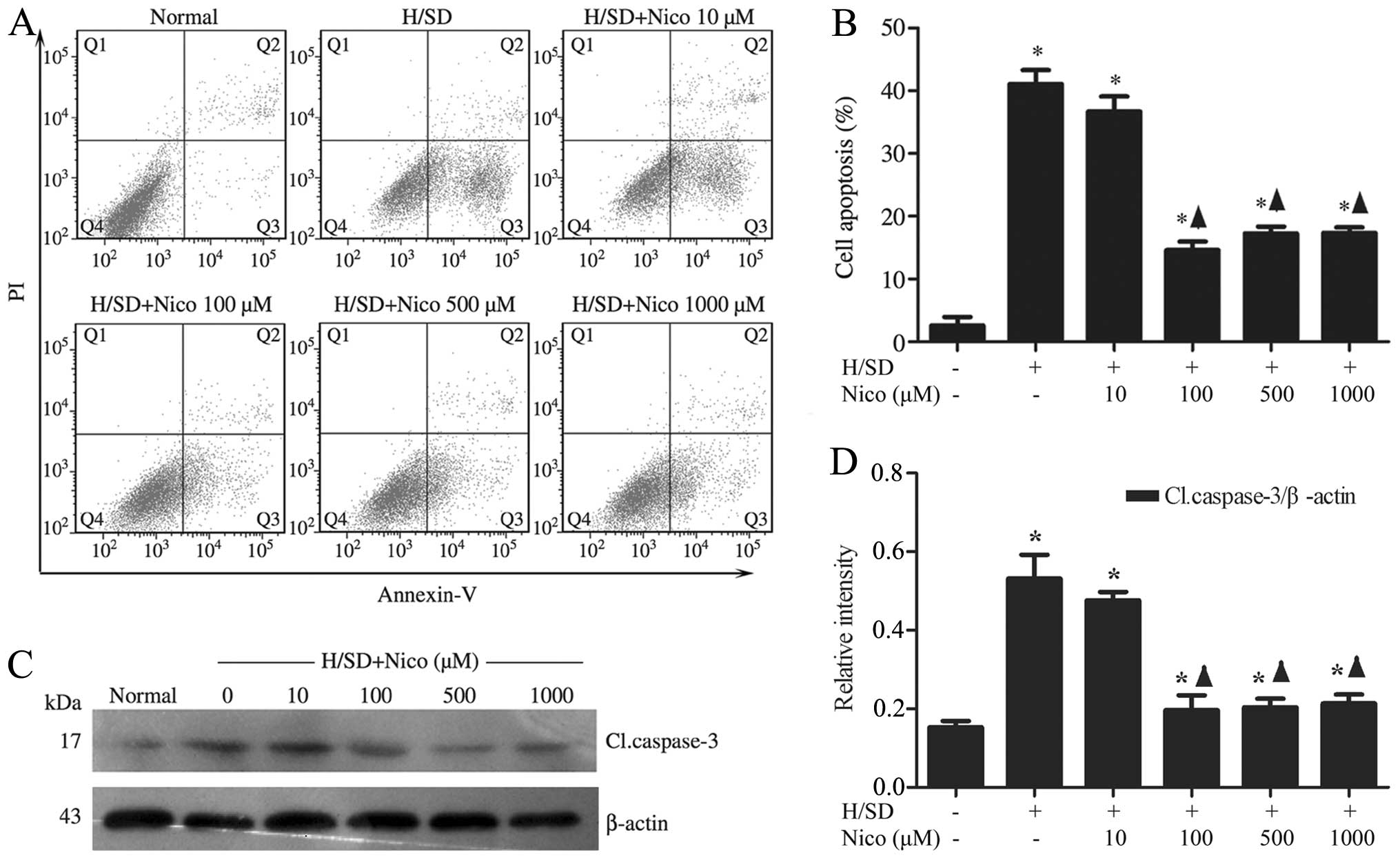
Nicorandil protects MSCs against
H/SD-induced apoptosis
To determine whether nicorandil blocks the apoptotic
process induced by H/SD, the MSCs were pre-treated with increasing
concentrations of the drug (10–1,000 μM) for 1 h, followed
by exposure to H/SD for 9 h. The Annexin
V+/PI− MSC population, identified by flow
cytometry, was reduced in the nicorandil-treated groups,
particularly in the group treated with 100 μM nicorandil
[treated cells, 14.60±1.37% vs. apoptotic control (untreated
cells), 41.10±2.20%], compared with the control group (Fig. 2A and B). In order to further
elucidate the mechanisms underlying the anti-apoptotic effects
exerted by nicorandil, western blot analysis was used to measure
the expression levels of caspase-3, a known key mediator of
apoptosis. Nicorandil significantly suppressed the cleavage of
caspase-3 under H/SD conditions in a concentration-dependent
manner, with the highest inhibitory effect observed in the group
treated with 100 μM nicorandil (treated cells, 0.20±0.04 vs.
apoptotic control, 0.53±0.06; Fig.
2C and D).
Nicorandil activates the PI3K/Akt
signaling pathway in MSCs under H/SD conditions
Following validation of the anti-apoptotic effect of
nicorandil on MSCs under H/SD conditions, the underlying mechanisms
were explored. In view of the finding that PI3K/Akt signaling
protects the heart against ischemic injury, we examined the
association between nicorandil and the PI3K/Akt pathway. The MSCs
were treated with nicorandil (100 μM) for the indicated
periods of time (0, 30, 60, 90 and 120 min). Compared with the
control group, we observed that the activation of Akt, as evident
from the increased levels of p-Akt at Ser473 by nicorandil in a
time-dependent manner, peaked at 90 min [Akt (Ser473) 90 min,
0.81±0.05 vs. 0 min, 0.25±0.60] (Fig.
3A and C). To further confirm the role of this pathway in the
anti-apoptotic effects of nicorandil, PI3K/Akt was blocked using
the PI3K-specific inhibitor, LY294002. The results from western
blot analysis revealed that the inhibition of Akt expression by
LY294002 [Akt (Ser473) with LY294002, 0.30±0.04 vs. without
LY294002, 0.55±0.06] in the group treated with 100 μM
nicorandil (Fig. 3B and D).
Moreover, co-incubation with LY294002 partially abrogated the
anti-apoptotic effects of nicorandil on the MSCs, as evidenced by
the increase in the number of Annexin V+/PI−
cells (with LY294002, 21.99±1.83% vs. without LY294002,
14.07±1.52%; Fig. 3E and F).
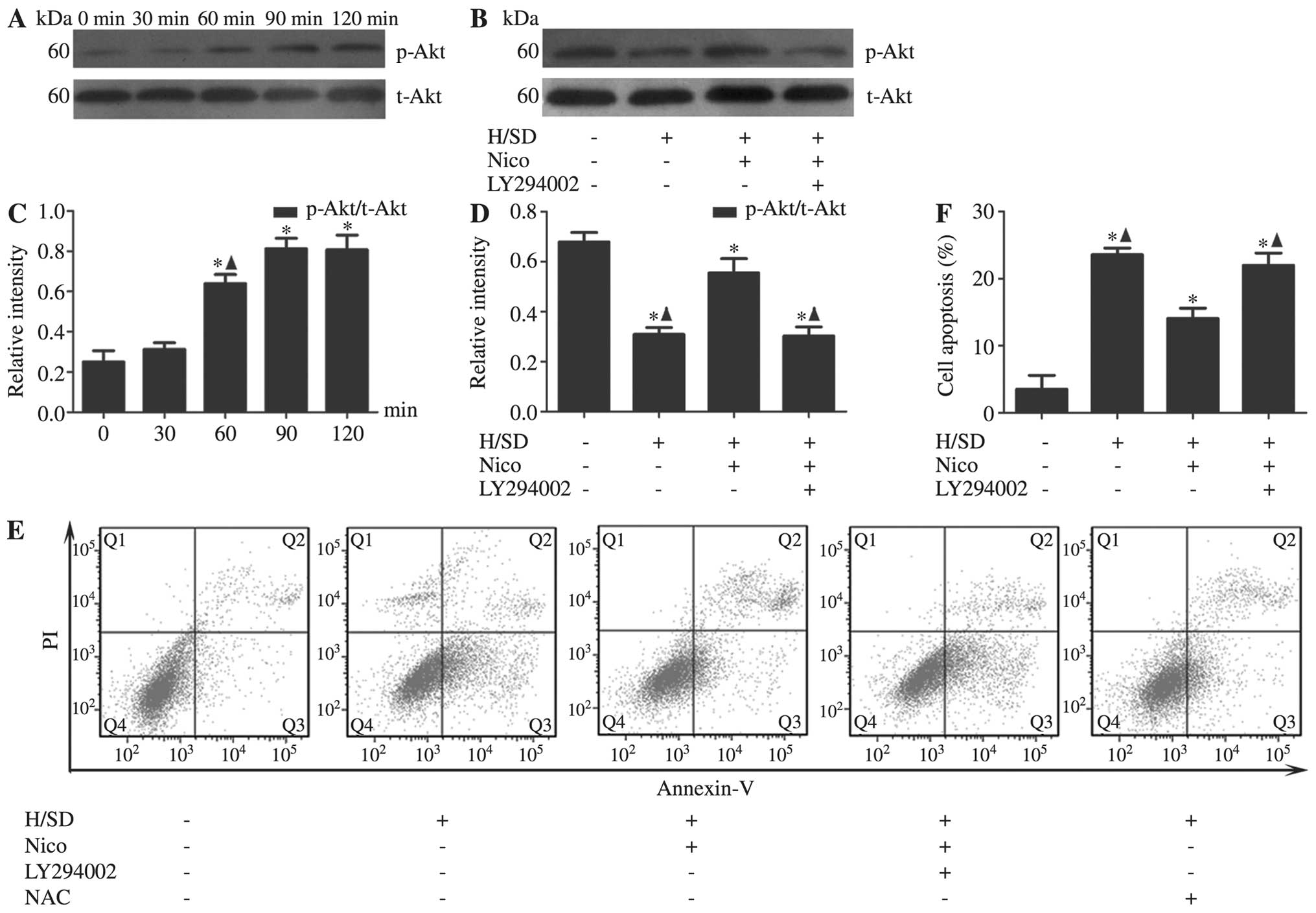 | Figure 3Nicorandil activates the PI3K/Akt
signaling pathway under H/SD conditions. MSCs were treated with
nicorandil (100 μM) for the indicated periods of under H/SD
conditions for 9 h. (A and C) The levels of p-Akt was upregulated
by nicorandil in a time-dependent manner, peaking at 90 min. Data
are presented as the means ± SD of 3 separate experiments.
*P<0.05, compared with the normal group;
▲P<0.05, compared with the group treated with
nicorandil for 90 min. (B and D) The inhibition of PI3K with
LY294002 triggered p-Akt inactivation. (E and F) Nicorandil induced
a significant decrease in the apoptotic rate of MSCs under H/SD
conditions, which was reversed by LY294002. Data are presented as
the means ± SD of 3 separate experiments. *P<0.05,
compared with the normal group; ▲P<0.05, compared
with the 100 μM nicorandil-treated group. PI3K,
phosphoinositide 3-kinase; MSCs, mesenchymal stem cells; H/SD,
hypoxia/serum deprivation; Nico, nicorandil; p-Akt, phosphorylated
Akt (Ser473); t-Akt, total Akt; NAC, N-acetyl-L-cysteine. |
Nicorandil exerts anti-apoptotic effects
by stabilizing the MMP
Depolarization of the inner MMP is a sign of cell
death (23). Therefore, in order
to ascertain whether nicorandil preserves mitochondrial integrity
through the maintenance of MMP, we performed JC-1 staining. As
shown in Fig. 4G, the red/green
ratio of JC-1 was decreased in the MSCs exposed to H/SD compared
with the normal group, and this effect was reversed by nicorandil,
particularly in the group treated with 100 μM nicorandil,
which is consistent with the Annexin V-PI measurements (Fig. 3E and F). These results confirm the
beneficial effects of nicorandil on mitochondrial function. The
integrity of the mitochondrial membrane affects the release of
pro-apoptotic cytochrome c from the mitochondria to the
cytosol (24). Western blot
analysis revealed the inhibition of the release of cytochrome
c in the nicorandil-treated group, compared with the group
exposed to H/SD (levels of mitochondrial cytochrome c:
treated cells, 1.41±0.10 vs. apoptotic control, 0.61±0.07; Fig. 4A and B). To further determine
whether the anti-apoptotic effects of nicorandil involve the
inhibition of the mitochondrial pathway in the MSCs exposed to
H/SD, the Bcl-2/Bax ratio, which plays an important role in
mitochondrial integrity (25),
was examined by western blot analysis (Fig. 4C and D). Compared with the cells
exposed to H/SD, the Bcl-2/Bax ratio was significantly increased in
the group treated with 100 μM nicorandil (treated cells,
1.61±0.10 vs. apoptotic control, 0.50±0.10; Fig. 4C and D), and the levels of
caspase-3, a key mediator of apoptosis, were decreased (treated
cells, 0.37±0.04 vs. apoptotic control, 0.54±0.06; Fig. 4E and F).
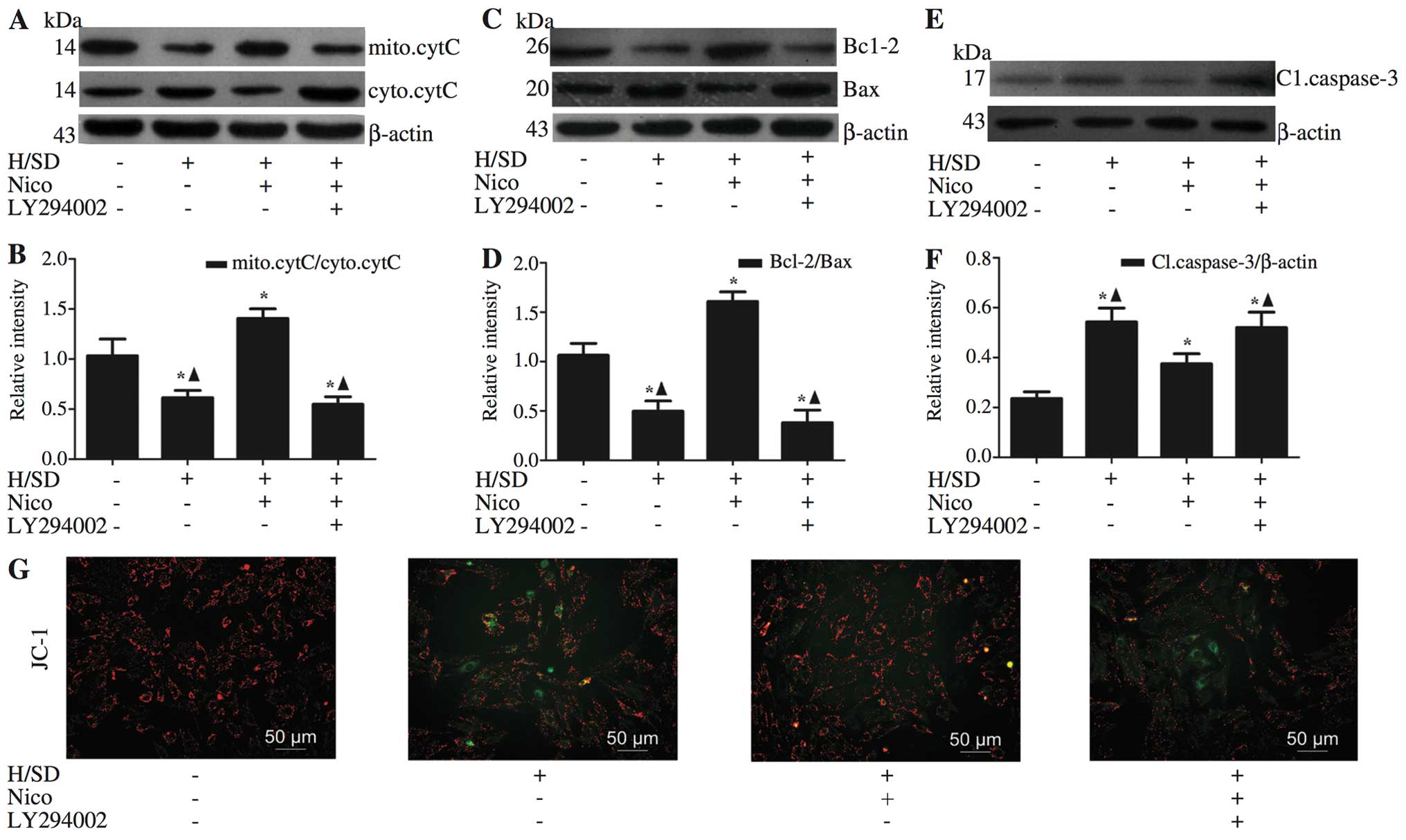 | Figure 4Nicorandil exerts anti-apoptotic
effects by stabilizing MMP. Western blot analysis revealed that
nicorandil induced a significant increase in (C and D) the
expression of anti-apoptotic Bcl-2, with a concomitant decrease in
(A–D) the pro-apoptotic proteins Bax and cytochrome c, as
well as apoptosis-related (E and F) capsase-3, and these effects
were reversed by LY294002 [an inhibitor of phosphoinositide
3-kinase (PI3K)]. (G) Nicorandil exerted a significant inhibitory
effect on mitochondrial dysfunction, as verified by JC-1 staining.
Data are presented as the means ± SD of 3 separate experiments.
*P<0.05, compared with the normal group;
▲P<0.05, compared with the 100 μM
nicorandil-treated group. H/SD, hypoxia/serum deprivation; Nico,
nicorandil; MMP, mitochondrial membrane potential; mito.cytC,
mitochondrial cytochrome c; cyto.cytC, cytosolic cytochrome
c; Cl.caspase-3, cleaved caspase-3. |
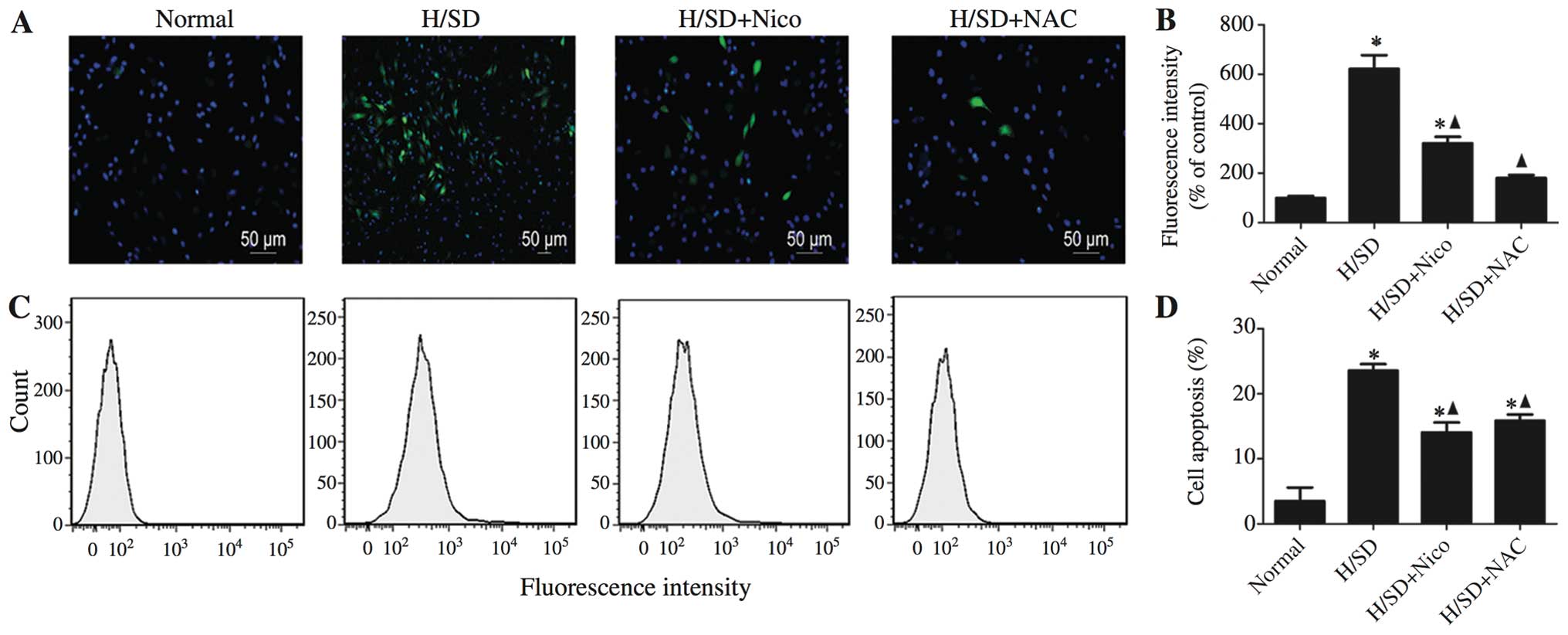
Involvement of ROS in the
nicorandil-mediated protective effects against the apoptosis of
MSCs exposed to H/SD
ROS function as pivotal components in pro-apoptotic
signaling cascades (26). In this
study, MSCs exposed to H/SD displayed an approximate 7-fold
increase in ROS production, compared with the untreated cells
(Fig. 5A and B). Nicorandil
induced a significant inhibition of ROS production, as was
determined by the DCFH oxidation assay. Similar results were
obtained with the general ROS scavenger, NAC (500 μM)
(nicorandil-treated cells, 321.00±25.71 vs. apoptotic control,
621.98±55.29; NAC-treated cells, 180.33±11.72 vs. apoptotic
control, 621.98±55.29; Fig. 5A
and B). Consistently, the results of flow cytometry revealed that
pre-treatment with either nicorandil or NAC for 1 h prior to
exposure to H/SD, induced a significant decrease in apoptosis
(nicorandil-treated cells, 14.07±1.51% vs. apoptotic control,
23.57±1.00%; NAC-treated cells, 15.87±0.95% vs. apoptotic control,
23.57±1.00%; Figs. 3E and
5C and D), indicating that
nicorandil confers its protective effects partly by decreasing ROS
production.
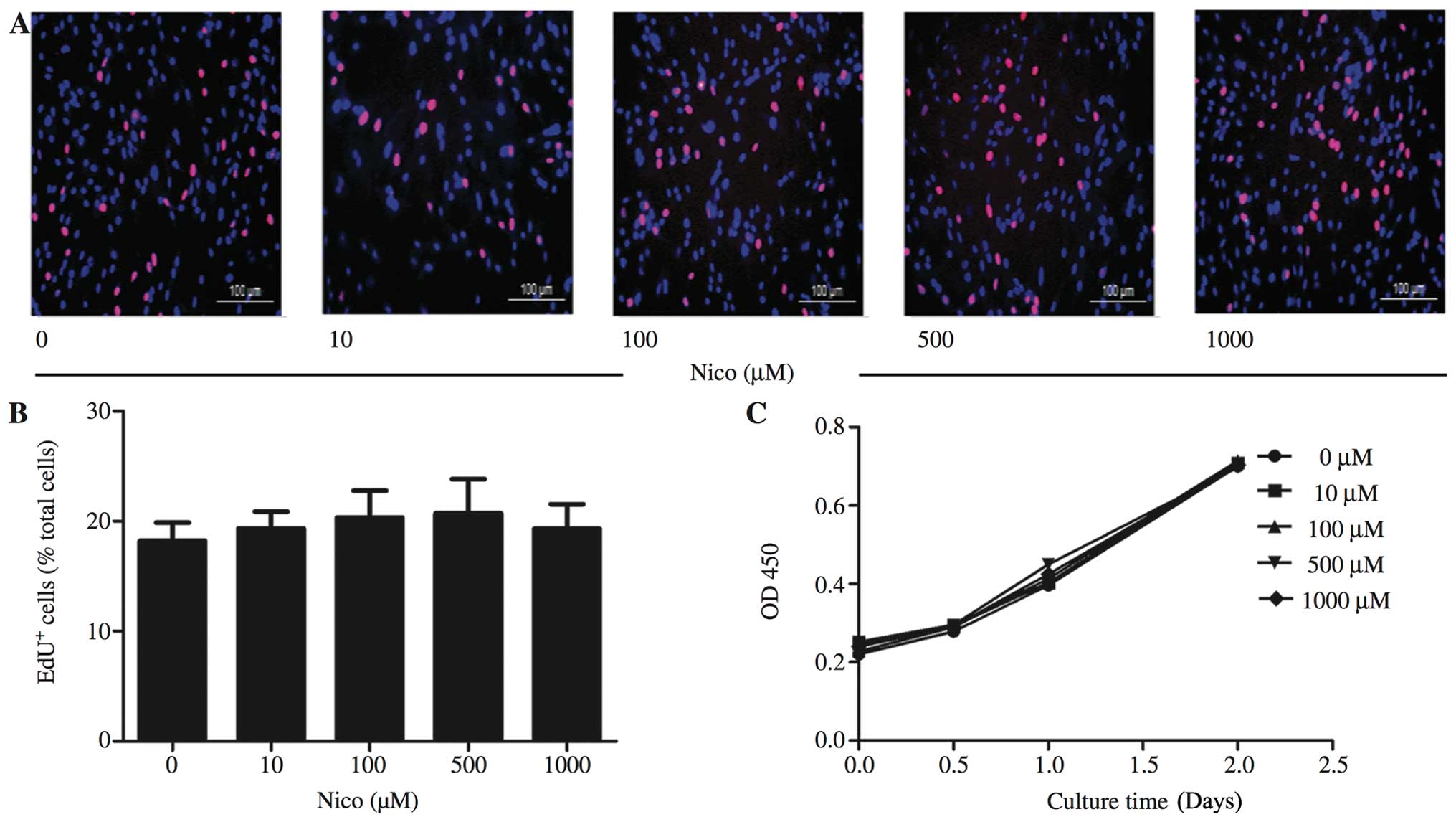
Nicorandil has little effect on MSC
proliferation
To the best of our knowledge, the effects of
nicorandil on MSC proliferation have not been documented to date.
Therefore, we examined the effects of nicorandil at the
aforementioned range of concentrations on MSC viability using the
CCK-8 assay and EdU staining. Data from EdU staining indicated that
nicorandil had little effect on MSC proliferation (Fig. 6A and B). In accordance with the
results of EdU staining, the proliferation growth curves of the
MSCs obtained by CCK-8 assay did not show any significant changes
during the 2 days of nicorandil treatment at any of the
concentrations used (Fig.
6C).
Discussion
In the present study, we demonstrated that
nicorandil suppressed the apoptosis of MSCs exposed to H/SD in a
dose-dependent manner. Moreover, the anti-apoptotic effects of
nicorandil were shown to be mediated through the PI3K/Akt,
mitochondrial and ROS signaling pathways. To the best of our
knowledge, this is the first study to report the protective effects
of nicorandil on MSCs under H/SD conditions in vitro and the
underlying mechanisms.
Since MSCs are easily obtained and exhibit
impressive paracrine ability, multilineage differentiation
potential (27) and the capacity
to activate endogenous cardiac progenitor cells (CPCs) (28), they represent one of the most
ideal seed cell candidates for tissue regeneration in several
clinical diseases, including myocardial infarction (3). However, the therapeutic potential of
MSCs is limited by their poor survival rate following
transplantation into the deleterious infarcted myocardium (29). Zhang et al (30) demonstrated that stem cell therapy
only slightly improved cardiac function, and the majority of the
implanted MSCs in the infarcted myocardium died within 7 days.
Therefore, the high rate of attrition of transplanted MSCs is a
primary concern. Studies have demonstrated that apoptosis is the
main pattern of cell death when MSCs are exposed to H/SD in
vitro, mimicking the in vivo microenvironment of
ischemic injury (6,31).
Nicorandil is the only mitoKATP channel
opener currently used in clinical practice as a cardioprotective
drug. Experimental and clinical studies have described the
protective effects of nicorandil against ischemic heart disease
(14,15). Recently, other cytoprotective
effects of nicorandil have been reported. Izumiya et al
(18) demonstrated that the
treatment of ApoE− deficient mice on an atherogenic diet
for 8 weeks with nicorandil significantly reduced atherosclerotic
lesions and plaque necrosis. Moreover, nicorandil attenuated
tunicamycin-induced C/EBP homologous protein (CHOP) expression in
cultured THP-1 macrophages (18).
Nicorandil has also been shown to reduce the activation of the
inflammasome and, subsequently, the release of caspase-1 and the
levels of IL-1β and IL-18 (20).
The majority of these studies have demonstrated that the opening of
mitoKATP channels is one of the most important
mechanisms underlying the protective effects of nicorandil. Kim
et al (15) reported that
nicorandil inhibited apoptosis induced by oxidative stress through
the activation of mitoKATP channels, partly by
preserving ΔΨm. The mitoKATP channel opener, diazoxide,
has recently been shown to improve the survival ability of MSCs
in vivo and in vitro when these are transplanted into
rats with acute myocardial infarction or are subjected to oxidative
stress with 100 μM H2O2
(preconditioned MSCs) by improving mitochondrial function (32,33). Based on these findings, we
hypothesized that nicorandil may exert protective effects on MSCs
under H/SD conditions by activating the mitoKATP
channels.
In our experiments, the apoptosis of the MSCs
exposed to H/SD significantly increased, compared to the normal
group. Notably, pre-treatment with nicorandil at concentrations
ranging from 10 to 1,000 μM for 1 h and exposure to H/SD for
9 h led to decreased apoptosis in a concentration-dependent manner.
Hoever, further studies are required to explore the molecular
mechanisms underlying the regulation of apoptosis by
nicorandil.
We further examined whether the PI3K/Akt signaling
pathway is involved in the anti-apoptotic activity of nicorandil in
MSCs. The PI3K/Akt pathway plays an essential role in promoting
survival in various cell types (34,35). Our results demonstrated that
nicorandil increased Akt phosphorylation and these protective
effects were effectively blocked by a PI3K inhibitor (LY294002),
strongly suggesting an essential role for the PI3K/Akt signaling
pathway in the nicorandil-mediated protection of MSCs exposed to
H/SD.
As an inevitable by-product of mitochondrial
respiration, ROS are mainly produced in the mitochondria under
normal conditions, and moderate amounts are necessary for cell
survival, proliferation and pro-longevity (36). However, during hypoxia, an
imbalance between the formation and scavenging of free radicals
leads to the overproduction of electrons. These electrons react
with remnant molecular oxygen, leading to ROS generation (37). Plethoric ROS may result in the
loss of MMP, the release of pro-apoptotic molecules and the
initiation of caspase-dependent apoptosis (26). Previous studies have demonstrated
that the regulation of the mitochondrial potassium membrane
permeability contributes to the mitoKATP opener-mediated
suppression of ROS production (38). Our findings indicated that H/SD
induced the production of excess ROS, and this effect was
suppressed by treatment with nicorandil.
Our study had several limitations. Firstly, the H/SD
model is an in vitro experimental representation of acute
myocardial infarction, which fails to completely mimic the in
vivo ischemic and inflammatory microenvironment. Further
studies are warranted to ensure the comprehensive understanding of
the cellular mechanisms involved, both in vitro and in
vivo. Secondly, apoptosis is only one of the processes that
damage MSCs during H/SD. Other factors involving inflammation may
also lead to the deterioration of the survival ability of MSCs.
Recently, nicorandil was reported to reduce the activation of the
inflammasome and the release of caspase-1, IL-1β and IL-18,
following oxygen-glucose deprivation-induced inflammation in BV-2
cells (20). Therefore, we need
to focus on the role of inflammasomes in MSCs under H/SD conditions
and on the related effects of nicorandil. Thirdly, multiple
mechanisms and pathways, such as 5′ adenosine
monophosphate-activated protein kinase (AMPK)/endothelial nitric
oxide synthase (eNOS), participate in sustaining and mediating
apoptosis (39), and thus require
further investigation.
In conclusion, the results of the present study
provide preliminary evidence indicating that nicorandil promotes
MSC survival under conditions mimicking the myocardial ischemia.
The pro-survival effects of nicorandil against H/SD-induced
mitochondrial apoptosis are possibly a result of the activation of
the PI3K/Akt signaling pathway and the reduction of ROS
production.
Acknowledgments
We would like to thank Dr Bo Sun for assisting with
the FACS analysis and Xiaojing Liu for assisting with the
revisions. Dr Bo Sun is a member of the Key Laboratory of
Myocardial Ischemia Mechanism and Treatment (Harbin Medical
University, Ministry of Education). This study was supported by
grants from the National Natural Science Foundation of China (to
B.Y.; grant nos. 81171430 and 81330033).
References
|
1
|
Moran AE, Forouzanfar MH, Roth GA, et al:
Temporal trends in ischemic heart disease mortality in 21 world
regions, 1980 to 2010: the Global Burden of Disease 2010 study.
Circulation. 129:1483–1492. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Quevedo HC, Hatzistergos KE, Oskouei BN,
et al: Allogeneic mesenchymal stem cells restore cardiac function
in chronic ischemic cardiomyopathy via trilineage differentiating
capacity. Proc Natl Acad Sci USA. 106:14022–14027. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Karantalis V, DiFede DL, Gerstenblith G,
et al: Autologous mesenchymal stem cells produce concordant
improvements in regional function, tissue perfusion, and fibrotic
burden when administered to patients undergoing coronary artery
bypass grafting: The Prospective Randomized Study of Mesenchymal
Stem Cell Therapy in Patients Undergoing Cardiac Surgery
(PROMETHEUS) trial. Circ Res. 114:1302–1310. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Toma C, Pittenger MF, Cahill KS, Byrne BJ
and Kessler PD: Human mesenchymal stem cells differentiate to a
cardiomyocyte phenotype in the adult murine heart. Circulation.
105:93–98. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pagani FD, DerSimonian H, Zawadzka A, et
al: Autologous skeletal myoblasts transplanted to ischemia-damaged
myocardium in humans. Histological analysis of cell survival and
differentiation. J Am Coll Cardiol. 41:879–888. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhu W, Chen J, Cong X, Hu S and Chen X:
Hypoxia and serum deprivation-induced apoptosis in mesenchymal stem
cells. Stem Cells. 24:416–425. 2006. View Article : Google Scholar
|
|
7
|
Nichols CG: KATP channels as
molecular sensors of cellular metabolism. Nature. 440:470–476.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Costa AD, Quinlan CL, Andrukhiv A, West
IC, Jabůrek M and Garlid KD: The direct physiological effects of
mitoK(ATP) opening on heart mitochondria. Am J Physiol Heart Circ
Physio. 290:H406–H415. 2006. View Article : Google Scholar
|
|
9
|
Yamada M: Mitochondrial ATP-sensitive
K+ channels, protectors of the heart. J Physiol.
588:283–286. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Barajas-Martinez H, Hu D, Ferrer T, et al:
Molecular genetic and functional association of Brugada and early
repolarization syndromes with S422L missense mutation in KCNJ8.
Heart Rhythm. 9:548–555. 2012. View Article : Google Scholar :
|
|
11
|
van Bon BW, Gilissen C, Grange DK, et al:
Cantú syndrome is caused by mutations in ABCC9. Am J Hum Genet.
90:1094–1101. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Harakalova M, van Harssel JJ, Terhal PA,
et al: Dominant missense mutations in ABCC9 cause Cantu syndrome.
Nat Genet. 44:793–796. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Miura T and Miki T: ATP-sensitive
K+ channel openers: old drugs with new clinical benefits
for the heart. Curr Vasc Pharmacol. 1:251–258. 2003. View Article : Google Scholar
|
|
14
|
Akao M, Teshima Y and Marban E:
Antiapoptotic effect of nicorandil mediated by mitochondrial
atp-sensitive potassium channels in cultured cardiac myocytes. J Am
Coll Cardiol. 40:803–810. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kim JH, Jeong MH, Yun KH, et al:
Myocardial protective effects of nicorandil during percutaneous
coronary intervention in patients with unstable angina. Circ J.
69:306–310. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tsuchida A, Miura T, Tanno M, et al:
Infarct size limitation by nicorandil: roles of mitochondrial
K(ATP) channels, sarcolemmal K(ATP) channels, and protein kinase C.
J Am Coll Cardiol. 40:1523–1530. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Iwakura K: Modulation of individual
susceptibility to the no-reflow phenomenon after acute myocardial
infarction. Curr Pharm Des. 19:4519–4528. 2013. View Article : Google Scholar
|
|
18
|
Izumiya Y, Kojima S, Kojima S, et al:
Long-term use of oral nicorandil stabilizes coronary plaque in
patients with stable angina pectoris. Atherosclerosis. 214:415–421.
2011. View Article : Google Scholar
|
|
19
|
Lee TM, Lin MS, Tsai CH and Chang NC:
Effect of ischaemic preconditioning on regional release of
inflammatory markers. Clin Sci (Lond). 109:267–276. 2005.
View Article : Google Scholar
|
|
20
|
Zhao AP, Dong YF, Liu W, Gu J and Sun XL:
Nicorandil inhibits inflammasome activation and Toll-like
receptor-4 signal transduction to protect against oxygen-glucose
deprivation-induced inflammation in BV-2 cells. CNS Neurosci Ther.
20:147–153. 2014. View Article : Google Scholar
|
|
21
|
Hou M, Cui J, Liu J, et al:
Angiopoietin-like 4 confers resistance to hypoxia/serum
deprivation-induced apoptosis through PI3K/Akt and ERK1/2 signaling
pathways in mesenchymal stem cells. PLoS One. 9:e858082014.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang Q, Yang YJ, Wang H, et al: Autophagy
activation: a novel mechanism of atorvastatin to protect
mesenchymal stem cells from hypoxia and serum deprivation via
AMP-activated protein kinase/mammalian target of rapamycin pathway.
Stem Cells Dev. 21:1321–1332. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gottlieb E, Armour SM, Harris MH and
Thompson CB: Mitochondrial membrane potential regulates matrix
configuration and cytochrome c release during apoptosis. Cell Death
Differ. 10:709–717. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Garrido C, Galluzzi L, Brunet M, Puig PE,
Didelot C and Kroemer G: Mechanisms of cytochrome c release from
mitochondria. Cell Death Differ. 13:1423–1433. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ghavami S, Hashemi M, Ande SR, et al:
Apoptosis and cancer: mutations within caspase genes. J Med Genet.
46:497–510. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kanwar JR, Kamalapuram SK and Kanwar RK:
Survivin signaling in clinical oncology: a multifaceted dragon. Med
Res Rev. 33:765–789. 2013. View Article : Google Scholar
|
|
27
|
Frenette PS, Pinho S, Lucas D and
Scheiermann C: Mesenchymal stem cell: keystone of the hematopoietic
stem cell niche and a stepping-stone for regenerative medicine.
Annu Rev Immunol. 31:285–316. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ye L, Zhang P, Duval S, Su L, Xiong Q and
Zhang J: Thymosin β4 increases the potency of transplanted
mesenchymal stem cells for myocardial repair. Circulation.
128(Suppl 1): S32–S41. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Clifford DM, Fisher SA, Brunskill SJ, et
al: Stem cell treatment for acute myocardial infarction. Cochrane
Database Syst Rev. 2:CD0065362012.PubMed/NCBI
|
|
30
|
Zhang Z, Li S, Cui M, et al: Rosuvastatin
enhances the therapeutic efficacy of adipose-derived mesenchymal
stem cells for myocardial infarction via PI3K/Akt and MEK/ERK
pathways. Basic Res Cardiol. 108:3332013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Deuse T, Peter C, Fedak PW, et al:
Hepatocyte growth factor or vascular endothelial growth factor gene
transfer maximizes mesenchymal stem cell-based myocardial salvage
after acute myocardial infarction. Circulation. 120(Suppl 11):
S247–S254. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Afzal MR, Haider HK, Idris NM, Jiang S,
Ahmed RP and Ashraf M: Preconditioning promotes survival and
angiomyogenic potential of mesenchymal stem cells in the infarcted
heart via NF-kappaB signaling. Antioxid Redox Signal. 12:693–702.
2010. View Article : Google Scholar :
|
|
33
|
Suzuki Y, Kim HW, Ashraf M and Haider H:
Diazoxide potentiates mesenchymal stem cell survival via
NF-kappaB-dependent miR-146a expression by targeting Fas. Am J
Physiol Heart Circ Physiol. 299:H1077–H1082. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Mangi AA, Noiseux N, Kong D, et al:
Mesenchymal stem cells modified with Akt prevent remodeling and
restore performance of infarcted hearts. Nat Med. 9:1195–1201.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yee C, Yang W and Hekimi S: The intrinsic
apoptosis pathway mediates the pro-longevity response to
mitochondrial ROS in C. elegans. Cell. 157:897–909. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Acín-Pérez R, Carrascoso I, Baixauli F, et
al: ROS-triggered phosphorylation of complex II by Fgr kinase
regulates cellular adaptation to fuel use. Cell Metab.
19:1020–1033. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Gao L, Liu R, Gao F, Wang Y, Jiang X and
Gao X: Plasmon mediated generation of reactive oxygen species from
near-infrared light excited gold nanocages for photodynamic therapy
in vitro. ACS Nano. 8:7260–7271. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ozcan C, Bienengraeber M, Dzeja PP and
Terzic A: Potassium channel openers protect cardiac mitochondria by
attenuating oxidant stress at reoxygenation. Am J Physiol Heart
Circ Physiol. 282:H531–H539. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Xu L, Wang S, Li B, Sun A, Zou Y and Ge J:
A protective role of ciglitazone in ox-LDL-induced rat
microvascular endothelial cells via modulating PPARγ-dependent
AMPK/eNOS pathway. J Cell Mol Med. 19:92–102. 2015. View Article : Google Scholar :
|