Introduction
The disease with the second highest rate of
mortality, for women, is human cervical carcinoma (HCC), which is
prevalent in developing countries (1,2).
The majority of HCCs derive from epithelial cells that have been
persistently infected with high-risk oncogenic human
papillomaviruses (HPVs), which express the E6 and E7 mRNAs, and
proteins binding/neutralizing anti-oncogenic p53 and pRb.
Furthermore, other co-factors, which have been less thoroughly
studied, also promote HCC development (3,4).
Notably, a small percentage (10%) of HCCs develops independently of
a persistent high-risk oncogenic HPV infection (5). The complex molecular mechanisms
responsible for the development of HCCs thus remain partially
unclear, and this hinders the identification of predictive markers
for the early detection of malignancy development and the discovery
of novel, effective therapies.
Proteomics and bioinformatics analysis have allowed
researchers to identify differential protein expression patterns
and the molecular mechanisms responsible for the development of HCC
(6). A previous study identified
a consensus of 66 proteins termed the 'central core of HCC protein
expression̓ which was deduced from the proteomes of 6 HCC cell
lines (not comprising the C4-I cell line); this differed from the
proteomic profile corresponding to the non-tumorigenic cell line,
HaCaT keratinocytes (7).
Conversely, a comparative transcriptomics study carried out on 9
HCC cell lines singled out C4-I cells as those which mimicked most
closely late-stage invasive in vivo HCCs (8). This prompted us to select the C4-I
cells as our experimental model in order to study the signaling of
protein kinase C (PK)C isoforms at the nuclear envelope (NE) under
conditions of spontaneous growth or apoptosis.
PKCs include 12 gene-related serine/threonine PK
isoforms, which play crucial roles in cell survival,
differentiation, polarity regulation, gene transcription, mitotic
cycle control, malignant initiation and promotion, and drug- or
irradiation-elicited apoptosis (1,9–13).
On the basis of their activation requirements, the PKC isoforms are
divided as follows: i) classical PKCs [cPKCs, i.e., α, βI, βII and
γ, the activation of which requires calcium, diacylglycerol (DAG)
and phosphatidylserine (PS)]; ii) novel PKCs (nPKCs, i.e., δ, ε, η
and θ, the activation of which requires DAG and PS, but not
calcium); and iii) atypical PKCs (aPKCs, i.e., ζ and ι/λ, the
activation of which requires only PS). Each PKC isoform has unique
structural properties and different subcellular locations, and each
has a different signaling role according to cell type and
subcellular compartment, e.g., the endoplasmic reticulum (ER),
mitochondria and NE (9–13).
The many functions of the NE are of great
pathophysiological importance: it has been shown to play important
roles in the upkeep of the nuclear integrity, in chromatin
organization control and maintenance, the sequestration of
transcription factors, gene transcription, DNA replication, cell
development, cell differentiation, signaling molecule production
and activity, and it is also involved in protecting cells against
stress (14). However, the roles
of PKC isoform(s) signaling at the NE, particularly in apoptotic
cells, are not yet fully understood. Previously, using pyF111
cells, i.e., polyomavirus (the rodent HPV counterpart)-infected rat
embryo fibroblasts, we demonstrated that various PKC isoforms
(e.g., βI, βII and δ) translocated to the NE and there exhibit an
increase or decrease in activity during apoptosis induced by
etoposide (also known as VP-16, a topoisomerase-II inhibitor) or
photoexcited calphostin-C (15–18). Etoposide is a drug that is used to
treat several types of cancer, including cervical carcinoma;
several clinical studies have suggested that the combination of
etoposide with cisplatin is effective for treating advanced or
recurrent cervical cancers (19).
Therefore, in a recent study of ours (20), we aimed to elucidate, by means of
proteomic and biochemical methods, the interactions of PKCζ
isoforms with protein partners at the nuclear membranes (NMs) of
both spontaneously proliferating (i.e., untreated) and apoptotic
(i.e., VP-16-exposed) HCC C4-I cells. Through the combined use of
two-dimensional electrophoresis (2-DE), matrix-assisted laser
desorption ionization (MALDI) time-of-flight (TOF) mass
spectrometry (MS), peptide mass fingerprinting (PMF), and
bioinformatics analysis, we selected 31 and 33 proteins which
co-immunoprecipitated with PKCζ from the NMs of untreated or
VP-16-exposed C4-I cells, respectively. These proteins pertained to
8 functional groups, the members and relative sizes of which
differed. Of the proteins detected, only 8 proteins, including
B-cell lymphoma 10 (Bcl10) were belonged to both subproteomes.
Therefore, given the highly dynamic complexity of the NM-linked
interactors/substrates, the molecular interactions and functional
roles of PKCζ changed remarkably, in keeping with the untreated or
apoptogen-treated cellular contexts (20). Moreover, we unexpectedly found
that at the NMs of VP-16-treated C4-I cells, PKCζ formed complexes
with and phosphorylated the Bcl10 protein, which prompted the
caspase-3-mediated pro-apoptotic inactivation of PKCζ (20).
The Bcl10 gene was identified from the
(1;14)(p22;q32) breakpoint in mucosa-associated lymphoid tissue
(MALT) lymphomas and is ubiquitously expressed in normal tissues
(21). It encodes a 233 amino
acid protein that has an N-terminal domain (amino acids 1–13)
necessary for transcriptional activation (22,23), a caspase recruitment domain (CARD;
amino acids 14–90) to which CARD motif-endowed proteins [e.g.,
CARD-containing MAGUK protein 1 (CARMA1)] bind (21), and a C-terminal region (amino
acids 91–233) rich in serines and threonines that undergoes
multiple phosphorylations (24).
The Bcl10 protein functions are manifold. Previous gene-knockout
research has identified Bcl10 as a positive regulator of
neural tube closure and of the antigen receptor-induced activation
of nuclear factor-κB (NF-κB), a transcription factor involved in
cell survival, lymphocyte proliferation and normal immune responses
(25). Wild-type (wt)
Bcl10 operates as a tumor suppressor, whereas mutated
Bcl10 acquires oncogenic properties that act to promote
various lymphomas and mesotheliomas (21,26). However, no Bcl10 mutations
have been detected in many other types of human solid tumors,
including HCCs (21,27). Conversely, the pathophysiological
mechanism(s) mediated by the Bcl10 protein during apoptosis has(ve)
not, as yet, been fully elucidated. It has been reported that the
enforced overexpression of wt Bcl10 protein weakly induces
apoptosis and activates NF-κB in cells; yet, the expression of a
Bcl10 mutant protein, in which the CARD domain is truncated, does
not trigger apoptosis but activates NF-κB (26,28,29). Evidence to date suggests that the
Bcl10 protein is involved in the auto-proteolytic activation of
pro-caspase-9 (28), in
dissociating caspases from their inhibitors, cellular inhibitor of
apoptosis proteins (cIAPs) (30),
and in the formation of pro-apoptotic complexes with PKCζ at the
NMs of HCC C4-I cells (20,31).
In our previous study, of the proteins that
co-immunoprecipitated with PKCζ from the NMs, we identified the
3-phosphoinositide-dependent protein kinase-1 (PDK1) (20), a master regulator of the AGC
family of PKs. PDK1 mediates various important cellular functions,
and it phosphorylates and activates several AGC-family members
involved in the modulation of cell growth, proliferation, survival
and metabolism (32). PDK1 is a
constitutively active kinase that phosphorylates other members of
the same family, i.e., PKCs, S6K, SGKs, p90 ibosomal protein S6
kinases (RSKs) and AKT at their activation loop (residue
Thr308 or equivalent). Therefore, the specificity of the
functions of PDK1 is ensured by its location near its downstream
targets and by protein-protein interactions (33–35). Previously, PDK1 has been shown to
play a crucial nucleating role in T cells by connecting the T cell
receptor (TCR) to two signaling complexes, PKCθ/IKK and
CARMA1/Bcl10/MALT1 (or CBM signalosome), triggering NF-κB
activation in response to TCR signaling (36).
The present study aimed to further define the
interactive roles Bcl10 plays not only with PKCζ, but also with
PDK1 and caspase-3 at the NMs of apoptotic C4-I cells. Our results
support the hypothesis that Bcl10 protein functions as an essential
'linchpin' which nucleates pro-apoptotic complexes comprising PDK1,
PKCζ and caspase-3 at the NMs of VP-16-treated C4-I cells.
Materials and methods
Cell culture
The HCC C4-I cells were a gift from Professor Nelly
Auersperg (University of British Columbia, Vancouver, BC, Canada).
The cells were plated in 175-cm2 plastic flasks
(Sarstedt, Verona, Italy) and incubated at 37°C in 95% air/5%
CO2 in complete medium consisting of 95% (v/v)
Dulbecco's modified Eagle's Minimum Essential Medium (MEM;
Sigma-Aldrich, Milan, Italy) and 5% (v/v) heat-inactivated (56°C
for 30 min) fetal bovine serum (Lonza Group, Ltd., Basel,
Switzerland) containing gentamycin (0.1 mg/ml; Lonza Group, Ltd.).
Prior to reaching confluence, the cell cultures were split at a
ratio of 1:6 by incubating them at 18±2°C with 0.025% (w/v) trypsin
(Sigma-Aldrich).
Cell apoptosis
The cells were seeded (0.8×106 cells) in
each of several 175-cm2 flasks. At the 0 h of the
experiment (i.e., 24 h later), the cells in some flasks were
sampled (untreated controls), while the topoisomerase-II inhibitor,
VP-16 (etoposide or
4-demethyl-epipodophyllotoxin-9-[4,6-O-ethylidene-β-D-glucopyranoside);
2.0 μg/ml; Sigma-Aldrich) was added to the remaining flasks.
The medium was not changed during the observation period (48 or 72
h). In order to assess cell damage by epifluorescence microscopy,
the cells were stained with acridine orange (AO) and ethidium
bromide (EB), which simultaneously revealed viable, apoptotic and
necrotic cells.
Isolation of nuclei and NM fractions
The cells were harvested by scraping them into cold
(4°C) phosphate-buffered saline (PBS) and centrifuging the
suspension at 200 x g for 10 min. To isolate the nuclei and NEs,
cell fractionations were carried out as previously described
(20). The integrity of the
nuclei was judged by phase contrast microscopy, and the purity of
the NMs was assessed by western blot analysis with an anti-lamin B1
(C-20) goat antibody (sc-6216; Santa Cruz Biotechnology, Inc.,
Heidelberg, Germany). The nuclei were suspended in an excess volume
of hypotonic buffer [10 mM Tris (pH 7.4), 10 mM
Na2HPO4, 5 mM sodium fluoride, 20 μM
sodium orthovanadate and complete EDTA-free protease inhibitor
mixture (Roche Diagnostics GmbH, Milan, Italy)] containing DNAse I
and heparin (0.2 and 5.0 mg/mg of nuclear protein, respectively).
This suspension was incubated at 4°C for 45 min and then
centrifuged at 9,500 x g for 15 min. The resulting pellet was the
NE-enriched NM fraction, while the supernatant was the
nucleoplasmic (NP) fraction. In this study, we focused on NMs, but
also probed total cell lysates (TCLs) as required.
Immunoprecipitation of NM-bound
proteins
Equal amounts of protein (150 μg) from the
NMs were used in the immunopre-cipitation experiments, which were
performed with Dynabeads Protein G (Life Technologies Italia,
Monza, Italy) and antibodies against PKCζ (sc-216), PDK1 (sc-17766)
or Bcl10 (sc-5273 mouse monoclonal or sc-5611 rabbit polyclonal;
Santa Cruz Biotechnology, Inc.) according to the manufacturer's
instructions. The immunocomplex-bearing beads were washed 5 times
with Tris-buffered saline [20 mM Tris (pH 7.4), 200 mM NaCl], 5 mM
sodium fluoride, 20 μM sodium orthovanadate, and complete
EDTA-free protease inhibitor mixture (Roche Diagnostics GmbH).
After a final wash, the immunocomplex-bearing beads were
resuspended in tris-buffered saline to measure PKCζ native activity
or in sample buffer for western blot analysis.
Western blot analysis
Equal amounts (10–20 μg) of protein from each
subcellular fraction were determined by means of Bio-rad Protein
Assay (Bio-rad Laboratories, Milan, Italy) and processed for
western blot analysis as previously described (15–18,20). Different antibodies (final
concentration, 1.0 μg/ml) were used to for antigen
detection: anti-PKCζ (C-20; sc-216), anti-[pho sphorylated
(p)-Thr410]-PKCζ (sc-101778), anti-Bcl10 (331.3 sc-5273
mouse monoclonal or H-197 sc-5611 rabbit polyclonal), anti-PDK1
(A-10; sc-17766) and anti-Lamin B1 (C-20; sc-6216; this antibody
was used to reveal equal loading of proteins) (all from Santa Cruz
Biotechnology, Inc.). The blots were then incubated with alkaline
phosphatase-conjugated anti-mouse, anti-rabbit or anti-goat IgGs
(Santa Cruz Biotechnology, Inc.) and stained with BCIP/NBT liquid
substrate reagent or CDP-Star (both from Sigma-Aldrich). The
developed blots were photographed using a digital camera, and the
Mr and density of each specific band were assessed using
SigmaGel™ software (Jandel Corp., San Rafael, CA, USA).
Assay for the detection of immunopurified
native PKCζ-specific activity
A fluorometric PKC activity assay kit, the Omnia™
Ser/Thr Recombinant Kit 8 (Life Technologies Italia), which
included the Omnia™ S/T-peptide-8 as a PKCζ-specific substrate, was
used. To measure the specific activity (μg/protein) of the
PKCζ isoforms immunoprecipitated from the NMs, the assay mixture
was set up as according to the manufacturer's instructions. No
co-factor was added to the immunocomplex-bearing beads, as the
detection of PKCζ basal activity does not need Ca2+, PS
or DAG. Each sample was incubated in the presence or absence of a
myristoylated PKCζ pseudosubstrate inhibitor (Myr-SIYRRGARRWRKL-OH,
100 μM; Sigma-Aldrich) to confirm the specificity of PKCζ
activity. The amounts of phosphorylated S/T-peptide-8 were
determined by measuring fluorescence (λex 360 nm;
λem 485 nm) according to the manufacturer's
instructions. The results were expressed in arbitrary units
calculated for each sample as ΔF μg−1
immunoprecipitated protein.
Assay for the detection of caspase-3
activity
The specific activity of caspase-3 was measured
using its specific 7-amido-4-methylcoumarin (AMC)-conjugated
substrate, Acetyl-Asp-Met-Gln-Asp-AMC (Ac-DMQD-AMC; 50 μM;
Bachem, Weil am Rhein, Germany) according to the procedure
described in our previous study (20). The results were expressed in
arbitrary units.
Isolation of p-caspase-3
Phosphoproteins were isolated using
PhosphoCruz-Agarose™ (Santa Cruz Biotechnology, Inc.) according to
the manufacturer's instructions. Briefly, 300 μg NM proteins
were diluted up to 1.0 ml with 50 mM MES, 1.0 M NaCl, 0.25% CHAPS
pH 6.6 (binding/washing buffer), and reacted with
PhosphoCruz-Agarose for 90 min at 4°C under swelling. Following 3
washes with binding/washing buffer, the phosphoproteins were eluted
from PhosphoCruz-Agarose in 100 mM ammonium bicarbonate, 0.25%
CHAPS pH 9.0 (elution buffer) and assessed by western blot analysis
using anti-caspase-3 antibody (clone 4-1-18; MAB4703; Merck
Millipore, Milan, Italy).
Isolation of cytoplasms and nuclei for
cell-free reconstituted N-C constructs
We followed our previously described procedure
(20). Untreated C4-I cells or
C4-I cells treated with VP-16 for 24 h were harvested by scraping
them into cold (4°C) PBS. The cells were then washed twice by
centrifugation/resuspension cycles in cold PBS, and incubated on
ice for 10 min at a density of 1.0×107 cells/ml in lysis
buffer [150 mM NaCl, 1.0 mM KH2PO4, 5.0 mM
MgCl2, 1.0 mM EGTA, 1.0 mM dithiothreitol, 20 μM
sodium orthovanadate, 5.0 mM sodium fluoride complete-EDTA-free
protease inhibitor mixture (Roche Diagnostics GmbH, Milan, Italy),
5.0 mM HEPES (pH 7.4), 10% glycerol and 0.3% Triton X-100]. The
lysates were then centrifuged (2,000 x g for 10 min at 4°C), and
their supernatants were the cytoplasmic extracts (C) from
either untreated (0 h) cells or cells treated with VP-16 for 24 h.
Intact nuclei (N) were isolated only from the untreated
cells, washed twice by centrifugation/resuspension in lysis buffer
without Triton X-100, and suspended at a final density of
1.0–2.0×107 nuclei/ml. Both types of cytoplasmic pools
(4 volumes) were then incubated at 30°C for 30 min with N
isolated from untreated cells (1 volume), thus forming the
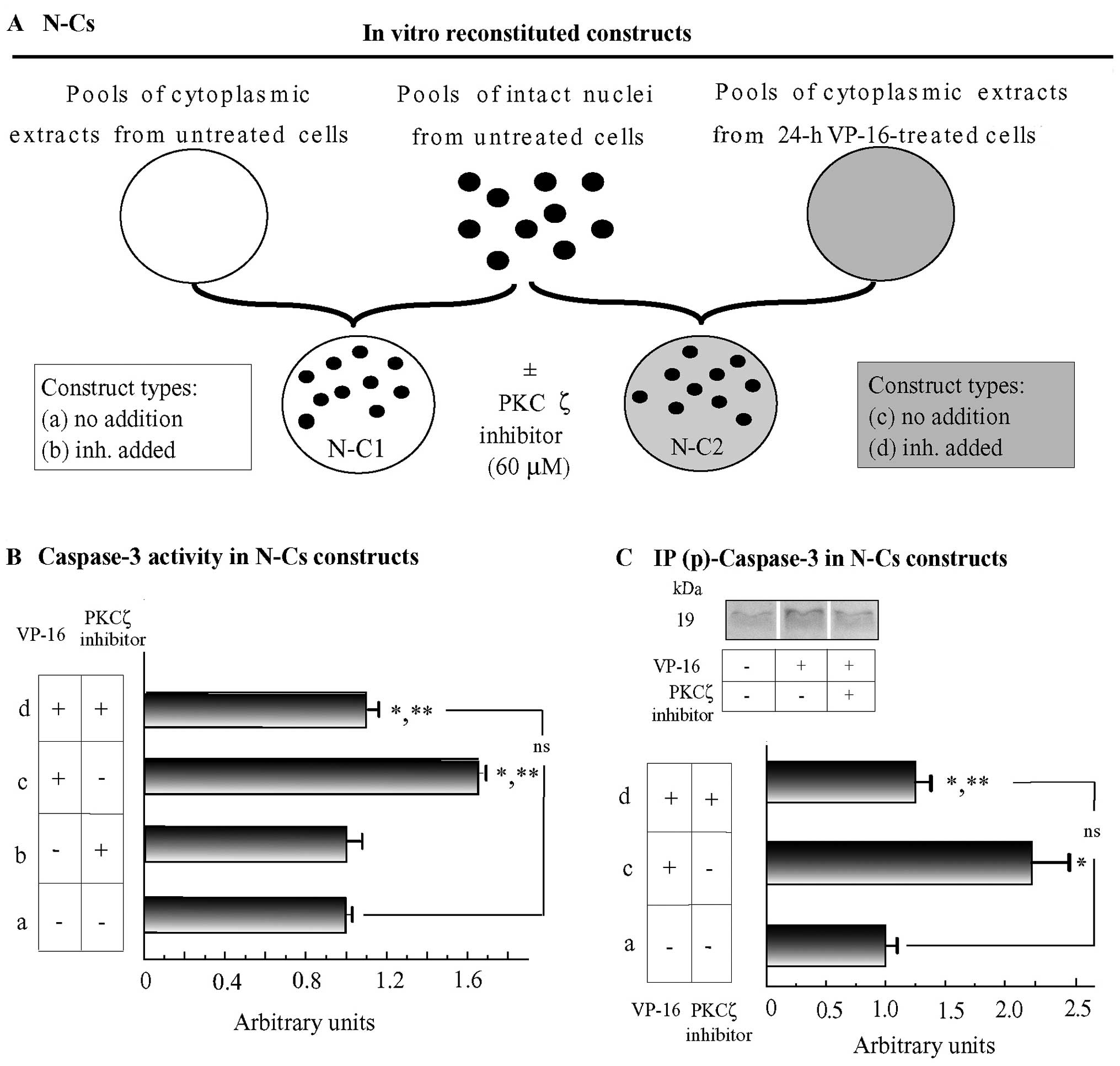
reconstituted constructs (N-Cs). Of these, we experimentally used 4
types i.e., types (a) and (b), untreated N-Cs
minus/plus (respectively) a specific inhibitor of PKCζ activity (60
μM); and types (c) and (d), VP-16-treated
C's plus untreated N minus/plus (respectively)
a specific inhibitor of PKCζ activity (60 μM) (Fig. 4A). p-Caspase-3 was assessed in the
NM-immunoprecipitated phosphoprotein fraction (using
PhosphoCruz-Agarose; Santa Cruz Biotechnology, Inc.) through
western blot analysis with anti-caspase-3 antibody.
Suppression of Bcl10 expression using
small interfering RNA (siRNA)
Chemically synthetic siRNA specifically suppressing
Bcl10 protein synthesis was purchased from Qiagen (Milan, Italy).
The sequences of this siRNA-Bcl10 were as follows: sense,
5′-GAA UCU AUU CGG CGA GAAA-3′, and antisense, 5′-UUU CUC GCC GAA
UAG AUUC-3′. The cells were plated in 6-well plates at a density of
45×103 cells/well, transfected with siRNA-Bcl10 (8.0 nM)
using HiPerFect reagent in siRNA buffer as the vehicle (HPF;
Qiagen), and cultured for 24 h according to the manufacturer's
instructions. Subsequently, a second dose of siRNA-Bcl10 (8.0 nM)
in HPF was administered simultaneously with VP-16 (2.0
μg/ml) at the experimental 0 h of the study and the cells
were sampled 24 h later. Relevant transfection controls were as
follows: i) cells were transfected with an Alexa Fluor 488-labeled
scrambled siRNA sequence (Qiagen) and ii) cells were treated only
with HPF. The entry of the transfection complexes into the C4-I
cells was confirmed through observation under a fluorescence
microscope (BX60™; Olympus Italia, Segrate, Milan, Italy) of the
cells that had been transfected with an Alexa Fluor 488-labeled
scrambled siRNA sequence (data not shown). The effectiveness of
silencing Bcl10 expression was determined by western blot analysis
of the NMs and TCLs with an a nti-Bcl10 monoclona l a ntibody (Sa
nt a Cr uz Biotechnology, Inc.).
Bcl10-suppressing lentiviral iRNA
(iBcl10) transactivation of C4-I cells
We used the human immunodeficiency virus type
1-derived lentiviral vector to generate a constitutively active
pLKO.1-puro/Bcl10 vector which would then be used for the specific
depletion of Bcl10. The most effective Bcl10-suppressing lentiviral
constructs had target sequences that differed from those of the
siRNA-Bcl10 (see above), that is: GTT GAA TCT ATT CGG CGA GAA or
GAA GTG AAG AAG GAC GCC TTA. The recombinant lentivirus was
produced by transducing HEK293FT cells using a ViraPower Lentiviral
Expression system (Life Technologies Italia). Briefly,
pLKO.1-puro/Bcl10 (3.0 μg) was co-transduced in subconfluent
HEK293FT cultures with the ViraPower Packaging Mix using
Lipofectamine 2000. Lentiviruses were harvested 48 h later,
filtered through a Millex-HV 0.45 μm and stored at −80°C,
according to the manufacturer's instructions. For RNA interference
experiments, the viral stocks were added to the C4-I cells
(0.8×106) supplemented with 4.0 μg/ml polybrene
(Santa Cruz Biotechnology, Inc.). Infected cells were selected by
incubation with 5.0 μg/ml puromycin (Sigma-Aldrich) for 4
days. A lentiviral non-target vector (nt) containing a short
hairpin that does not recognize any human or mouse gene (MISSION
pLKO.1-puro; Sigma-Aldrich) was used in parallel as a negative
control in all the transduction experiments. The effectiveness of
silencing Bcl10 expression was determined by western blot analysis
of the TCLs and/or NMs with an anti-Bcl10 monoclonal antibody
(Santa Cruz Biotechnology, Inc.).
Statistical analysis
One-way analysis of variance (ANOVA) with the post
hoc Holm-Sidak test (both pair-wise multiple comparison procedures)
were applied to the data, and a value of P<0.05 was considered
to indicate a statistically significant difference using the
Sigmastat 3.5™ software package (Systat Software Inc., Chicago, IL,
USA).
Results
Apoptotic effects of VP-16 on C4-I
cells
These effects have been previously detailed
(20). For the present aims, we
need only mention that the numbers of substrate-adherent viable
cells (i.e., those with a typically patterned AΟ-stained DNA; those
stained with EB were excluded) in the untreated cultures trebled
between the experimental 0 h (i.e., 24 h after plating in F-175
flasks and the addition of fresh medium at 24 h) and 72 h. C4-I
cell proliferation was inhibited immediately after the addition of
etoposide (VP-16; 2.0 μg/ml) and 24 h later the viable cell
numbers began to decline (72 h, −61% vs. 0 h; P<0.01).
PKCζ•Bcl10•PDK1 complexes increase at NMs
of apoptotic C4-I cells
PKCζ activation requires the phosphorylation of
Thr410 within its T-loop (which is effected by an
upstream kinase, PDK1) (37). We
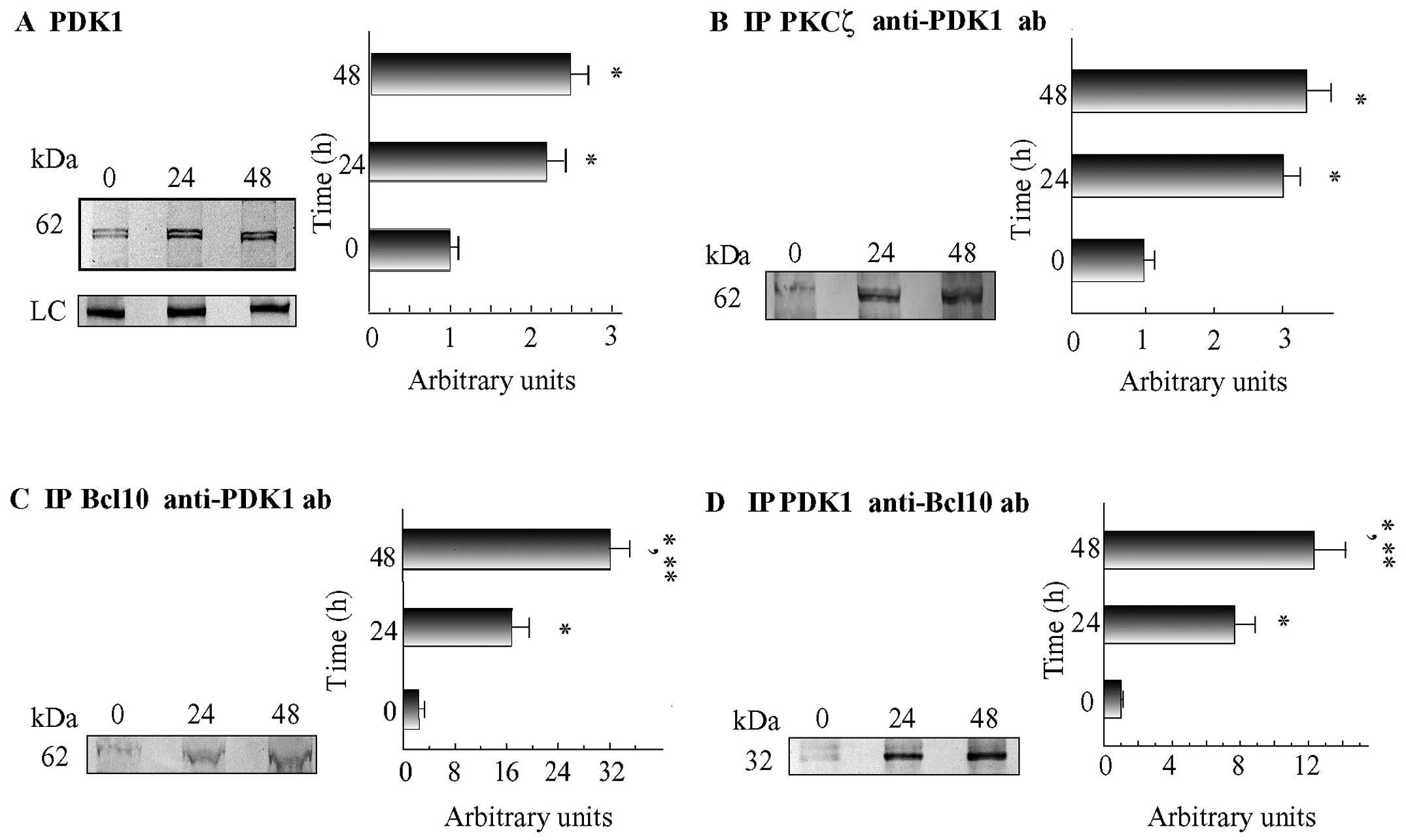
first confirmed by western blot analysis the previous
identification through PMF of PDK1 as an NM protein which
co-immunoprecipitates with PKCζ (20) (Fig.
1A). Furthermore, we found that following treatment with VP-16,
the levels of PDK1 at the NMs increased significantly [24 h,
2.2-fold; 48 h, 2.5-fold vs. 0 h; P<0.02 in both instances vs.
the 0 h (control) values] (Fig.
1A). Moreover, to determine the possible involvement of PDK1 in
the functioning of the PKCζ•Bcl10 complexes, we used
immunoprecipitation to examine whether PDK1 co-immunoprecipitates
with Bcl10 and PKCζ from the NMs of C4-I cells. We found that PKCζ
was associated with increasing levels of PDK1 (24 h, 3.0-fold; 48
h, 3.3-fold vs. 0 h; P<0.02 in both instances vs. 0 h values)
(Fig. 1B). Reverse
co-immunoprecipitation assays performed using an anti-PDK1 ab
confirmed PDK1's soaring association with PKCζ (data not shown). We
also observed that at the NMs of VP-16-treated cells, Bcl10
co-immmunoprecipitated with increasing amounts of PDK1 (24 h,
17.5-fold and 48 h, 32-fold vs. 0 h; P<0.001 in both instances
vs. the 0 h values) (Fig. 1C).
Finally, at the NMs of the VP-16-treated cells, PDK1 was associated
with increasing amounts of Bcl10 (24 h, 7.7-fold and 48 h,
12.3-fold vs. 0 h; P<0.05 in both instances vs. the 0 h values)
(Fig. 1D). Taken together, these
results indicate that PKCζ, Bcl10 and PDK1 are joined together in
heterotrimeric protein complexes that increasingly assemble over
time at the NMs of apoptotic C4-I cells.
Bcl10 knockdown prevents PDK1 from
linking with/activating PKCζ at NMs of apoptotic C4-I cells
The detected interaction of PDK1 with Bcl10 was
particularly noteworthy, as we had previously reported that
increasing amounts of assembled PKCζ•Bcl10 complexes accrued at the
NMs of VP16-treated C4-I cells (20,31). Therefore, in this study, we aimed
to establish whether the specific knockdown of Bcl10 expression
would hinder the assembly of PDK1•PKCζ complexes and, consequently,
hinder the increase in PKCζ activity at the NMs of apoptotic C4-I
cells. To this end, we used i) a specific siRNA targeting Bcl10
(siBcl10); and ii) a short hairpin Bcl10 mRNA-suppressing
lentiviral vector (iBcl10) targeting two nucleotide
sequences differing from that aimed at by the siBcl10 (see
Materials and methods).
siBcl10
As shown in Fig.
2A, similar levels of Bcl10 protein were found in the blots of
the TCLs of the untreated or VP-16-treated C4-I cells. A double
administration (at −24 and 0 h) of siBcl10 (8.0 nM) dissolved in
HPF suppressed, by 24 h, 78.0–80.3% of the Bcl10 protein level in
the TCLs from both the untreated and VP-16-exposed C4-I cells.
Notably, siRNA-mediated Bcl10 suppression brought about three
interrelated effects at the NMs of the C4-I cells treated with
VP-16 for 24 h: i) it prevented the increase in the assembly (0 h)
of PKCζ•PDK1 complexes, thereby suppressing the 3-fold increase
otherwise elicited by VP-16 in the Bcl10-expressing cells (Fig. 2B); ii) it reduced below basal
levels the PDK1-effected phosphorylation at Thr410 which
activated the PKCζ holoprotein, thereby obliterating the increase
in Thr410 phosphorylation occurring in VP-16-treated
Bcl10-expressing cells (Fig. 2C);
and iii) it completely blocked the surge, over basal levels, of
PKCζ activity observed in VP-16-exposed Bcl10-expressing cells
(Fig. 2D). Remarkably, these 3
effects were specific to the siRNA-mediated suppression of Bcl10,
since treatment with a scrambled siRNA neither modified basal PKCζ
activity nor hindered its VP-16-elicited increase (Fig. 2D).
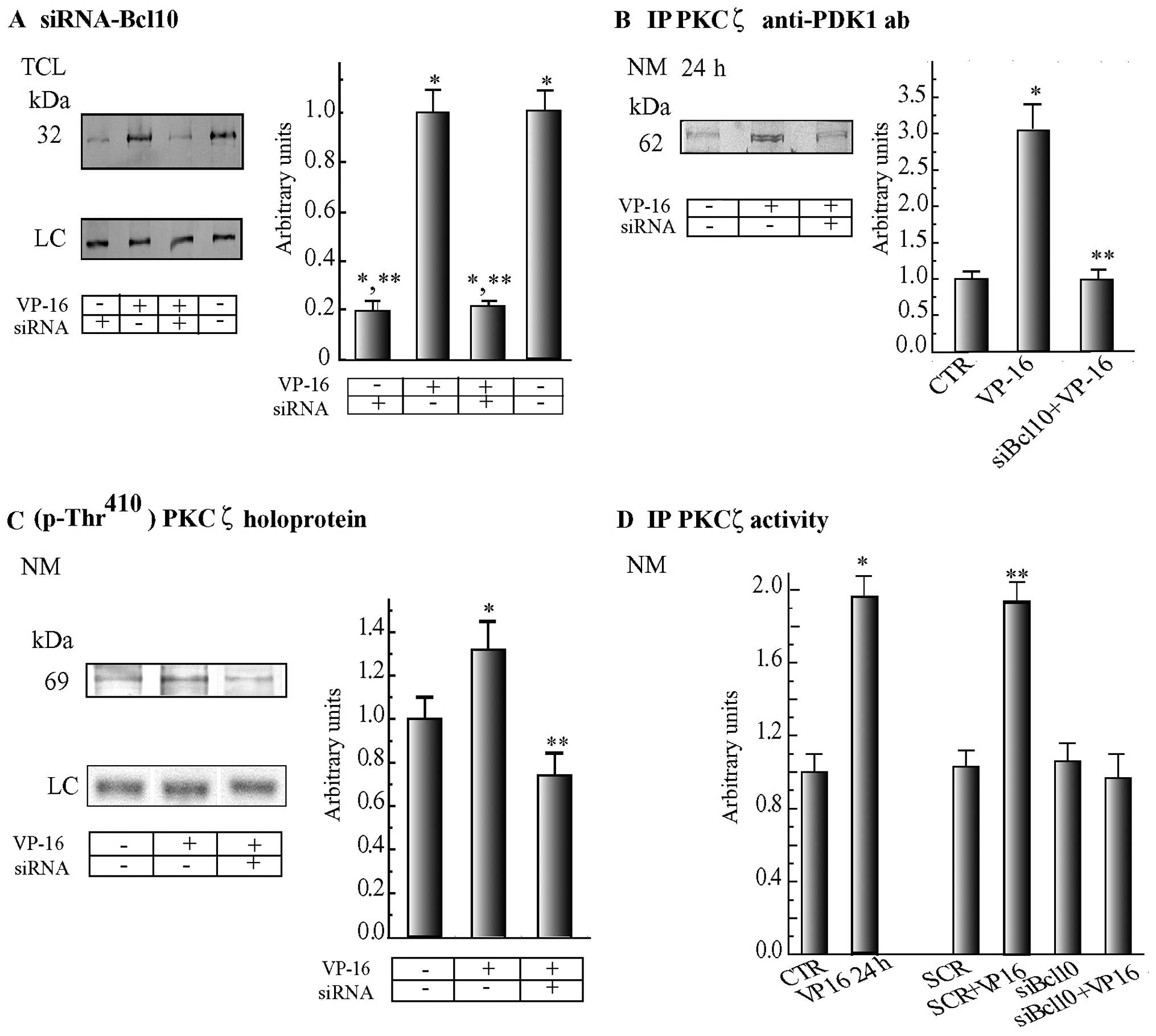 | Figure 2Suppression of B-cell lymphoma 10
(Bcl10) expression by siRNA prevents 3-phosphoinositide-dependent
protein kinase-1 (PDK1) from increasingly associating with protein
kinase (PK)Cζ at the NE (nuclear envelope), thereby hindering the
local surges in PKCζ phosphorylation and activity that would
otherwise occur in VP-16-treated C4-I cells. (A) The amount of
Bcl10 was similar in total cell lysates (TCLs) from both untreated
and VP-16 (2.0 μg/ml)-treated C4-I cells, but in both cases
they were reduced substantially and by similar degrees following a
double exposure (at −24 and 0 h) to a specific siRNA that mediated
Bcl10 suppression (siBcl10; 8.0 nM) dissolved in HiPerFect
reagent (HPF), as detailed in the Materials and methods. TCLs from
harvested cells were immunoblotted with an anti-Bcl10 monoclonal
antibody. (B) Bcl10 suppression by means of a specific siRNA
(siBcl10) prevented PDK1 from increasing its levels of association
with PKCζ at the nuclear membrane (NM) fractions of cells treated
with VP-16 for 24 h over the basal levels of untreated control
(CTR) cells. PKCζ was immunoprecipitated from equal aliquots of the
NM fractions, and the immunoprecipitation samples were blotted and
treated with an anti-PKD1 antibody. (C) Suppression of Bcl10 by
siRNA also decreased to below starting (0 h) levels the
phosphorylation at Thr410 of PKCζ holoproteins, caused
by PDK1, as revealed by a specific phosphorylated antibody (which,
however, did not bind p-Thr410-PKCζ catalytic
fragments). Conversely, by 24 h p-Thr410-PKCζ
holoprotein was beginning to surge in C4-I cells treated with VP-16
alone. Cells were processed as in (B). (D) Suppression of Bcl10 by
siRNA prevented the increase in NM-linked PKCζ native-specific
activity from happening by 24 h of VP-16 exposure (siBcl10 +
VP-16). By contrast, PKCζ native-specific activity increased at the
NMs of C4-I cells treated with VP-16 alone (VP-16 24 h). A
scrambled (SCR) siRNA did not change the basal (0 h) levels of
PKCζ-specific native activity assayed at the NM fractions of C4-I
cells and did not hinder its increase 24 h after the addition of
VP-16 (SCR + VP-16 24 h). CTR, untreated cells; siBcl10,
Bcl10-siRNA. Fluorometric assays of PKCζ activity were carried out
as detailed in Materials and methods. Results are expressed in
arbitrary units as ∆F μg/NM protein. Representative western
blots are shown on the left panels in (A–C). The corresponding
densitometric data of the specific protein bands were normalized,
taking as 1.0 the value of Bcl10 (A) or PDK1 (B) or
p-Thr410-PKCζ holoprotein (C) of the untreated (0 h)
control specimens, and expressed in arbitrary units. These data are
presented as the means ± SE (n=4 in all instances). (A–D)
*P<0.02 (at least) compared with untreated (0 h) cell
values (CTR), ** P<0.05 (at least) compared with
values of cells treated with VP-16 alone. LC, loading control; ab,
antibody. |
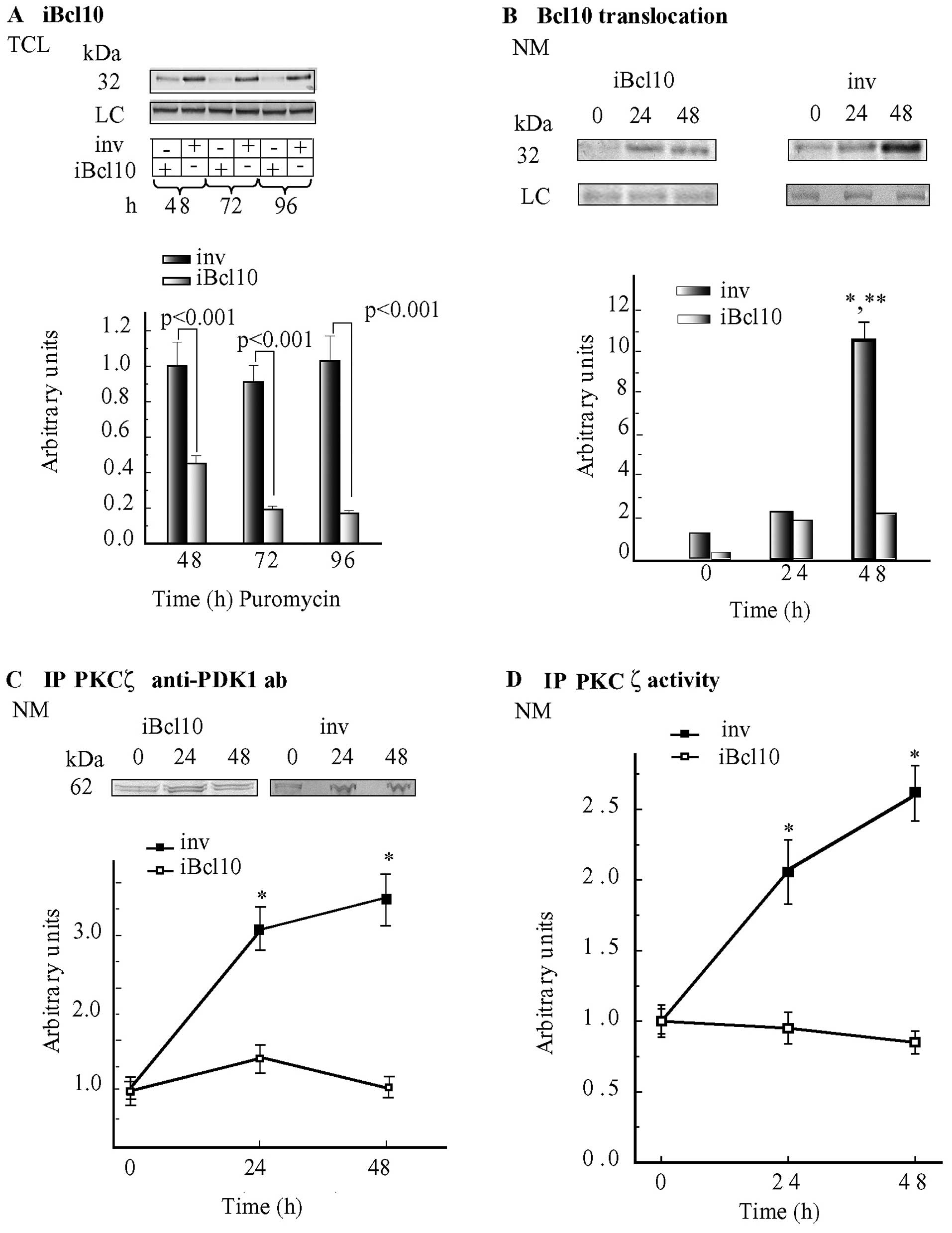
iBcl10
We also explored the role(s) of Bcl10 in the
association of PKCζ with PDK1 using a Bcl10-suppressing lentiviral
vector (iBcl10) and, as control, a non-target vector
(inv). In keeping with previous our findings (20), Bcl10 levels remained steady in the
TCL samples from the inv-transduced cells, whereas in the
TCL samples from iBcl10-transduced cells, which were not
treated with VP-16, these levels decreased by ~82% (P<0.001)
after 72 and 96 h of puromycin selection (Fig. 3A).
At the NM, samples from iBcl10-transduced
cells, not treated with VP-16 (puromycin selection 48 h =
experimental 0 h time point), the levels of Bcl10 protein plummeted
to 12.5% of those detected at NMs isolated from
inv-transduced cells (Fig.
3B). However, a significant loading (10.5-fold vs. 0 h;
P<0.001) of Bcl10 onto NMs took place 48 h after the addition of
VP-16 to the inv-transduced cells (Fig. 3B). Conversely, Bcl10 translocation
increased 13.5-fold at the NMs isolated from
iBcl10-transfected cells by 24 h vs. the corresponding 0 h
controls, but changed little between 24 and 48 h, and reached
maximum levels that were only ~20% of those found at the NMs of
VP-16-treated inv-transduced cells at 48 h (Fig. 3B). Hence, the suppression of Bcl10
significantly reduced Bcl10 translocation onto the NE.
The basal (0 h) levels of PDK1
co-immunoprecipitating with PKCζ were similar at the NMs isolated
from VP-16-untreated inv-transduced C4-I cells, or
iBcl10-transfected C4-I cells or wt C4-I cells (Fig. 3C and cf. 1B). However, at the NMs
isolated from VP-16-treated (24 and 48 h) iBcl10-transfected
cells, the levels of PDK1-linked PKCζ were not altered vs. the 0 h
levels, revealing that with respect to the starting levels, no
increase in the association between PKCζ and PDK1 had occurred;
this observation was at sharp variance with the increases in
PDK1-PKCζ complexes that were detected at the NMs from VP-16
treated inv-transduced and wt cells (Fig.x 3C and cf. 1B). Notably,
immunoprecipitated PKCζ native activity was not altered at the NMs
from VP-16-treated iBcl10-transfected cells, whereas it
increased significantly at the NMs from VP-16-treated
inv-transduced cells (Fig.
3D) and wt cells (data not shown).
PKCζ promotes caspase-3 phosphorylation,
enhancing its proteolytic activity at NMs of wt, but not
siBcl10-transfected or iBcl10-transfected apoptotic C4-I cells
Caspase-3 has been previously identified as a
protein that co-immunoprecipitates with the PKCζ•Bcl10 complexes at
the NMs of VP-16-treated cells (20), and actively cleaves PKCζ. Thus, in
this study, we aimed to define the interactions between caspase-3
and the PKCζ•Bcl10 complex and determine whether PKCζ-specific
activity has any influence on the association of caspase-3 with
PKCζ•Bcl10 complexes. To address this issue, we used a
PKCζ-specific pseudosubstrate inhibitor (60 μM). This
inhibitor curtailed 90% of PKCζ activity immunoprecipitated from
the NMs of VP-16-treated C4-I cells, but at the same time was
highly cytotoxic and caused massive cell death within 4 h, which
consequently obscured the results (data not shown). To overcome
this hurdle, we generated 4 types of cell-free N-Cs, to which the
PKCζ pseudosubstrate inhibitor (60 μM) was or was not added
(Fig. 4A). The cell-free N-C
constructs model consisted of intact nuclei from untreated (0 h)
cells mixed with either cytoplasms from untreated (0 h) cells
(N-C1) or apoptotic cytoplasms from cells previously exposed
to VP-16 for 24 h (N-C2). Note that the first 24 h are the
time point when PKCζ native-specific activity associated to Bcl10
doubled and showed a major increase in the VP-16-treated cells as
compared to the control cells (see Fig. 3D). Both these constructs were
incubated at 30°C for 30 min before their NMs were isolated. We
found that adding the PKCζ activity inhibitor wholly suppressed any
increase in caspase-3 activity occurring at the NMs from
N-C2 constructs with respect to N-C1 ones (Fig. 4B). Therefore, caspase-3
recruitment and activation at the NE specifically required PKC-ζ
phosphorylating activity at the NM of apoptotic C4-I cells.
To validate this result, we investigated the
phosphorylation levels of caspase-3 during VP16-induced apoptosis.
We immunoprecipitated the phosphorylated proteins of the NMs
isolated from N-C constructs and then probed the immunoblots of the
immunoprecipitated components with a specific anti-caspase-3
antibody. As shown in Fig. 4C,
the levels of p-caspase-3 (p19 large sub-unit) increased markedly
at the NMs of the VP16-treated C4-I cells, and this was prevented
from occurring by the specific PKC-ζ inhibitor.
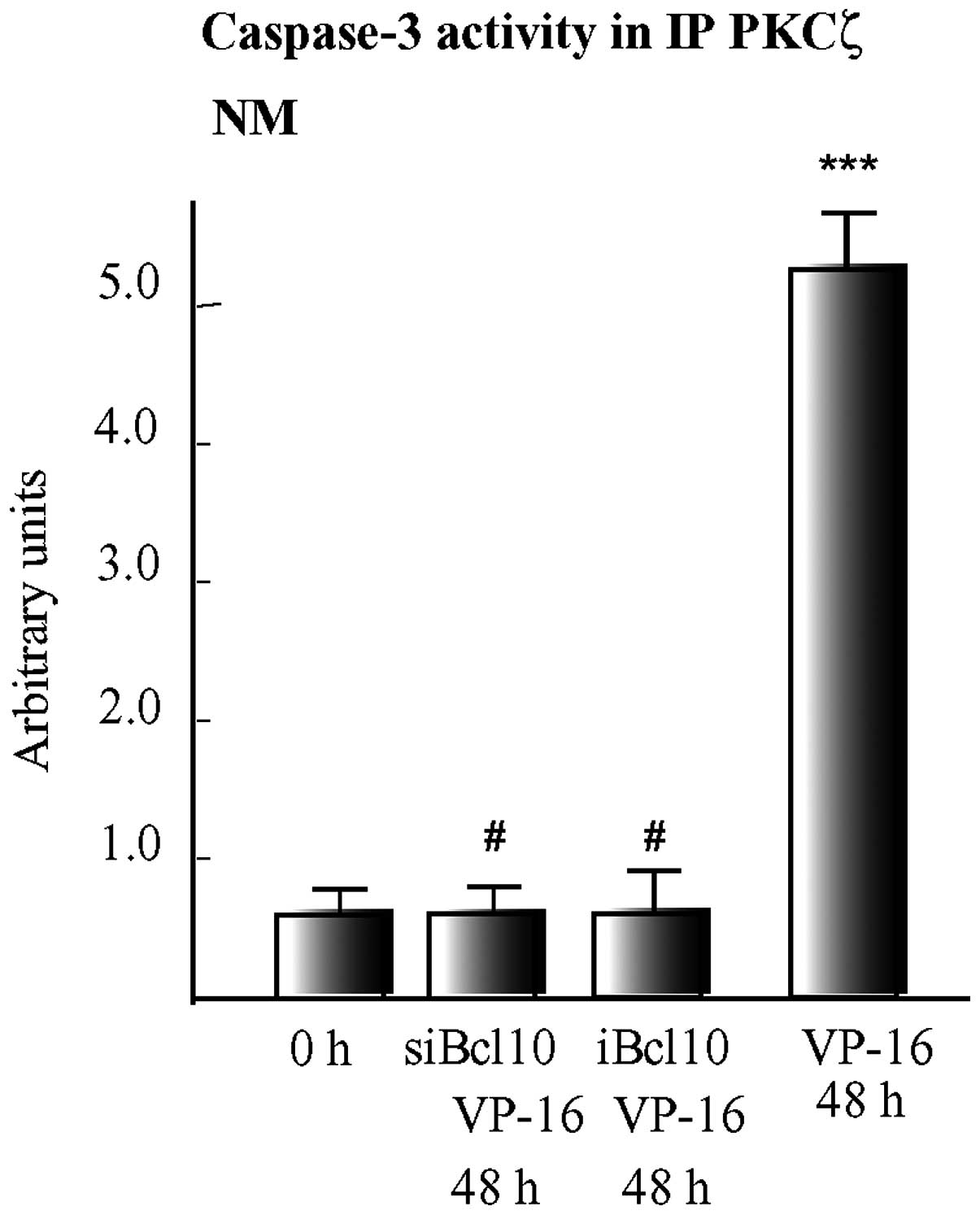
Finally, we examined the effects of suppressing
Bcl10 expression, using a specific siRNA or lentiviral vector on
caspase-3 activity. We found that 48 h after the addition of VP-16,
caspase-3 activity co-immunoprecipitating with PKCζ at the NMs of
wt cells increased 5-fold (P<0.001) vs. the 0 h values (Fig. 5), thus reaching its maximum value
and concurrently the amount of PKCζ catalytic fragments cleaved by
caspase-3 progressively increased at the NMs topping by 48 h and 72
h of VP-16 treatment [for details see (19)]. By contrast, the suppression of
Bcl10 by siBcl10 or iBcl10 prevented any increase in
PKCζ-associated caspase-3 activity from occurring in the apoptotic
cells. These results indicated that Bcl10 nucleated complexes
comprising PDK1, PKCζ and caspase-3, which eventually led to the
phosphorylation and enhancement of the activity of caspase-3, as
previously shown (20), and in
turn cleaved and inactivated the PKCζ holoproteins.
Taken together, these results prove that, under
VP-16 treatment, Bcl10 plays a crucial role at the NE by
influencing the interaction between PDK1 and PKCζ; this interaction
allows PDK1 to phosphorylate at Thr410 and activate
PKCζ, and active PKCζ to phosphorylate Bcl10. Next, phosphorylated
Bcl10 causes a further interaction of PKCζ with caspase-3 that
enhances the proteolytic activity of the latter and effects the
pro-apoptotic cleavage and inactivation of PKCζ (Fig. 5).
Discussion
PKCζ is overexpressed in squamous HCCs, HeLa cells
and C4-I cells (20,38). In a previous study (20), we investigated the role(s) that
PKCζ plays at the NE of C4-I cells by analyzing, using proteomics
analysis, the proteins with which PKCζ interacts. We found that 31
proteins and 33 proteins co-immunoprecipitated with PKCζ from the
NMs, respectively, of proliferating (untreated) and VP-16-treated
C4-I cells, respectively. Only 8 of these proteins, including the
Bcl10 protein, were present in both types of NM samples. The
identified PKCζ-interacting NM proteins were assigned to 8 main
functional groups, the relative percentage fractions of which
differed in the NMs isolated from untreated vs. VP-16-treated
cells. The protein groups related to transcription control, signal
transduction and mitotic cell cycle regulation were prominent in
the untreated NM samples, whereas in the VP-16-treated NM samples
the protein group regulating programmed cell death was most
prominent, followed by the signaling and transcription-regulating
protein group. However, as previously noted, 71% of the signaling
proteins, 86% of the apoptosis-regulating proteins, and 100% of the
cell cycle- and transcription regulation-related and
chaperone/adapter proteins were utterly dissimilar in the two
situations considered (20).
Contentious opinions have been expressed in the past
about the pro- or anti-apoptotic roles played by PKCζ (39–41). Our results from proteomics
analysis clearly demonstrated that PKCζ changed nearly 86% of its
interacting proteins at the NMs of C4-I cells. Therefore, we
surmised that PKCζ operates as a role-shifting kinase that
interacts with and phosphorylates widely differing sets of NM
proteins under conditions of growth or apoptosis (20).
In addition, in our previous study, we identified a
hitherto unreported PKCζ•Bcl10 complex, the amount of which surged
with time at the NMs of apoptotic C4-I cells (20). While selecting the
PKCζ-interacting NM proteins, we noted Bcl10 since i) it
co-immunoprecipitated with PKCζ from the NMs of both growing and
apoptotic cells; and ii) in 2-DE gels of the VP-16-treated samples,
we observed by 24 h a shift toward more acidic values of the
pIs of both PKCζ and Bcl10 vs. the 0 h values, which
suggested that post-translational changes, e.g., phosphorylations,
had occurred during this time lapse (20). We observed that PKCζ increasingly
interacted with Bcl10 at the NMs of apoptotic C4-I cells and
provided evidence that PKCζ must be added to the group of the known
Bcl10-phosphorylating kinases (20). Conversely, among the proteins
interacting with Bcl10•PKCζ complexes at the same NMs we did not
find any protein(s) already known to associate with Bcl10 or to
partake with it in the CBM signalosomes (20). Bcl10 has been described as an
apoptosis-regulating protein and tumor suppressor gene involved in
NF-κB-mediated functions (21,26,28,29,42,43). Hitherto, studies on Bcl10 have
mostly been carried out on various types of lymphomas (44). In such tumors, Bcl10 is
phosphorylated by several PKs, including
Ca2+-calmodulin-dependent PKII, AKT1, p38-MAPK, IκB
kinase and PKC (21,30). Recently Kuo et al (45) demonstrated that the suppression of
Bcl10 by siRNA hindered the growth of 4 otherwise untreated HCC
cell lines (i.e., C33A, CaSki, HeLa and SiHa) through the
NF-κB-dependent regulation of cyclin D1.
The results of our present study attest, to the best
of our knowledge, for the first time that another PK, PDK1,
associates with the Bcl10•PKCζ complexes, thus forming the
PDK1•Bcl10•PKCζ heterotrimer at the NMs of C4-I cells. Under basal
(untreated) conditions, these heterotrimers are scanty at NMs.
However, while undergoing VP-16-induced apoptosis, a massive
increase of Bcl10, PDK1 and PKCζ onto NMs and consequently an
increasing assembly of the PDK1•Bcl10•PKCζ complexes occur with
time. In keeping with the well-established view that PDK1
phosphorylates at Thr410 and activates PKCζ (37), we demonstrated in this study that
this happens also in the PDK1•Bcl10•PKCζ complexes at the NMs of
C4-I cells. However, we demonstrated in the present study that
during VP-16-induced apoptosis, PDK1 interacts with PKCζ only when
both proteins are complexed with Bcl10. The specific function of
PDK1 has been shown to be regulated not only by its own
phosphorylation, but also by protein-protein interactions and its
subcellular localizations (33–35). We demonstrated in this study that
the depletion of Bcl10 expression by either specific siRNA
suppression or lentiviral transactivation wholly prevented the
otherwise increasing interaction between PDK1 and PKCζ and, hence,
the PKCζ-activating phosphorylation at Thr410 by PDK1.
Therefore, according to our findings, during VP-16-induced
apoptosis, Bcl10 acts as the essential pivot which nucleates the
assembly of PDK1•Bcl10•PKCζ complexes at NMs.
The influence of Bcl10 at the NMs of apoptotic C4-I
cells is far reaching and is triggered by the phosphorylation of
Bcl10 on PDK1-activated PKCζ (20). Since the specific depletion of
PKCζ through lentiviral iRNA transactivation prevents the
accumulation of Bcl10 at NMs in apoptotic C4-I cells, it seems
likely that PKCζ first translocates to the NE; secondly, it enlists
Bcl10 there; thirdly, it is phosphorylated and activated by
Bcl10-recruited PDK1; and fourthly, it phosphorylates Bcl10
(20) (Fig. 6). p-Bcl10 then nucleates a second
complex (or expands the first one) comprising PKCζ and active
caspase-3. This is revealed by results gained by the specific
lentiviral-transduced or siRNA-mediated depletion of Bcl10,
demonstrating that under these conditions the progressive
proteolytic cleavage and functional inactivation of PKCζ by active
caspase-3 no longer take place (20). Of note, Yan et al (28) reported that in 293T and MCF7
cells, Bcl10 interacts via a specific CARD with the upstream
pro-caspase-9, thus mediating the autoproteolytic activation of the
latter through oligomerization (another proapototic activity of
Bcl10). Conversely, in the same cell types, the downstream effector
caspase-3 failed to co-immunoprecipitate with Bcl10 (28). The authors pointed out that their
evidence had intrinsic flaws since it was based upon ectopically
overexpressed proteins, and that the operative mechanism(s) through
which Bcl10 induced apoptosis remained unclear (28). In our previous study (20), the C4-I cells did not overexpress
any protein, nor did we detect caspase-9 association with Bcl10. By
contrast, we found that caspase-3 co-immunoprecipitated with the
PKCζ complexes from the NMs of VP-16-treated cells. It is
increasingly recognized that the activation of caspases is
controlled by several mechanisms, including protein-protein
interactions, post-translational modifications and phosphorylations
by specific kinases (46,47). Our findings indicate that
caspase-3 co-immunoprecipitated with PKCζ complexes from the NMs of
VP-16-treated cells. The association with PKCζ suggests that
caspase-3 is one of the main substrates for Bcl10-bound
PDK1-activated PKCζ during apoptosis. We have not directly proved
that caspase-3 is a PKCζ substrate in vitro, but by using
cell-free in vitro reconstituted N-C constructs we provide
evidence that the enhancement of caspase-3 activity requires
functionally mature PKCζ. The suppression of PKCζ activity by a
specific inhibitor (highly cytotoxic to whole cells) prevented the
increased phosphorylation of 19-kDa caspase-3 and the enhancement
of its activity in the N-C constructs. In this regard, Voss et
al (48) demonstrated in
human monocytes that PKCδ, another PKC isoform, associated with
caspase-3 and phosphorylated it in vitro, thereby enhancing
caspase-3 activity. Moreover, in our model, when Bcl10 expression
was suppressed, caspase-3 did not co-immunoprecipitate with the
Bcl10•PKCζ complexes. Therefore, it seems feasible that
PKCζ-phosphorylated Bcl10 either directly or through one or more
yet to be identified go-between(s) links caspase-3 moieties to the
phosphorylated Bcl10•PKCζ complexes (Fig. 6). This would allow PKCζ to
phosphorylate and activate caspase-3, and in turn, activated
caspase-3 would cleave and inactivate PKCζ (Fig. 6).
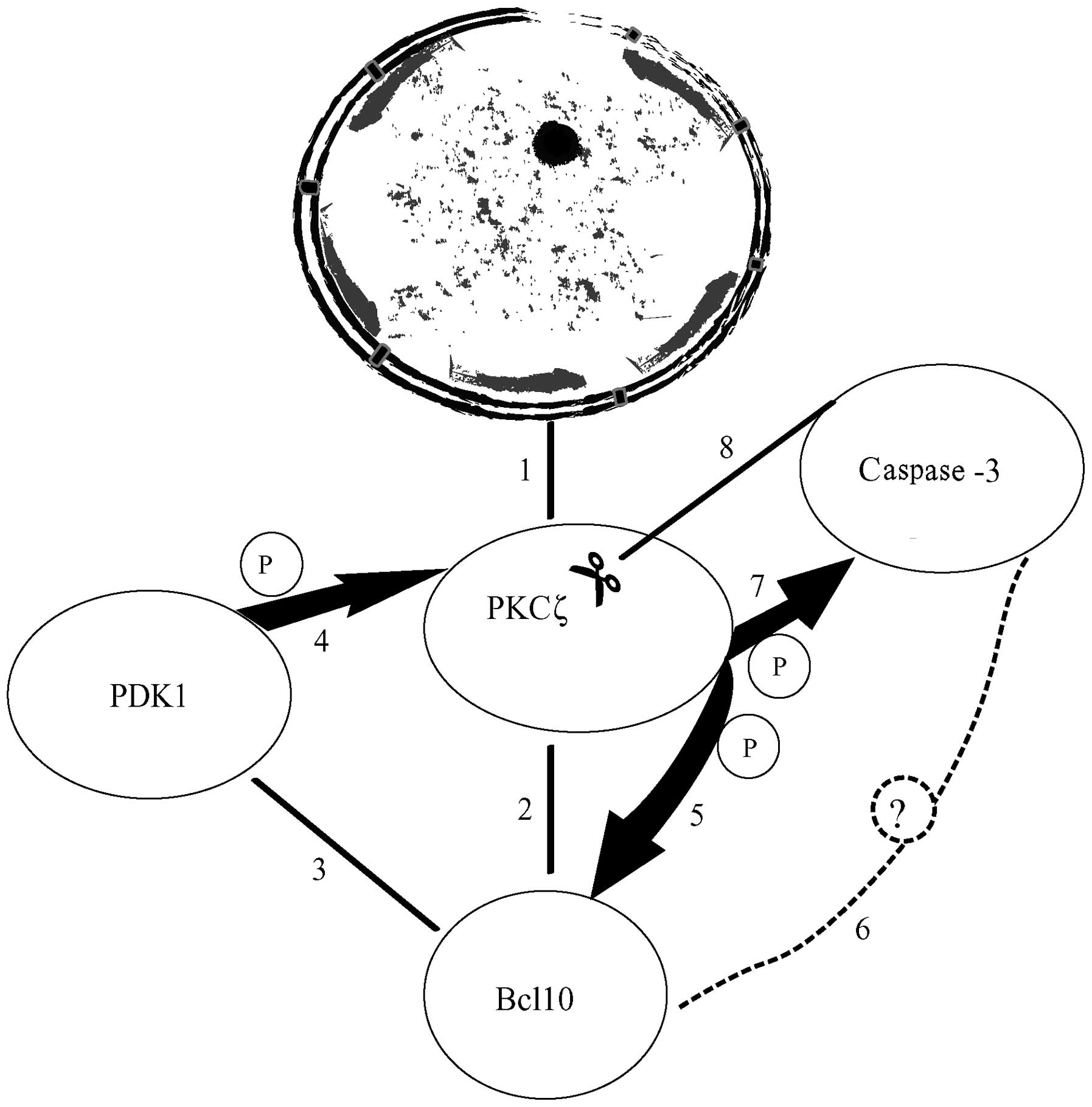 | Figure 6Depiction of the sequence of events
leading to the assembly of a pro-apoptotic protein complex at the
nuclear membranes (NMs) of C4-I cells following treatment with
VP-16. (1) Initially, protein
kinase (PK)Cζ increasingly connects with the nuclear envelope (NE).
(2) B-cell lymphoma 10 (Bcl10)
associates with NE-linked PKCζ. (3) Subsequently,
3-phosphoinositide-dependent protein kinase-1 (PDK1) associates
with PKCζ•Bcl10, thus forming a heterotrimeric complex. (4) PDK1 then phosphorylates and activates
PKCζ. (5) Activated
phosphorylated (p)-PKCζ in turn phosphorylates Bcl10. (6) p-Bcl10 is now capable of forming a
new complex or enlarging the existing complex (as shown here),
likely by incorporating a still unidentified member (dashed circle
with question mark) that in turn binds to 19-kDa caspase-3.
(7) 19-kDa caspase-3 would then
be phosphorylated by Bcl10-linked PKCζ, an event enhancing its
proteolytic activity. (8)
Finally, caspase-3 (whose activity is enhanced) cleaves the
Bcl10-bound PKCζ holoprotein. Reportedly, PKCζ proteolysis causes a
transient increase followed by a decrease in enzyme activity
levels: this decrease in activity was observed between 48 and 72 h
of VP-16 treatment in our model [Chiarini et al (19)]. An effective lentiviral or siRNA
suppression of Bcl10 expression prevents the assembly of the
multiprotein complex(es) and its consequences (cf. Figs. 2 and 3). Therefore, Bcl10 plays a pivotal
role, mediating the unidirectional regulation of PKCζ by PDK1 and
the bidirectional pro-apoptotic interactions between 19 kDa
caspase-3 and active PKCζ. For clarity, the size of the
multiprotein complexes shown has been enlarged out of proportion
with respect to the nucleus it binds. |
Hence, the assembled phosphorylated Bcl10•PKCζ
complexes associate with caspase-3, and thus it is clear that they
play a pro-apoptotic role at the NMs of VP-16-treated C4-I cells.
These results permit us to surmise that both PKCζ and Bcl10 are
potential targets for novel approaches to HCC therapy.
Acknowledgments
This study was partly supported by the Italian
Ministry of Education, University and Research (MIUR; FUR 2013/4).
We are deeply grateful to Professor Nicole Murray and Professor
Alan Fields (Mayo Clinic, Jacksonville, Florida, USA) for their
useful comments on lentiviral transduction experiments and to Dr
Maddalena Marconi for her skillful technical assistance.
Abbreviations:
|
inv
|
non-target vector
|
|
HCC
|
human cervical carcinoma
|
|
iBcl10
|
short hairpin Bcl10 mRNA-suppressing
lentiviral vector
|
|
NE
|
nuclear envelope
|
|
NM
|
nuclear membrane
|
|
PDK1
|
3-phosphoinositide-dependent protein
kinase-1
|
|
PK
|
protein kinase
|
|
siBcl10
|
small interfering RNA mediating Bcl10
suppression
|
|
TCL
|
total cell lysate
|
|
wt
|
wild-type
|
References
|
1
|
Mukherjee S, Dey S, Bhattacharya RK and
Roy M: Isothiocyanates sensitize the effect of chemotherapeutic
drugs via modulation of protein kinase C and telomerase in cervical
cancer cells. Mol Cell Biochem. 330:9–22. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Scarinci IC, Garcia FA, Kobetz E,
Partridge EE, Brandt HM, Bell MC, Dignan M, Ma GX, Daye JL and
Castle PE: Cervical cancer prevention: new tools and old barriers.
Cancer. 116:2531–2542. 2010.PubMed/NCBI
|
|
3
|
Molano M, Van den Brule A, Plummer M,
Weiderpass E, Posso H, Arslan A, Meijer CJ, Muñoz N and Franceschi
S; HPV Study Group: Determinants of clearance of human
papillomavirus infections in Colombian women with normal cytology:
a population-based, 5-year follow-up study. Am J Epidemiol.
158:486–494. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Perez-Plasencia C, Duenas-Gonzalez A and
Alatorre-Tavera B: Second hit in cervical carcinogenesis process:
involvement of wnt/beta catenin pathway. Int Arch Med. 1:102008.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kwasniewska A, Postawski K,
Gozdzicka-Jozefiak A, Kwasniewski W, Grywalska E, Zdunek M and
Korobowicz E: Estrogen and progesterone receptor expression in
HPV-positive and HPV-negative cervical carcinomas. Oncol Rep.
26:153–160. 2011.PubMed/NCBI
|
|
6
|
Zhu X, Lv J, Yu L, Zhu X, Wu J, Zou S and
Jiang S: Proteomic identification of differentially-expressed
proteins in squamous cervical cancer. Gynecol Oncol. 112:248–256.
2009. View Article : Google Scholar
|
|
7
|
Higareda-Almaraz JC, Enríquez-Gasca MR,
Hernández-Ortiz M, Resendis-Antonio O and Encarnación-Guevara S:
Proteomic patterns of cervical cancer cell lines, a network
perspective. BMC Syst Biol. 5:962011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Carlson MW, Iyer VR and Marcotte EM:
Quantitative gene expression assessment identifies appropriate cell
line models for individual cervical cancer pathways. BMC Genomics.
8:117–129. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dempsey EC, Newton AC, Mochly-Rosen D,
Fields AP, Reyland ME, Insel PA and Messing RO: Protein kinase C
isozymes and the regulation of diverse cell responses. Am J Physiol
Lung Cell Mol Physiol. 279:L429–L438. 2000.PubMed/NCBI
|
|
10
|
Reyland ME: Protein kinase C isoforms:
multi-functional regulators of cell life and death. Front Biosci
(Landmark Ed). 14:2386–2399. 2009. View
Article : Google Scholar
|
|
11
|
Breitkreutz D, Braiman-Wiksman L, Daum N,
Denning MF and Tennenbaum T: Protein kinase C family: on the
crossroads of cell signaling in skin and tumor epithelium. J Cancer
Res Clin Oncol. 133:793–808. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Diaz-Meco MT, Dominguez I, Sanz L, Dent P,
Lozano J, Municio MM, Berra E, Hay RT, Sturgill TW and Moscat J:
zeta PKC induces phosphorylation and inactivation of I kappa
B-alpha in vitro. EMBO J. 13:2842–2848. 1994.PubMed/NCBI
|
|
13
|
Filomenko R, Poirson-Bichat F, Billerey C,
Belon JP, Garrido C, Solary E and Bettaieb A: Atypical protein
kinase C zeta as a target for chemosensitization of tumor cells.
Cancer Res. 62:1815–1821. 2002.PubMed/NCBI
|
|
14
|
Malhas AN and Vaux DJ: The nuclear
envelope and its involvement in cellular stress responses. Biochem
Soc Trans. 39:1795–1798. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Dal Pra I, Whitfield JF, Chiarini A and
Armato U: Changes in nuclear protein kinase C-delta holoenzyme, its
catalytic fragments, and its activity in polyomavirus-transformed
pyF111 rat fibroblasts while proliferating and following exposure
to apoptogenic topoisomerase-II inhibitors. Exp Cell Res.
249:147–160. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chiarini A, Whitfield JF, Armato U and Dal
Pra I: Protein kinase C-beta II Is an apoptotic lamin kinase in
polyomavirus-transformed, etoposide-treated pyF111 rat fibroblasts.
J Biol Chem. 277:18827–18839. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chiarini A, Whitfield JF, Armato U and Dal
Pra I: VP-16 (etoposide) and calphostin C trigger different nuclear
but akin cytoplasmic patterns of changes in the distribution and
activity of protein kinase C-betaI in polyomavirus-transformed
pyF111 rat fibroblasts. Int J Mol Med. 17:111–120. 2006.
|
|
18
|
Chiarini A, Whitfield JF, Pacchiana R,
Armato U and Dal Pra I: Photoexcited calphostin C selectively
destroys nuclear lamin B1 in neoplastic human and rat cells - a
novel mechanism of action of a photodynamic tumor therapy agent.
Biochim Biophys Acta. 1783:1642–1653. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Watanabe Y, Hoshiai H, Nakanishi T,
Kawamura N, Tanaka N, Isaka K, Kamiura S, Ohmichi M, Hatae M and
Ochiai K: Evaluation of oral etoposide in combination with
cisplatin for patients with recurrent cervical cancer: long-term
follow-up results of a Japanese multicenter study. Anticancer Res.
31:3063–3067. 2011.PubMed/NCBI
|
|
20
|
Chiarini A, Marconi M, Pacchiana R, Dal
Prà I, Wu J and Armato U: Role-shifting PKCζ fosters its own
proapoptotic destruction by complexing with Bcl10 at the nuclear
envelope of human cervical carcinoma cells: a proteomic and
biochemical study. J Proteome Res. 11:3996–4012. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Willis TG, Jadayel DM, Du M-Q, Peng H,
Perry AR, Abdul-Rauf M, Price H, Karran L, Majekodunmi O, Wlodarska
I, et al: Bcl10 is involved in t(1;14)(p22;q32) of MALT B cell
lymphoma and mutated in multiple tumor types. Cell. 96:35–45. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen M, Li LY and Qi Y-P: Bcl10 protein
can act as a transcription activator in yeast. Mol Cell Biochem.
246:97–103. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Liu Y, Dong W, Chen L, Zhang P and Qi Y:
Characterization of Bcl10 as a potential transcriptional activator
that interacts with general transcription factor TFIIB. Biochem
Biophys Res Commun. 320:1–6. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Thome M and Weil R: Post-translational
modifications regulate distinct functions of CARMA1 and BCL10.
Trends Immunol. 28:281–288. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ruland J, Duncan GS, Elia A, del Barco
Barrantes I, Nguyen L, Plyte S, Millar DG, Bouchard D, Wakeham A,
Ohashi PS and Mak TW: Bcl10 is a positive regulator of antigen
receptor-induced activation of NF-kappaB and neural tube closure.
Cell. 104:33–42. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhang Q, Siebert R, Yan M, Hinzmann B, Cui
X, Xue L, Rakestraw KM, Naeve CW, Beckmann G, Weisenburger DD, et
al: Inactivating mutations and overexpression of BCL10, a caspase
recruitment domain-containing gene, in MALT lymphoma with
t(1;14)(p22;q32). Nat Genet. 22:63–68. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lambers AR, Gumbs C, Ali S, Marks JR,
Iglehart JD, Berchuck A and Futreal PA: Bcl10 is not a target for
frequent mutation in human carcinomas. Br J Cancer. 80:1575–1576.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yan M, Lee J, Schilbach S, Goddard A and
Dixit V: mE10, a novel caspase recruitment domain-containing
proapoptotic molecule. J Biol Chem. 274:10287–10292. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Koseki T, Inohara N, Chen S, Carrio R,
Merino J, Hottiger MO, Nabel GJ and Núñez G: CIPER, a novel NF
kappaB-activating protein containing a caspase recruitment domain
with homology to Herpesvirus-2 protein E10. J Biol Chem.
274:9955–9961. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yui D, Yoneda T, Oono K, Katayama T,
Imaizumi K and Tohyama M: Interchangeable binding of Bcl10 to TRAF2
and cIAPs regulates apoptosis signaling. Oncogene. 20:4317–4323.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Pacchiana R, Abbate M, Armato U, Dal Prà I
and Chiarini A: Combining immunofluorescence with in situ proximity
ligation assay: a novel imaging approach to monitor protein-protein
interactions in relation to subcellular localization. Histochem
Cell Biol. 142:593–600. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Bayascas JR: Dissecting the role of the
3-phosphoinosi-tide-dependent protein kinase-1 (PDK1) signalling
pathways. Cell Cycle. 7:2978–2982. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Casamayor A, Morrice NA and Alessi DR:
Phosphorylation of Ser-241 is essential for the activity of
3-phosphoin-ositide-dependent protein kinase-1: identification of
five sites of phosphorylation in vivo. Biochem J. 342:287–292.
1999. View Article : Google Scholar
|
|
34
|
Kikani CK, Dong LQ and Liu F: 'New̓-clear
functions of PDK1: beyond a master kinase in the cytosol? J Cell
Biochem. 96:1157–1162. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sephton CF, Zhang D, Lehmann TM,
Pennington PR, Scheid MP and Mousseau DD: The nuclear localization
of 3′-phosphoinositide-dependent kinase-1 is dependent on its
association with the protein tyrosine phosphatase SHP-1. Cell
Signal. 21:1634–1644. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lee KY, D'Acquisto F, Hayden MS, Shim JH
and Ghosh S: PDK1 nucleates T cell receptor-induced signaling
complex for NF-kappaB activation. Science. 308:114–118. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hodgkinson CP and Sale GJ: Regulation of
both PDK1 and the phosphorylation of PKC-zeta and -delta by a
C-terminal PRK2 fragment. Biochemistry. 41:561–569. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yu LR, Lv JQ, Jin LY, Ding SD, Ma XY, Wang
JJ and Zhu XQ: Over-expression of protein kinase C isoforms (α, δ,
θ and ζ) in squamous cervical cancer. Neoplasma. 58:491–498.
2011.
|
|
39
|
Hirai T and Chida K: Protein kinase Czeta
(PKCzeta): Activation mechanisms and cellular functions. J Biochem.
133:1–7. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Xin M, Gao F, May WS, Flagg T and Deng X:
Protein kinase Czeta abrogates the proapoptotic function of Bax
through phosphorylation. J Biol Chem. 282:21268–21277. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Nazarenko I, Jenny M, Keil J, Gieseler C,
Weisshaupt K, Sehouli J, Legewie S, Herbst L, Weichert W,
Darb-Esfahani S, et al: Atypical protein kinase C zeta exhibits a
proapoptotic function in ovarian cancer. Mol Cancer Res. 8:919–934.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Yoneda T, Imaizumi K, Maeda M, Yui D,
Manabe T, Katayama T, Sato N, Gomi F, Morihara T, Mori Y, et al:
Regulatory mechanisms of TRAF2-mediated signal transduction by
Bcl10, a MALT lymphoma-associated protein. J Biol Chem.
275:11114–11120. 2000. View Article : Google Scholar
|
|
43
|
Rosebeck S, Rehman AO, Lucas PC and
McAllister-Lucas LM: From MALT lymphoma to the CBM signalosome:
three decades of discovery. Cell Cycle. 10:2485–2496. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Yeh PY, Kuo S-H, Yeh K-H, Chuang SE, Hsu
C-H, Chang WC, Lin H-I, Gao M and Cheng A-L: A pathway for tumor
necrosis factor-alpha-induced Bcl10 nuclear translocation. Bcl10 is
up-regulated by NF-kappaB and phosphorylated by Akt1 and then
complexes with Bcl3 to enter the nucleus. J Biol Chem. 281:167–175.
2006. View Article : Google Scholar
|
|
45
|
Kuo SH, Chou CH, Cheng AL, Wang CW, Chen
YH and Chen RJ: Expression of BCL10 in cervical cancer has a role
in the regulation of cell growth through the activation of
NF-κB-dependent cyclin D1 signaling. Gynecol Oncol. 126:245–251.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Parrish AB, Freel CD and Kornbluth S:
Cellular mechanisms controlling caspase activation and function.
Cold Spring Harb Perspect Biol. 5:a0086722013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Kurokawa M and Kornbluth S: Caspases and
kinases in a death grip. Cell. 138:838–854. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Voss OH, Kim S, Wewers MD and Doseff AI:
Regulation of monocyte apoptosis by the protein kinase
Cdelta-dependent phosphorylation of caspase-3. J Biol Chem.
280:17371–17379. 2005. View Article : Google Scholar : PubMed/NCBI
|