Introduction
Ischemia-reperfusion injury (IRI), in particular
renal IRI, is an unavoidable event and a critical clinical problem
in organ transplantation. Renal IRI is a major cause of kidney
graft dysfunction eventually leading to graft loss (1,2).
IRI is also involved in graft malfunction following transplantation
in cardiac and aortic surgery (3,4).
The physiological response of tubular epithelial cells (TECs) has a
pivotal role in kidney IRI etiology (5–7),
which involves a cascade of events, including oxidative stress and
inflammation that in turn trigger wound healing by restoration of
blood flow to the ischemic tissue (6,8).
Following ischemia, oxidative stress leads to the production of
tumor necrosis factor-α and interferon-γ, cytokines associated with
TEC injury (9,10). Following exposure to these
cytokines, TECs trigger kidney malfunction (5,6).
Accumulating evidence suggests that kidney IRI is a
complex event involving innate and adaptive immune cells, including
cluster of differentiation 4+ (CD4+) T cells,
B cells, resident dendritic cells, natural killer (NK) cells and
neutrophils (11–13), although kidney IRI can develop in
T and B cell-deficient Rag1−/− mice (14,15). The present study focused on the
role of innate immune cells, specifically NK cells, in renal IRI.
Normally, NK cells recognize and lyse virus-infected and tumor
cells; however recently, NK cells have been shown to be involved in
renal IRI through direct killing of TECs (16,17). In mice, induced expression of the
NK group 2 member D (NKG2D) ligand Rae-1 on TECs has been observed
during kidney IRI (18). From
these observations, NK cells appear to be major participants in
kidney IRI.
NK cell activity is triggered by its recognition of
a target cell through integration of signals from inhibitory and
activating receptors (19,20).
For example, activation of NK cells is dependent on NK cell
expression of NKG2D, an activating receptor, and tumor cell
expression of NKG2D ligands, together with the presence of a danger
signal, such as cellular stress and/or viral infection (21). In humans, the NKG2D ligands major
histocompatibility complex class I-related chain molecules A and B
(MICA/B) and UL16-binding proteins (ULBPs), are upregulated in
response to various signals, including cellular and genotoxic
stress, but are not expressed under normal conditions. The
interaction of MICA/B and NKG2D on NK cells triggers the cytotoxic
function of NK cells. Thus, NKG2D ligands are considered useful
therapeutic targets for the elimination of tumor and virus-infected
cells by NK cells (22,23). Certain studies have reported that
tumor cells can produce transforming growth factor-β (TGF-β)
leading to a decrease in surface NKG2D expression on NK and
CD8+ T cells (24,25).
TGF-β is a well-characterized cytokine that can
induce cell apoptosis, proliferation and differentiation (26). Certain studies of renal IRI have
found that TGF-β expression is strongly induced in renal tubules,
as well as in biopsies of kidney grafts following ischemic injury
(27,28). In addition, several studies have
reported that TGF-β detected in TECs was a signal indicating
recovery in post-ischemic kidneys (29,30); however, the functional
implications of upregulated TGF-β expression in TECs during renal
IRI are not clear.
Notably, certain previous studies have reported that
TECs produce TGF-β following renal IRI, suggesting that TGF-β may
inhibit NK cell activation via downregulation of the NKG2D
receptor. By contrast, other studies have shown that NK cells kill
TECs through upregulation of NKG2D ligands during renal IRI
(18,31). On the basis of these observations,
we hypothesized that TGF-β regulates NKG2D ligand expression in
renal IRI. In the present study, whether the expression of NKG2D
ligands on human TECs (HK-2) is regulated by TGF-β was
investigated. The functional consequences of TGF-β-induced NKG2D
ligand expression on TECs during the interaction between TECs and
NK cells were also evaluated. The results of the present study
examining TGF-β modulation of the NK cell-TEC interaction in renal
IRI may lead to novel insights and useful therapeutic approaches to
treat renal inflammatory injury, including renal
transplantation.
Materials and methods
Cell culture and reagents
The human renal proximal TEC line (HK-2) was
purchased from American Type Culture Collection (Manassas, VA, USA)
and cultured in Dulbecco's modified Eagle medium (Invitrogen,
Carlsbad, CA, USA) with non-essential amino acids, 0.05 mg/ml
bovine pituitary extract, 50 ng/ml human recombinant epidermal
growth factor, 100 U/ml penicillin, 100 µg/ml streptomycin
and 10% fetal bovine serum (FBS) under a 5% CO2
atmosphere at 37°C. NK92-MI cells were cultured in α-modification
of Eagle's Minimum Essential medium supplemented with 2 mM
L-glutamine, 0.2 mM inositol, 20 mM folic acid, 12.5% FBS and 12.5%
horse serum. Anti-NKG2D antibodies (#FAB139A) and anti-NKG2D ligand
antibodies (#MAB13001, #MAB1380, #MAB1298, #MAB1517) were purchased
from R&D Systems (Minneapolis, MN, USA).
Isolation and culture of human peripheral
blood lymphocytes
Peripheral blood lymphocytes (PBLs) from buffy coats
of healthy donors were prepared by Ficoll-Hypaque density gradient
centrifugation (GE Healthcare, Little Chalfont, UK). The study was
approved by the Institutional Bioethics Review Board at the Medical
College of Inje University (Busan, Korea), and all the donors
provided informed written consent prior to their participation in
the study. Cells were stimulated with human interleukin (IL)-2 (500
U/ml; a quantity commonly used to stimulate primary NK cells) for
cytokine production (32).
Flow cytometry analysis
Surface antigen fluorescence-activated cell sorting
(FACS) analysis was performed to detect NKG2D ligands on HK-2
cells. Cells were washed twice with ice-cold phosphate-buffered
saline (PBS), and incubated with mouse anti-MICA antibody
(#MAB13001) or anti-human ULBP monoclonal antibodies [anti-ULBP1
(#MAB1380), anti-ULBP2 (#MAB1298) and anti-ULBP3 (#MAB1517);
R&D Systems} for 30 min on ice. After 2 washes, cells were
incubated with an appropriate fluorescein isothiocyanate
(FITC)-conjugated secondary antibody diluted in PBS for 30 min on
ice and analyzed using a FACSCalibur flow cytometer.
Intracellular FACS analysis was performed to detect
intracellular NKG2D ligand protein levels in HK-2 cells. Cells were
washed twice with ice-cold PBS containing 0.05% bovine serum
albumin (BSA) and 0.02% sodium azide (PBS-BSA), and were fixed in
2% paraformaldehyde in PBS for 15 min on ice. The cells were
subsequently washed once in cold PBS-BSA, and resuspended in PBS
containing 0.1% saponin and 0.05% sodium azide (permeabilization
buffer) for 15 min, followed by incubation with anti-MICA and
anti-ULBPs antibodies for 30 min on ice. Following 2 washes, cells
were incubated with an appropriate FITC-conjugated secondary
antibody in permeabilization buffer for 30 min on ice. Samples were
analyzed using a FACSCalibur flow cytometer (BD Biosciences,
Franklin. Lakes, NJ, USA).
Reverse transcription-polymerase chain
reaction (RT-PCR)
RNA was isolated from HK-2 cells using RNeasy
(Qiagen, Valencia, CA, USA), according to the manufacturer's
instructions, and reverse transcribed into cDNA. The following
primers were used for PCR amplification: TGF-β sense, 5′-GCC CTG
GAC ACC AAC TAT TGC-3′ and antisense, 5′-GCT GCA CTT GCA GGA GCG
CAC-3′; and MICA sense, 5′-TGC TTC TGG CTG GCA TCT TC-3′ and
antisense, 5′-TAG TTC CTG CAG GCA GTC TG-3′. Cycling conditions for
MICA and TGF-β were 1 min at 94°C, 1 min at 60°C and 1 min at 72°C
for 32 cycles. β-actin was amplified as a control using a sense,
5′-GTG GGG CGC CCC AGG CAC CA-3′ and antisense primer, 5′-CTC CTT
AAT GTC ACG CAC GAT TTC-3′; in the RT-PCR experiments. Cycling
conditions for β-actin were 1 min at 94°C, 1 min at 55°C and 1 min
at 72°C for 25 cycles. The PCR products were analyzed by ethidium
bromide-stained 1.2% agarose gel electrophoresis
Small interfering RNA (siRNA)
transfection
TGF-β expression was blocked by siRNA. Cell
transfection was performed using Lipofectamine 2000 (Invitrogen)
for 48 h. The siRNA sequences were as follows: TGF-β siRNA sense,
5′-CAG AGU ACA CAC AGC AUA U-dTdT-3′ and antisense, 5′-AUA UGC UGU
GUG UAC UCU G-dTdT-3′; and non-related control siRNA
(GL3-luciferase) for TGF-β siRNA transfection sense, 5′-AGG GAU ACA
UAC GUG CAC G-dTdT-3′ and antisense, 5′-CGU GCA CGU AUG UAU CCC
U-dTdT-3′. RT-PCR was performed to determine the level of silencing
following transfection.
NK cell cytotoxicity assays
NK cell cytotoxic activity was determined by
calcein-AM assays, as previously described (33). Briefly, target cells were washed
twice with PBS and incubated with 5 mM calcein-AM (Molecular
Probes, Eugene, OR, USA) in serum-free RPMI-1640 medium for 10 min
at 37°C. Labeled target cells were distributed into U-bottom
microtiter plates at a concentration of 2×104
cells/well. Effector cells (PBL or the NK92MI NK cell line) were
added at various effector to target ratios in quadruplicate. Target
cells (HK-2 cells) in complete RPMI-1640 medium alone were used to
determine spontaneous calcein-AM retention. Maximal lysis was
determined by solubilizing 3 wells of the target cells in lysis
buffer (0.1% Triton X-100). After incubation for 5 h, the assays
were analyzed using a fluorescence reader. The percentage of
specific cytotoxicity was calculated as follows: % specific release
= [(retention of experimental well - retention of spontaneous
well)/(retention of maximal lysis well - retention of spontaneous
well)] ×100.
Enzyme-linked immunosorbent assay
(ELISA)
The concentrations of TGF-β and soluble MICA in
culture supernatants were determined using human TGF-β and MICA
ELISA kits (R&D Systems) following the manufacturer's
instructions. Briefly, cell culture supernatant was added to
microplate wells coated with either anti-TGF-β or anti-MICA
antibody. After incubation with the cultured supernatant for 2 h,
the wells were washed 4 times with washing buffer. Horseradish
peroxidase-conjugated detection antibodies were added for 2 h,
followed by the addition of the substrate solution for 30 min.
Samples were read at 450 nm with an ELISA reader (Molecular
Devices, Sunnyvale, CA, USA).
Statistical analysis
Statistical analysis was performed using GraphPad
Prism (GraphPad Software, La Jolla, CA, USA). Student's t-test was
used for assessing statistical significance for comparisons between
two groups. Intergroup differences were assessed by one-way
analysis of variance. P<0.05 was considered to indicate a
statistically significant difference.
Results
TGF-β enhances MICA expression on renal
proximal TECs
To evaluate NKG2D ligand expression in human renal
proximal tubular epithelial (HK-2) cells, surface and intracellular
expression of MICA and ULBP was analyzed. HK-2 cells expressed
MICA, ULBP2 and ULBP3, but not ULBP1, on the cell surface. However,
all these NKG2D ligands were detected intracellularly (Fig. 1A).
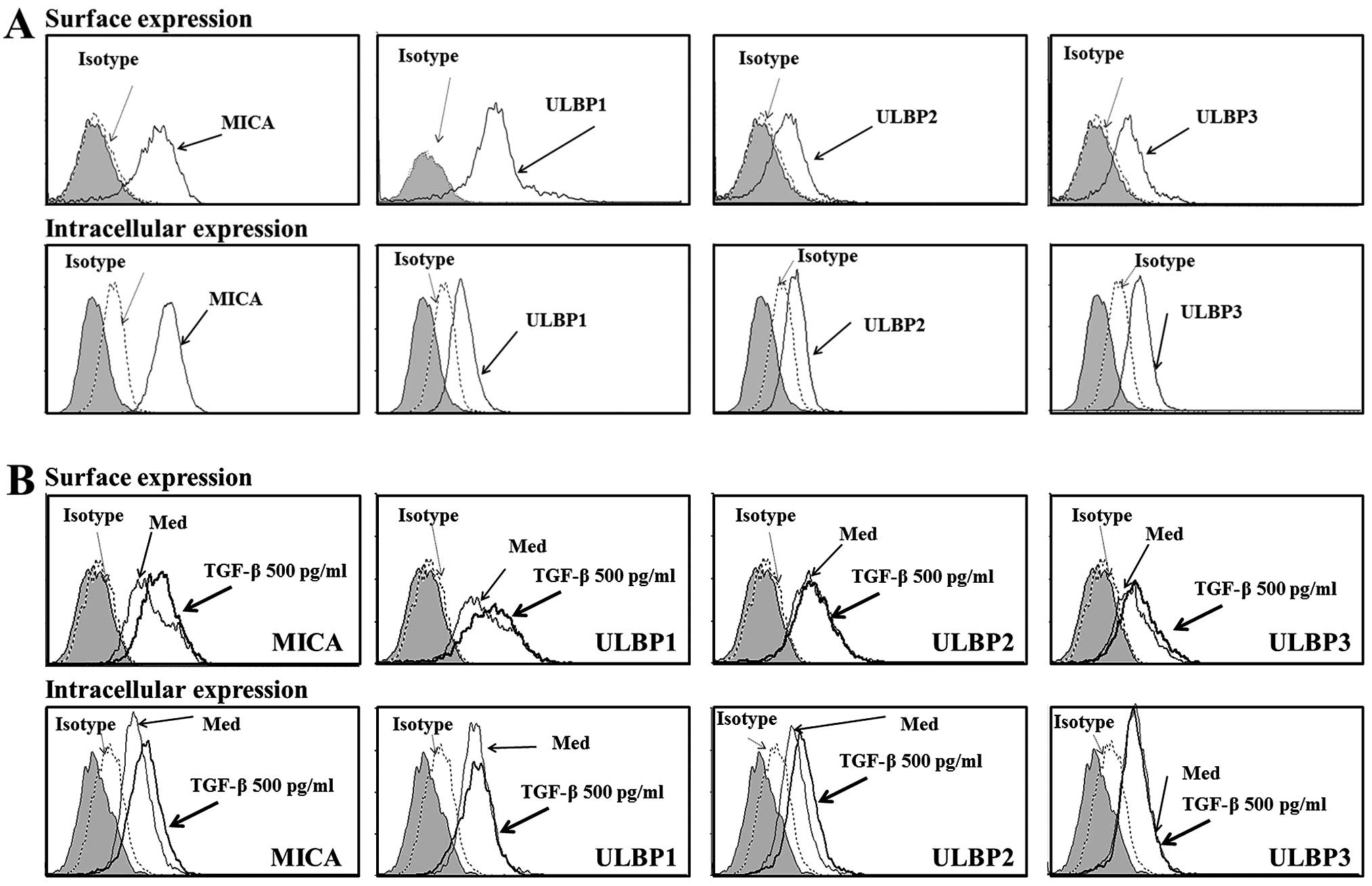 | Figure 1Expression of NKG2D ligands by renal
proximal tubular epithelial cell. (A) Surface and intracellular
staining of NKG2D ligand proteins. Human renal proximal TECs (HK-2)
were stained with control normal mouse immunoglobulin G (iso) or
anti-MICA, and anti-ULBP1, 2 or 3 antibodies, and analyzed by flow
cytometry. In the histograms, the gray peak represents the
unstained control. (B) TGF-β-induced expression of NKG2D ligands.
Renal proximal TECs (HK-2) were incubated in the presence of TGF-β
(500 pg/ml) for 48 h, and surface and intracellular expression of
NKG2D ligands were analyzed by flow cytometry. Results are
representative of three independent experiments. NKG2D, NK group 2
member D; TECs, tubular epithelial cells; MICA, major
histocompatibility complex class I-related chain molecules A; ULBP,
UL16-binding proteins; TGF, transforming growth factor. |
Several studies have reported TGF-β expression in
renal proximal TECs (30), and in
general, TGF-β has been shown to inhibit NK cell function via
downregulation of NKG2D (24–25,34). To investigate the association
between expression of NKG2D ligands and TGF-β in renal IRI, NKG2D
ligand expression was measured using FACS analysis following
treatment with exogenous TGF-β. Treatment with exogenous TGF-β
induced the intracellular expression of MICA and ULBP2 in HK-2
cells; however, it did not induce the intracellular expression of
other NKG2D ligands (Fig. 1B).
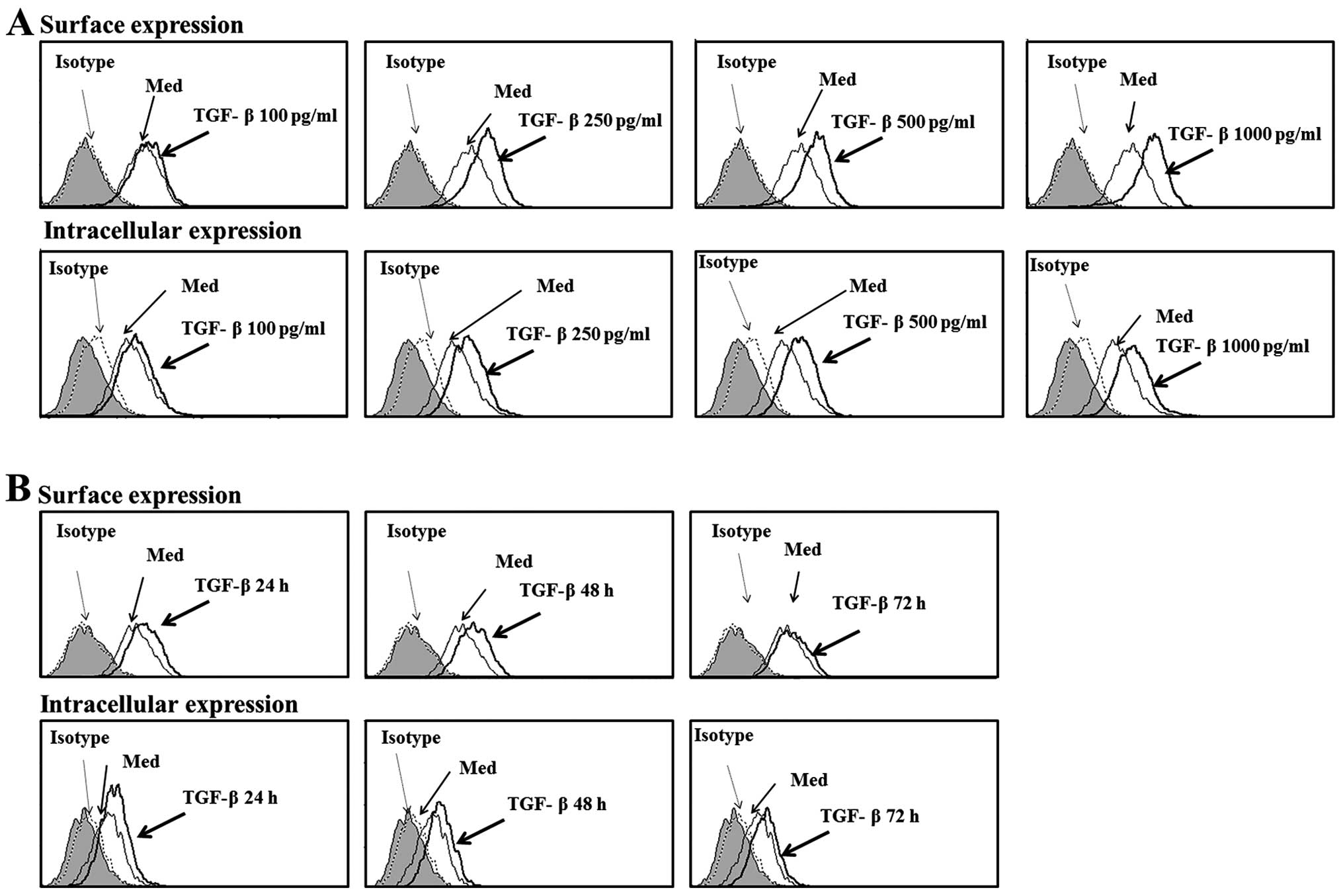
Notably, treatment with TGF-β induced MICA surface expression in
HK-2 cells, but induced little or no surface expression of
ULBP2.
To confirm these findings, the effects of different
concentrations of TGF-β on MICA expression in HK-2 cells were
evaluated. TGF-β treatment significantly induced surface and
intracellular expressions of MICA in a dose-dependent manner
(Fig. 2A). Time-course
experiments revealed that 48 h of incubation in the presence of
TGF-β was optimal for the maximum induction of intracellular and
surface expression of MICA in HK-2 cells (Fig. 2B). These results show that MICA
expression increases in HK-2 cells following treatment with TGF-β
in a dose- and time-dependent manner. Taken together, these data
suggest that TGF-β is a regulator of MICA expression in human
TECs.
TGF-β regulates expression of MICA in
renal proximal TECs via the ataxia telangiectasia mutated
(ATM)/ATM- and Rad3-related (ATM/ATR) pathway
Several studies in various cell types have revealed
that NKG2D ligand expression is primarily controlled by the ATM and
ATR protein kinase pathway (35,36). Kirshner et al (37) reported that TGF-β is involved in
the ATM/ATR kinase pathway in response to genotoxic stress. To
determine which pathway mediates the upregulation of MICA in HK-2
cells, an ATM/ATR kinase inhibitor was tested for its ability to
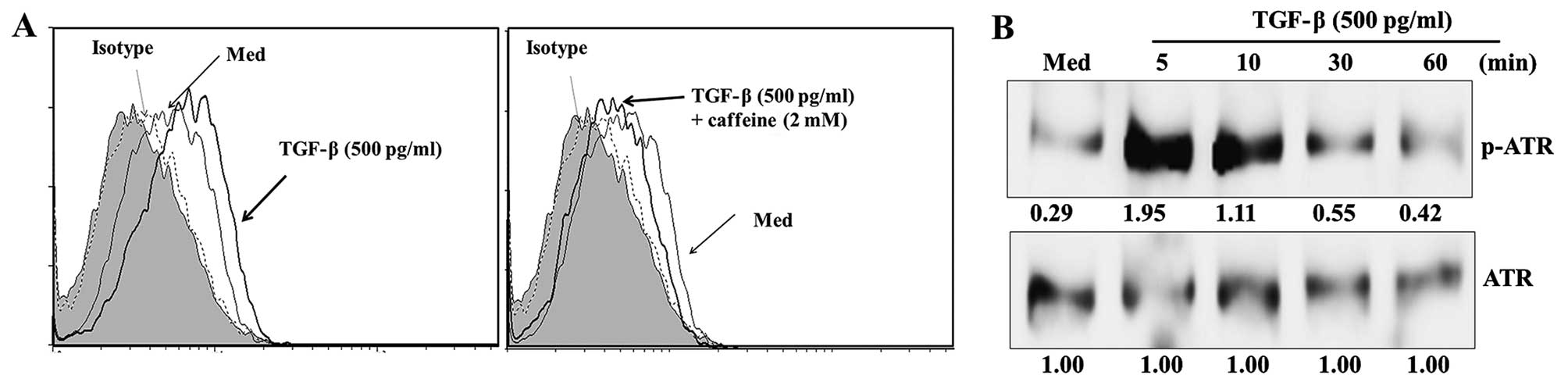
block MICA expression following treatment with TGF-β. The ATM/ATR
kinase inhibitor caffeine significantly blocked the expression of
MICA induced by TGF-β (Fig. 3A).
Additionally, ATM/ATR kinase activity and ATR kinase
phosphorylation were markedly increased in HK-2 cells following
treatment with TGF-β (Fig. 3B).
These results suggest that the ATM/ATR pathway has a critical role
in controlling the expression of MICA following TGF-β
treatment.
Functional consequences of TGF-β-induced
soluble and membrane-bound MICA expression in HK-2 cells
To examine whether membrane-bound MICA expression on
HK-2 cells influences cytotoxic function in primary NK cells, the
cytotoxicity of PBL, isolated from healthy individuals, against
HK-2 cells treated with or without TGF-β was evaluated.
IL-2-activated effector cells exhibited significantly higher
cytotoxicity against HK-2 cells treated with TGF-β compared with
the naïve HK-2 cells (Fig. 4A).
Subsequently, whether the observed TGF-β-mediated increase in the
susceptibility of HK-2 cells to NK cell cytotoxicity directly
involves NKG2D was examined. Neutralizing NKG2D with an anti-NKG2D
antibody moderately reduced PBL cytotoxicity against TGF-β-treated
HK-2 target cells, while treatment with anti-Fas or isotype control
antibodies did not (Fig. 4B).
These results suggest that TGF-β enhances NK cell cytotoxicity via
an NKG2D-mediated pathway.
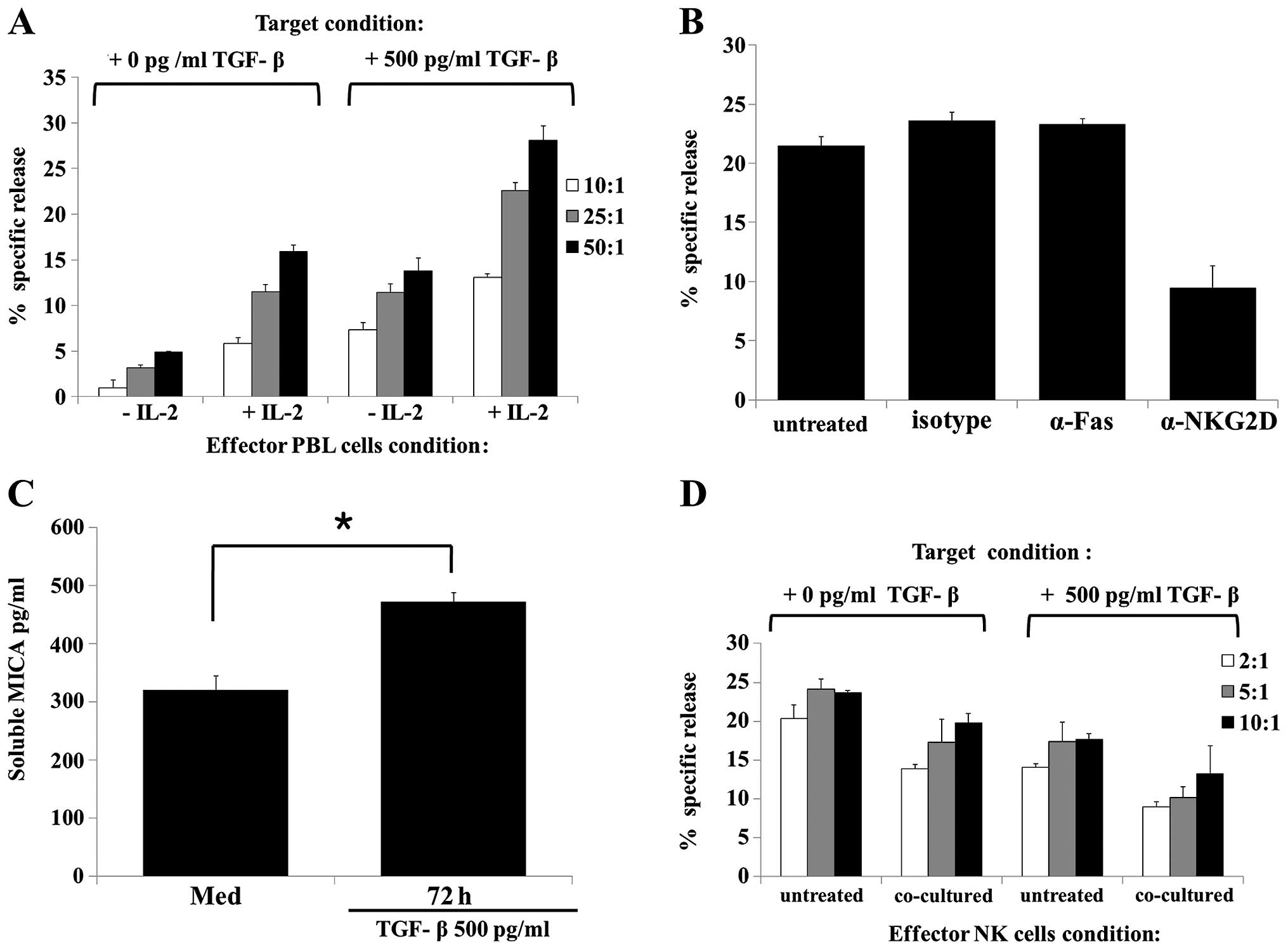 | Figure 4TGF-β treatment increased HK-2 cells
susceptibility to NK-mediated lysis via NKG2D mediated pathway. (A)
PBL cytotoxicity induced by treatment of HK-2 cells with TGF-β (500
pg/ml) for 48 h. HK-2 cells were incubated with or without TGF-β
(500 pg/ml) for 48 h. Target cells were subsequently harvested,
labeled with calcein-AM for 10 min, washed with phosphate-buffered
saline, and loaded at an effector to target cell ratio of 50:1
(black-filled bars), 25:1 (gray-filed bars), and 10:1 (open bars).
After a 4-h incubation period, the specific lysis activity of the
PBL was analyzed using a fluorescence reader. (B) Cytotoxicity of
PBL against HK-2 [treated with TGF-β (500 pg/ml)] was assessed
after pre-incubating PBL (5×106 cells/well) with IL-2
(500 U/ml) for 24 h. PBL cytotoxicity was induced by treatment of
HK-2 cells with TGF-β (500 pg/ml) for 48 h. Target HK-2 cells were
incubated with TGF-β (500 pg/ml) for 48 h. Target cells were
harvested, labeled, washed and loaded at an effector to target cell
ratio of 50:1, as described above. PBL and target cell mixtures
were subsequently cultured with the indicated antibodies (1
µg/ml) for an additional 4 h. (C) HK-2 cells were treated
with or without TGF-β (500 pg/ml) for 48 h. The MICA secretion
level in culture supernatants was analyzed by an enzyme-linked
immunosorbent assay. (D) Cytotoxicity of NK92-MI cells against HK-2
[treated with or without TGF-β (500 pg/ml)] was assessed after
pre-incubating NK92MI cells (2×106 cells/well) with
complete media (untreated) or with TGF-β (500 pg/ml) plus HK-2
cells (2×106 cells/well) for 48 h. TGF-β treated and
untreated HK-2 cells and NK cells were co-cultured using a
Transwell system. The HK-2 cells treated or untreated with TGF-β
for 48 h were used as the target cells. Results are representative
of three independent experiments. *P<0.05 vs. control. TGF,
transforming growth factor; NKG2D, NK group 2 member D; PBL,
peripheral blood lymphocytes; IL, interleukin; MICA, major
histocompatibility complex class I-related chain molecules A. |
As MICA is expressed on the cell surface and
released by metalloproteases (38,39), the effect of metalloprotease
treatment on TGF-β-induced MICA surface expression was evaluated.
ELISA analysis of culture supernatants revealed that the shedding
of MICA from TGF-β-treated HK-2 cells was significantly enhanced at
72 h compared to that of the cells treated with media alone
(Fig. 4C).
To determine the function of soluble MICA released
from TGF-β-treated HK-2 cells, whether co-culture of TGF-β-treated
HK-2 cells and NK cells affected the cytotoxicity of NK cells was
evaluated using a Transwell system. Co-cultivation of NK cells with
TGF-β-treated HK-2 cells resulted in significantly lower NK cell
cytotoxicity compared with the TGF-β-untreated HK-2 cells (Fig. 4D). Additionally, NK cells
co-cultured with TGF-β-treated HK-2 cells, in contrast to the NK
cells that were not co-cultured, showed reduced NK activity against
TGF-β-untreated HK-2 target cells. These results suggest that
soluble MICA released from TGF-β-treated HK-2 cells can reduce NK
cell cytotoxicity via downregulation of NKG2D expression.
Hypoxia-induced TGF-β promotes secretion
of soluble MICA from renal proximal TECs
To confirm that the secreted soluble MICA observed
in the HK-2 cell culture supernatant was due to hypoxia-induced
TGF-β expression, a hypoxia-mimetic chemical [200 µM cobalt
chloride (CoCl2)] was added to the HK-2 cells (40). CoCl2 enhanced
expression of TGF-β and MICA, as well as the release of soluble
MICA from HK-2 cells (Fig. 5A and
B). To elucidate the direct effect of TGF-β on soluble MICA
secretion, TGF-β gene silencing was performed using siRNA
transfection (Fig. 5C) and
confirmed by RT-PCR. Soluble MICA secretion corresponded with the
level of TGF-β silencing and was lower in TGF-β siRNA transfected
cells compared to the control and negative control
siRNA-transfected cells (Fig.
5D). This result indicated that hypoxia-enhanced TGF-β
expression causes an increase in soluble MICA secretion, which in
turn directly regulates IRI.
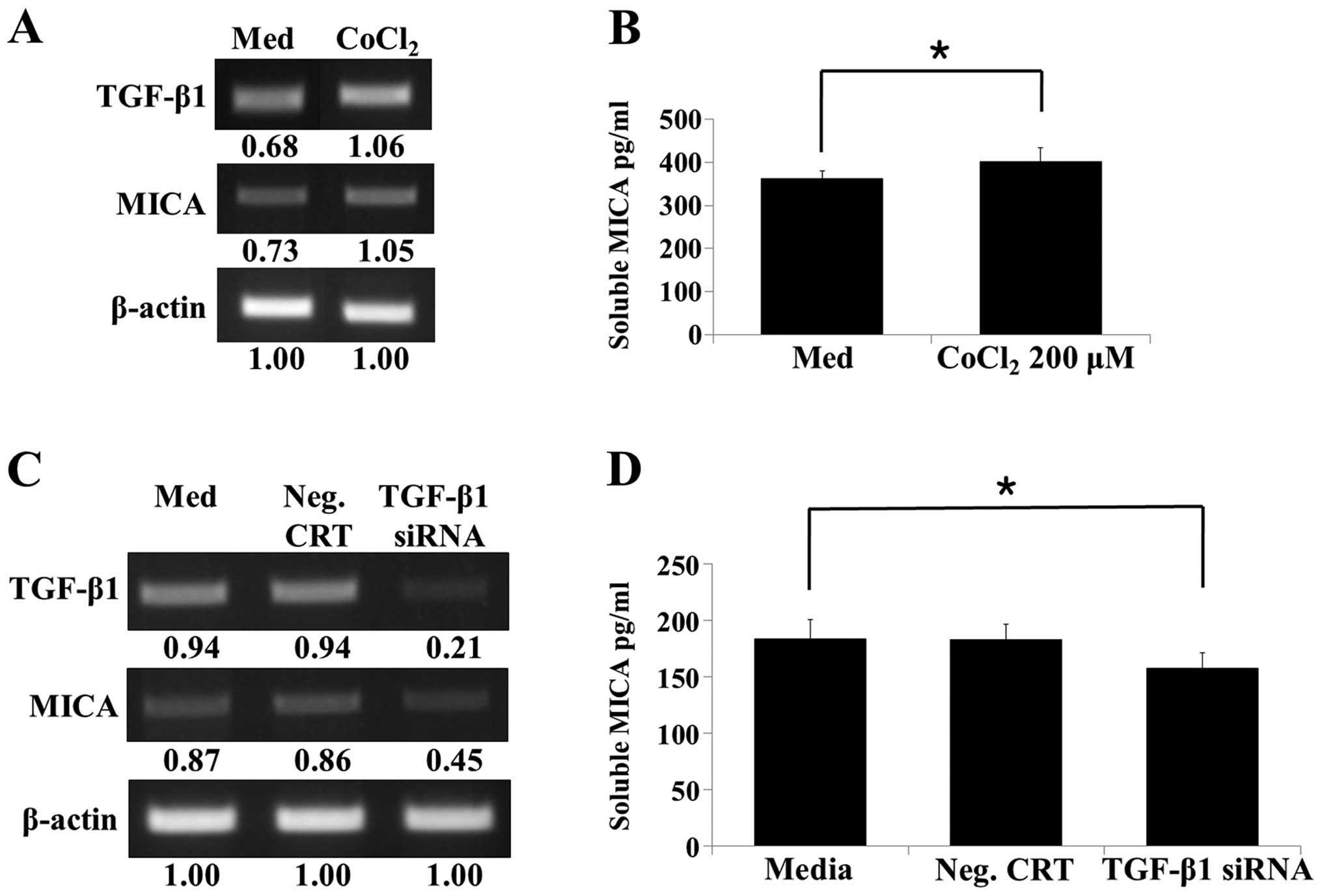 | Figure 5Production of soluble MICA from human
renal proximal tubular epithelial cells following treatment with
the hypoxia-mimetic chemical (CoCl2) and TGF-β gene
silencing. (A) HK-2 cells were incubated with or without 200
µM CoCl2 for 48 h, and subsequently the TGF-β and
MICA mRNA levels were analyzed by RT-PCR. (B) HK-2 cells were
treated with or without CoCl2 (200 µM) for 48 h.
MICA secretion into the culture supernatants was analyzed by ELISA.
(C) HK-2 cells were transfected with a control siRNA or TGF-β
siRNA, and RT-PCR was performed to determine the levels of TGF-β
and MICA. (D) HK-2 cells were transfected with a control siRNA or
TGF-β siRNA and maintained for 48 h. Following incubation, culture
supernatants were harvested, and the level of soluble MICA in the
culture supernatants was analyzed by ELISA. Results are
representative of three independent experiments.
*P<0.05 vs. control. MICA, major histocompatibility
complex class I-related chain molecules A; CoCl2, cobalt
chloride; TGF, transforming growth factor; RT-PCR, reverse
transcription polymerase chain reaction; ELISA, enzyme-linked
immunosorbent assay; siRNA, small interfering RNA. |
Discussion
TGF-β is a multifunctional cytokine that induces
cell apoptosis, differentiation and proliferation (26). Previous studies have reported that
TGF-β expression is induced throughout the kidney following renal
IRI (27), and TGF-β and its
receptor system have been predominantly detected in local
regenerative tubule cells (27,28). Furthermore, several growth
factors, including epidermal growth factor, platelet-derived growth
factor, and basic fibroblast growth factor, have been shown to
induce TGF-β production in various renal cell cultures (41,42). In addition, auto-induction of
TGF-β has also been observed in renal TECs (43); however, the function of TGF-β in
renal injury is not well understood. Recently, expression of TGF-β
detected in TECs has been shown to increase TEC survival following
ischemic injury via upregulation of anti-apoptotic gene expression
(30). Lee et al (44) reported that TGF-β is involved in
the mechanism of protection against
H2O2-induced HK-2 cells death and the
reduction of cellular events associated with renal IRI. These
studies suggest that TGF-β has a critical role in renal protection
during renal IRI.
Recently, NK cells have been linked to kidney IRI
through direct targeting and lysis of TECs (16,17), and induction of NKG2D ligand Rae-1
expression on TECs has been observed during kidney IRI (18). The present study shows that TGF-β
provides protection against NK cell killing, suggesting that TGF-β
modulates NK cell function in kidney IRI. It is well known that
TGF-β downregulates surface expression of NKG2D in NK cells
(24,25). In addition, Park et al
(34) recently reported that
TGF-β decreased DAP10, which in turn led to the downregulation of
NKG2D expression. Additionally, numerous cancer cells possess the
ability to internalize the expression of NKG2D through binding of
soluble MICA, representing another mechanism by which cancer cells
can evade NK cell cytotoxicity (38,39). Although TGF-β-induced
membrane-bound MICA expression on HK-2 cells is critical for their
susceptibility to NK cells, shedding soluble MICA can lead to
internalization of NKG2D on NK cells allowing HK-2 cell survival
(Figs. 4 and 5). Additionally, TGF-β knockdown
decreased production of soluble MICA in HK-2 cells (Fig. 5). On the basis of the direct
effects of TGF-β-induced soluble MICA on NKG2D expression, we
speculate that TGF-β has an important role in the survival of renal
proximal TECs through regulation of NKG2D expression in NK
cells.
While induction of NKG2D ligands expression on renal
proximal TECs during renal IRI has been reported previously in
mouse models (17,45), this event has not yet been
observed in a human model of IRI. In general, kidney recipient
patients are treated with immunosuppressive agents, including
cyclosporine A, prior to kidney transplantation. As the
immunological responses of kidney recipients are suppressed by the
immunosuppressive agents, the levels of TGF-β and soluble MICA that
are detected within the serum of kidney transplantation patients
cannot be considered accurate. However, Nakamura et al
(40) have reported that the
hypoxia-mimetic chemical CoCl2 can directly induce the
production of angiogenic factors in cultured renal proximal tubular
epithelial cells and that these factors have a critical role in the
suppression of progressive renal damage. The present study showed
that a hypoxia-mimetic chemical directly induced TGF-β and MICA
expression and enhanced production of soluble MICA in HK-2 cells
(Fig. 5A and B). These results
suggest that the induction of TGF-β and MICA expression by hypoxia
is a critical link between hypoxia and immune regulation during
IRI.
Expression of NKG2D ligands can generally be induced
in response to stress-associated signaling, including viral
infection and malignant transformation (46,47). Notably, several studies have
reported that tissue ischemia can lead to upregulation of NKG2D
ligands (17,18); however, the signaling pathways
responsible for this upregulation following renal IRI are only
partially understood. Numerous studies have shown that NKG2D ligand
expression can be regulated at the transcriptional and
posttranscriptional levels (48).
Previously, several studies have shown that the ATM/ATR pathway is
involved in the upregulation of NKG2D ligands following the DNA
damage response (35,36). Zhang et al (49) reported that ATM and p53 activation
by TGF-β occurred during apoptosis of epithelial cells. In
addition, TGF-β-regulated ATM activity in response to genotoxic
stress has also been reported (37). In the present study, expression of
MICA was upregulated in renal TECs following treatment with TGF-β,
while caffeine, an ATM/ATR kinase inhibitor, strongly inhibited
this expression (Fig. 3). These
findings suggest that TGF-β regulates expression of MICA through
activation of the ATM/ATR pathway in HK-2 cells.
Although membrane-bound MICA is critical for
elimination of MICA-expressing cells through activation of NKG2D on
NK, CD8+ T and γδ T cells, numerous types of cell can
affect survival through shedding of surface MICA by various
proteases (38,39). TGF-β enhances the production of
extracellular matrix proteins, and certain studies have reported
that TGF-β can also promote the expression of various proteases in
HK-2 cells (50,51). In the present study, soluble MICA
secretion was strongly increased in the culture supernatant of
TGF-β-treated HK-2 cells (Fig.
4C). Therefore, it is possible that TGF-β triggers the release
of soluble MICA from HK-2 cells, although this should be
investigated further in future studies.
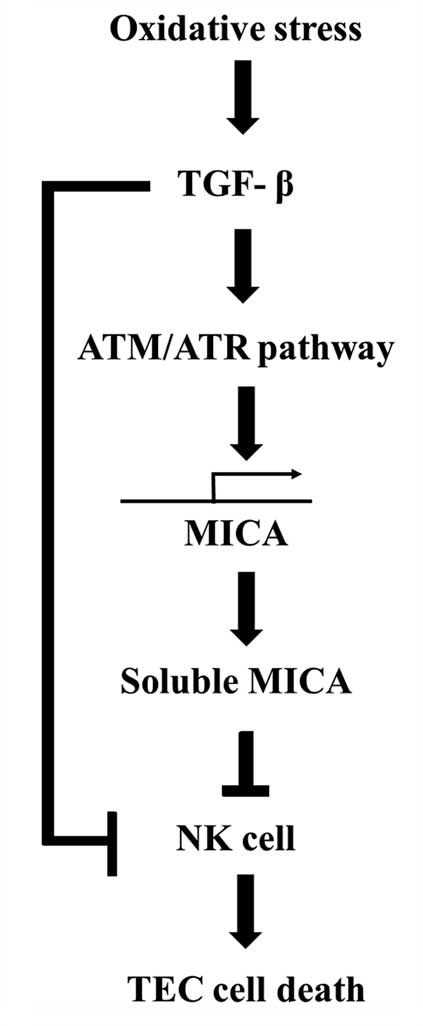
In conclusion, TGF-β induces MICA expression and its
release from human renal proximal TECs, thereby regulating NKG2D
expression on NK cells. The stability of MICA cell surface
expression and the amount of soluble MICA released appear to affect
the susceptibility of the target cells to NK cytotoxicity during
renal IRI (Fig. 6). Modulation of
surface and soluble MICA expression may provide a novel therapeutic
target to improve graft survival following transplantation during
renal IRI.
Abbreviations:
|
TGF-β
|
transforming growth factor-β
|
|
NK
|
natural killer
|
|
IRI
|
ischemia-reperfusion injury
|
|
TECs
|
tubular epithelial cells
|
|
NKG2D
|
NK group 2 member D
|
|
PBLs
|
peripheral blood lymphocytes
|
|
FACS
|
fluorescence-activated cell
sorting
|
|
FITC
|
fluorescein isothiocyanate
|
|
ATM
|
ataxia telangiectasia mutated
|
|
ATR
|
ATM- and Rad3-related
|
Acknowledgments
The present study was supported by a grant from the
National R&D Program for Cancer Control, Ministry for Health,
Welfare and Family Affairs, Republic of Korea (grant no. 0920060)
and by the Basic Science Research Program through the National
Research Foundation of Korea (NRF) funded by the Ministry of
Education, Science and Technology (grant no. 2011–0010291).
References
|
1
|
Shoskes DA and Halloran PF: Delayed graft
function in renal transplantation: Etiology, management and
long-term significance. J Urol. 155:1831–1840. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Thadhani R, Pascual M and Bonventre JV:
Acute renal failure. N Engl J Med. 334:1448–1460. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Nigwekar SU, Kandula P, Hix JK and Thakar
CV: Off-pump coronary artery bypass surgery and acute kidney
injury: A meta-analysis of randomized and observational studies. Am
J Kidney Dis. 54:413–423. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Perico N, Cattaneo D, Sayegh MH and
Remuzzi G: Delayed graft function in kidney transplantation.
Lancet. 364:1814–1827. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bonventre JV and Weinberg JM: Recent
advances in the pathophysiology of ischemic acute renal failure. J
Am Soc Nephrol. 14:2199–2210. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Boros P and Bromberg JS: New cellular and
molecular immune pathways in ischemia/reperfusion injury. Am J
Transplant. 6:652–658. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Molitoris BA and Sutton TA: Endothelial
injury and dysfunction: Role in the extension phase of acute renal
failure. Kidney Int. 66:496–499. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kaminski KA, Bonda TA, Korecki J and
Musial WJ: Oxidative stress and neutrophil activation–the two
keystones of ischemia/reperfusion injury. Int J Cardiol. 86:41–59.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Du C, Jiang J, Guan Q, Yin Z, Masterson M,
Parbtani A, Zhong R and Jevnikar AM: Renal tubular epithelial cell
self-injury through Fas/Fas ligand interaction promotes renal
allograft injury. Am J Transplant. 4:1583–1594. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Du C, Wang S, Diao H, Guan Q, Zhong R and
Jevnikar AM: Increasing resistance of tubular epithelial cells to
apoptosis by shRNA therapy ameliorates renal ischemia-reperfusion
injury. Am J Transplant. 6:2256–2267. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ascon DB, Lopez-Briones S, Liu M, Ascon M,
Savransky V, Colvin RB, Soloski MJ and Rabb H: Phenotypic and
functional characterization of kidney-infiltrating lymphocytes in
renal ischemia reperfusion injury. J Immunol. 177:3380–3387. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Burne-Taney MJ, Ascon DB, Daniels F,
Racusen L, Baldwin W and Rabb H: B cell deficiency confers
protection from renal ischemia reperfusion injury. J Immunol.
171:3210–3215. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Dong X, Swaminathan S, Bachman LA, Croatt
AJ, Nath KA and Griffin MD: Resident dendritic cells are the
predominant TNF-secreting cell in early renal ischemia-reperfusion
injury. Kidney Int. 71:619–628. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Burne-Taney MJ, Yokota-Ikeda N and Rabb H:
Effects of combined T- and B-cell deficiency on murine ischemia
reperfusion injury. Am J Transplant. 5:1186–1193. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Park P, Haas M, Cunningham PN, Bao L,
Alexander JJ and Quigg RJ: Injury in renal ischemia-reperfusion is
independent from immunoglobulins and T lymphocytes. Am J Physiol
Renal Physiol. 282:F352–F357. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang ZX, Shek K, Wang S, Huang X, Lau A,
Yin Z, Sun H, Liu W, Garcia B, Rittling S, et al: Osteopontin
expressed in tubular epithelial cells regulates NK cell-mediated
kidney ischemia reperfusion injury. J Immunol. 185:967–973. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang ZX, Wang S, Huang X, Min WP, Sun H,
Liu W, Garcia B and Jevnikar AM: NK cells induce apoptosis in
tubular epithelial cells and contribute to renal
ischemia-reperfusion injury. J Immunol. 181:7489–7498. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Feng L, Cheng F, Ye Z, Li S, He Y, Yao X,
Tang Q and Li Y: The effect of renal ischemia-reperfusion injury on
expression of RAE-1 and H60 in mice kidney. Transplant Proc.
38:2195–2198. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Moretta A, Marcenaro E, Parolini S,
Ferlazzo G and Moretta L: NK cells at the interface between innate
and adaptive immunity. Cell Death Differ. 15:226–233. 2008.
View Article : Google Scholar
|
|
20
|
Schmitt C, Ghazi B and Bensussan A: NK
cells and surveillance in humans. Reprod Biomed Online. 16:192–201.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Raulet DH: Interplay of natural killer
cells and their receptors with the adaptive immune response. Nat
Immunol. 5:996–1002. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Raulet DH: Roles of the NKG2D
immunoreceptor and its ligands. Nat Rev Immunol. 3:781–790. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Welte SA, Sinzger C, Lutz SZ, Singh-Jasuja
H, Sampaio KL, Eknigk U, Rammensee HG and Steinle A: Selective
intracellular retention of virally induced NKG2D ligands by the
human cytomegalovirus UL16 glycoprotein. Eur J Immunol. 33:194–203.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Crane CA, Han SJ, Barry JJ, Ahn BJ, Lanier
LL and Parsa AT: TGF-beta downregulates the activating receptor
NKG2D on NK cells and CD8+ T cells in glioma patients.
Neuro Oncol. 12:7–13. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lee JC, Lee KM, Kim DW and Heo DS:
Elevated TGF-beta1 secretion and down-modulation of NKG2D underlies
impaired NK cytotoxicity in cancer patients. J Immunol.
172:7335–7340. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Siegel PM and Massagué J: Cytostatic and
apoptotic actions of TGF-beta in homeostasis and cancer. Nat Rev
Cancer. 3:807–821. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Basile DP, Rovak JM, Martin DR and
Hammerman MR: Increased transforming growth factor-beta 1
expression in regenerating rat renal tubules following ischemic
injury. Am J Physiol. 270:F500–F509. 1996.PubMed/NCBI
|
|
28
|
Gobé G, Willgoss D, Hogg N, Schoch E and
Endre Z: Cell survival or death in renal tubular epithelium after
ischemia-reperfusion injury. Kidney Int. 56:1299–1304. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Docherty NG, Pérez-Barriocanal F, Balboa
NE and López-Novoa JM: Transforming growth factor-beta1
(TGF-beta1): A potential recovery signal in the post-ischemic
kidney. Ren Fail. 24:391–406. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Guan Q, Nguan CY and Du C: Expression of
transforming growth factor-beta1 limits renal ischemia-reperfusion
injury. Transplantation. 89:1320–1327. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Luo L, Lu J, Wei L, Long D, Guo JY, Shan
J, Li FS, Lu PY, Li PY and Feng L: The role of HIF-1 in
up-regulating MICA expression on human renal proximal tubular
epithelial cells during hypoxia/reoxygenation. BMC Cell Biol.
11:912010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Huenecke S, Zimmermann SY, Kloess S, Esser
R, Brinkmann A, Tramsen L, Koenig M, Erben S, Seidl C, Tonn T, et
al: IL-2-driven regulation of NK cell receptors with regard to the
distribution of CD16+ and CD16−
subpopulations and in vivo influence after haploidentical NK cell
infusion. J Immunother. 33:200–210. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lichtenfels R, Biddison WE, Schulz H, Vogt
AB and Martin R: CARE-LASS (calcein-release-assay), an improved
fluorescence-based test system to measure cytotoxic T lymphocyte
activity. J Immunol Methods. 172:227–239. PubMed/NCBI
|
|
34
|
Park YP, Choi SC, Kiesler P, Gil-Krzewska
A, Borrego F, Weck J, Krzewski K and Coligan JE: Complex regulation
of human NKG2D-DAP10 cell surface expression: Opposing roles of the
γc cytokines and TGF-β1. Blood. 118:3019–3027. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Cerboni C, Zingoni A, Cippitelli M,
Piccoli M, Frati L and Santoni A: Antigen-activated human T
lymphocytes express cell-surface NKG2D ligands via an
ATM/ATR-dependent mechanism and become susceptible to autologous
NK− cell lysis. Blood. 110:606–615. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lu X, Ohata K, Kondo Y, Espinoza JL, Qi Z
and Nakao S: Hydroxyurea upregulates NKG2D ligand expression in
myeloid leukemia cells synergistically with valproic acid and
potentially enhances susceptibility of leukemic cells to natural
killer cell-mediated cytolysis. Cancer Sci. 101:609–615. 2010.
View Article : Google Scholar
|
|
37
|
Kirshner J, Jobling MF, Pajares MJ, Ravani
SA, Glick AB, Lavin MJ, Koslov S, Shiloh Y and Barcellos-Hoff MH:
Inhibition of transforming growth factor-beta1 signaling attenuates
ataxia telangiectasia mutated activity in response to genotoxic
stress. Cancer Res. 66:10861–10869. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Salih HR, Goehlsdorf D and Steinle A:
Release of MICB molecules by tumor cells: Mechanism and soluble
MICB in sera of cancer patients. Hum Immunol. 67:188–195. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Salih HR, Rammensee HG and Steinle A:
Cutting edge: Down-regulation of MICA on human tumors by
proteolytic shedding. J Immunol. 169:4098–4102. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Nakamura M, Yamabe H, Osawa H, Nakamura N,
Shimada M, Kumasaka R, Murakami R, Fujita T, Osanai T and Okumura
K: Hypoxic conditions stimulate the production of angiogenin and
vascular endothelial growth factor by human renal proximal tubular
epithelial cells in culture. Nephrol Dial Transplant. 21:1489–1495.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Di Paolo S, Gesualdo L, Ranieri E,
Grandaliano G and Schena FP: High glucose concentration induces the
overexpression of transforming growth factor-beta through the
activation of a platelet-derived growth factor loop in human
mesangial cells. Am J Pathol. 149:2095–2106. 1996.PubMed/NCBI
|
|
42
|
Yamabe H, Osawa H, Kaizuka M, Tsunoda S,
Shirato K, Tateyama F and Okumura K: Platelet-derived growth
factor, basic fibroblast growth factor, and interferon gamma
increase type IV collagen production in human fetal mesangial cells
via a transforming growth factor-beta-dependent mechanism. Nephrol
Dial Transplant. 15:872–876. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Dockrell ME, Phanish MK and Hendry BM:
Tgf-beta auto-induction and connective tissue growth factor
expression in human renal tubule epithelial cells requires N-ras.
Nephron, Exp Nephrol. 112:e71–e79. 2009. View Article : Google Scholar
|
|
44
|
Lee HT, Kim M, Kim J, Kim N and Emala CW:
TGF-beta1 release by volatile anesthetics mediates protection
against renal proximal tubule cell necrosis. Am J Nephrol.
27:416–424. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Chen GE, Wu H, Ma J, Chadban SJ and
Sharland A: Toll-like receptor 4 engagement contributes to
expression of NKG2D ligands by renal tubular epithelial cells.
Nephrol Dial Transplant. 26:3873–3881. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Diefenbach A, Jensen ER, Jamieson AM and
Raulet DH: Rae1 and H60 ligands of the NKG2D receptor stimulate
tumour immunity. Nature. 413:165–171. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Groh V, Bahram S, Bauer S, Herman A,
Beauchamp M and Spies T: Cell stress-regulated human major
histocompatibility complex class I gene expressed in
gastrointestinal epithelium. Proc Natl Acad Sci USA.
93:12445–12450. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
López-Soto A, Folgueras AR, Seto E and
Gonzalez S: HDAC3 represses the expression of NKG2D ligands ULBPs
in epithelial tumour cells: Potential implications for the
immunosurveillance of cancer. Oncogene. 28:2370–2382. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Zhang S, Ekman M, Thakur N, Bu S,
Davoodpour P, Grimsby S, Tagami S, Heldin CH and Landström M:
TGFbeta1-induced activation of ATM and p53 mediates apoptosis in a
Smad7-dependent manner. Cell Cycle. 5:2787–2795. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Deng B, Yang X, Liu J, He F, Zhu Z and
Zhang C: Focal adhesion kinase mediates TGF-beta1-induced renal
tubular epithelial-to-mesenchymal transition in vitro. Mol Cell
Biochem. 340:21–29. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Wang QL, Tao YY, Yuan JL, Shen L and Liu
CH: Salvianolic acid B prevents epithelial-to-mesenchymal
transition through the TGF-beta1 signal transduction pathway in
vivo and in vitro. BMC Cell Biol. 11:312010. View Article : Google Scholar : PubMed/NCBI
|