Introduction
During the switching from a
differentiated/contractile to a dedifferentiated/synthetic
phenotype, the increased proliferation and migration of vascular
smooth muscle cells (VSMCs) are key events in the development of
artery restenosis following percutaneous coronary intervention
(1–3). All these events may be induced by
cytokines, such as platelet-derived growth factor BB (PDGF-BB)
(4). PDGF-BB initiates a
multitude of biological effects through the activation of
intracellular signal transduction pathways that contribute to VSMC
phenotypic modulation, proliferation, migration and collagen
synthesis. The importance of PDGF-BB in the development of
neointima formation has been established in models of arterial
injury (5). Therefore, the
inhibition of PDGF-stimulated VSMC proliferation, migration and
phenotypic modulation may represent an important point of
therapeutic intervention in restenosis following angioplasty.
Digoxin has been used as an effective therapy to
treat patients with congestive heart failure (CHF) for decades
(6,7). Previous studies have highlighted a
new aspect of the biology of cardiac glycosides as versatile signal
transducers (8,9) in that they control the transcription
of specific genes (10). Cardiac
glycosides (endogenous and exogenous) have been implicated in the
regulation of a number of important physiological and pathological
states (11–13). Furthermore, unexpected results
from epidemiological studies describing significantly lower
mortality rates of patients with cancer receiving cardiac
glycosides have sparked new interest into the anticancer properties
of these drugs. Subsequent in vitro and in vivo
studies verified these initial observations (14–16). Another study demonstrated that
digoxin inhibited the growth of neuroblastoma tumor xenografts in
mice and angiogenesis in chick chorioallantoic membrane assays
(17). In addition, in a previous
study, Yoshida et al (18)
demonstrated that digoxin suppressed retinal and choroidal
neovascularization, which blocks several proangiogenic pathways.
However, the role of digoxin in regulating VSMC activation is not
yet clearly understood. Although digoxin has been found to
attenuate the development of right ventricle hypertrophy and
prevent pulmonary vascular remodeling, as well as the increase in
pulmonary artery smooth muscle cell [Ca2+]i and pH
levels that occur in mice exposed to chronic hypoxia (19), little is known about the role of
digoxin in regulating aortic VSMC proliferation and migration and
its effectiveness in the prevention of restenosis.
In this study, we demonstrate that digoxin exerts an
inhibitory effect on the PDGF-BB-induced proliferation, migration
and phenotypic modulation of VSMCs, and prevents neointima
formation induced by balloon injury. We also demonstrate that the
digoxin-induced growth inhibition is associated with the
downregulation of CDK activation and the restoration of p27Kip1
levels in PDGF-stimulated VSMCs. This effect of digoxin is
mediated, at least in part, through an increase in integrin linked
kinase (ILK)/Akt signaling and a decrease in glycogen synthase
kinase (GSK)-3β signaling in PDGF-BB-stimulated VSMCs.
Materials and methods
Ethics statement
Animal experiments were carried out in accordance
with the Guide for the Care and Use of Laboratory Animals published
by the US National Institutes of Health (DHWE publication no.
96–01, revised in 2002) and was approved by the Ethics Review Board
for Animal Studies of Institute of Southeast University, Nanjing,
China.
Reagents
Recombinant human PDGF-BB, trypan blue reagent, the
phosphoinositide 3-kinase (PI3K) specific inhibitor, LY294002, the
GSK-3β antagonist, SB415286, and cell proliferation reagent
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
were purchased from Sigma, St. Louis, MO, USA. The proliferating
cell nuclear antigen (PCNA) antibody was purchased from Cell
Signaling Technology (Product no. 2586s).
Trypsin-ethylenediaminetetraacetic acid (EDTA) (0.25%), Dulbecco's
modified Eagle's medium/F12 (DMEM/F12) and fetal bovine serum (FBS)
were from PromoCell (Heidelberg, Germany). The digoxin injection
was acquired from Minsheng Pharmaceutical Group Co., Ltd.
(Hangzhou, China). Digoxin was purchased from J&K Scientific
Ltd. (Beijing, China) and dissolved in dimethyl sulfoxide (DMSO),
and the concentration of DMSO was <0.8% in the control and
drug-containing medium.
Cell proliferation assay
Proliferation was measured using cell counts and MTT
assay, as previously described. For cell counts: VSMCs were seeded
onto 96-well plates (4×103 cells/well) and treated with
various concentrations of digoxin for 24 h prior to stimulation
with or without PDGF-BB (25 µg/l). The cells were then
trypsinized with 0.1% trypsin-EDTA and counted using a
hemocytometer under a microscope. For MTT assay: briefly, the cells
at 60% confluency were plated in a 96-well microplate, and growth
was arrested by serum deprivation for 24 h. The cells were then
treated with PDGF-BB (25 µg/l) in the presence/absence of
digoxin for 24 h and loaded with MTT for the last 3 h.
Subsequently, the cells were dissolved by DMSO, and the color
intensity was read at 540 nm.
Evaluation of cell viability
Trypan blue exclusion was used to determine the
viability of the VSMCs and human umbilical vein endothelial cells
(HUVECs; purchased from PromoCell, Heidelberg, Germany; Cat. no.
C-12200). Following treatment with various concentrations of
digoxin for 24 or 48 h, the VSMCs or HUVECs were trypsinized and
incubated with 0.4% trypan blue dye. Cell viability was assessed by
the automated determination of the percentage of cells that were
able to exclude trypan blue using a Countess Automated Cell Counter
(Invitrogen).
Assays of cell cycle progression
Cell cycle progression was assessed using propidium
iodide (PI) staining with fluorescence-activated cell sorting
(FACS) analysis. Briefly, cells at 70% confluence were
pre-incubated in the presence or absence of digoxin (100 nM) in
serum-free medium for 24 hand and then stimulated with PDGF-BB (25
µg/l) for 24 h. The cells were then trypsinized and fixed
with ethanol at 4°C overnight. The fixed cells were collected by
centrifugation (1000 × g, 4°C), washed twice in phosphate-buffered
saline (PBS) and incubated with 600 µl PI staining buffer
(20 mg/ml PI and 50 mg/ml RNaseA), and then analyzed with FACS. The
cell cycle distributions were analyzed using MultiCycle AV software
(Phoenix Flow Systems, San Diego, CA, USA).
Western blot analysis
The VSMCs were cultured in a 9-cm diameter dish,
grown to 70–80% confluency, and then starved in serum-free medium
for 24 h. The cells were lysed in radioimmunoprecipitation assay
(RIPA) buffer with protease and phosphatase cocktails. Equal
amounts of protein (60–100 µg) were separated by 10% sodium
dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and
electrotransferred onto PVDF membranes (Millipore, Billerica, MA,
USA). The membranes were blocked, and then incubated with various
antibodies overnight, such as anti-CDK4 (Cat. no. SC23896, 1:500),
anti-CDK6 (Cat. no. SC53638, 1:500), anti-p27Kip1 (Cat. no. SC1641,
1:500) (all from Santa Cruz Biotechnology, Inc., Santa Cruz, CA,
USA), anti-SM22a (Cat. no. A5228, 1:1,000), anti-calponin (Cat. no.
C2687, 1:1,000), anti-smooth muscle (SM) α-actin (Cat. no. A2172,
1:1,000) (all from Sigma), anti-phospho-c-Jun NH2-terminal kinase
(p-JNK; Tyr183/185; Cat. no. 5135, 1:1,000), anti-JNK (Cat. no.
9258 1:1,000), anti-p38 mitogen-activated protein kinase (MAPK;
Cat. no. 9228, 1:1,000), anti-phospho-p38 MAPK (Thr180/Tyr182; Cat.
no. 4092, 1:1,000; p-MAPK), anti-extracellular signal-regulated
kinase (ERK)1/2 (Cat. no. 4696, 1:1,000), anti-phospho-ERK1/2
(p-ERK, Tyr204; Cat. no. 4374, 1:1000), anti-phospho-Akt (p-Akt,
S473; Cat. no. 12694, 1:1,000), anti-Akt (Cat. no. 2920, 1:1,000),
anti-phospho-GSK-3β (p-GSK-3β, S9; Cat. no. 14630, 1:1,000),
anti-GSK-3β (Cat. no. 9832, 1:1,000) (all from Cell Signaling
Technology), anti-ILK (Cat. no. 61183, 1:1,000; BD Biosciences,
Franklin Lakes, NJ, USA), anti-intercellular adhesion molecule-1
(ICAM-1; Cat. no. 4915, 1:500), anti-vascular cell adhesion
molecule-1 (VCAM-1; Cat. no. 13662, 1:500) (both from Cell
Signaling Technology), anti-matrix metalloproteinase (MMP)-2 (Cat.
no. SC13594, 1:500), anti-MMP-9 (Cat. no. SC21733, 1:500)
anti-tissue inhibitors of metalloproteinase (TIMP)-1 (Cat. no.
SC21734, 1:500), anti-TIMP-2 (Cat. no. SC365671, 1:500) (all from
Santa Cruz Biotechnology, Inc.), anti-glyceraldehyde 3-phosphate
dehydrogenase (GAPDH; Cat. no. 5174, 1:2,500) and anti-β-actin
(Cat. no. 8457, 1:2,500) (both from Cell Signaling Technology), and
then with the horseradish peroxidase-conjugated secondary antibody
(Beijing TDY Biotech Co., Ltd.) (1:5,000) for 2 h. Specific protein
expression levels were normalized to GAPDH or β-actin for total
protein analyses or to total proteins for phosphorylated protein
measurements. The blots were analyzed using the ChemiDoc™ MP
imaging system (Bio-Rad Laboratories, Inc., Hercules, CA, USA). The
experiments were replicated a number of times.
VSMC culture and treatment
Primary VSMCs were obtained from the thoracic aortas
of male Sprague-Dawley rats weighing between 150 and 180 g (3 rats
per experiment for the VSMCs), and were grown in DMEM/F12 medium
containing 10% FBS, 25 mmol/l HEPES (pH 7.4) at 37°C in a
humidified atmosphere of 95% air and 5% CO2, as
previously described (20). The
purity of the VSMCs was assessed by cell morphological observations
as the characteristic 'hill and valley' growth pattern and positive
immunocytochemical staining with a monoclonal antibody against
smooth muscle α-actin. All the experiments were performed using
VSMCs at passages 3–8.
Migration assay
Migration assay was performed using the Transwell
system (a 6.5-mm polycarbonate membrane with 8.0-µm pores;
Corning, Inc., Corning, NY, USA) (20). Cells suspensions
(5×104), containing fresh serum free medium, were seeded
on the upper chamber. PDGF-BB with or without digoxin was added to
the bottom chamber as the chemoattractant. The cells were allowed
to migrate through the membrane to the lower surface for 6 h. Cells
on the upper surface of the membrane that had not migrated were
scraped off with cotton swabs, and cells that had migrated to the
lower surface were fixed by 3.7% paraformaldehyde and stained with
0.1% crystal violet/20% methanol and counted. The migrated cell
numbers were calculated as the number of migrated cells/5 different
random high-power fields (at ×200 magnification).
Immunofluorescence staining
Following treatment, the cells were washed with PBS,
fixed with 3.7% paraformaldehyde for 15 min at room temperature,
and then treated with 0.1% Triton X-100 in PBS for 5 min. After
washing, the cells were blocked with 2% bovine serum albumin (BSA)
in PBS for 30 min. The cells were then incubated with primary
antibodies for 1 h at room temperature, washed with PBS, and then
incubated with FITC-conjugated secondary antibodies for 1 h in the
dark. Cells were mounted with 90% glycerol-PBS and examined under a
fluorescence microscope (Nikon, Tokyo, Japan).
Balloon injury and morphometric analysis
of neointima formation
Balloon denudation of the left common carotid artery
of the male Sprague-Dawley rats was performed, as previously
described (20). Sprague-Dawley
rats (n=48, weighing 300–350 g) were anesthetized using chloral
hydrate (350 mg/kg, i.p.), and the left common and external carotid
arteries were exposed and isolated. A 1.5 F Fogarty catheter
(Edwards Lifesciences, Irvine, CA, USA) was introduced into the
common carotid artery through an arteriotomy in the external
carotid artery and inflated to 2.0–3.0 atm and withdrawn repeatedly
3 times. The external carotid artery was then ligated, and the
blood flow was restored. Following balloon injury, digoxin (1.0
mg/kg/day) was intraperitoneal injected for 14 days. The control
rats also received a similar volume of distilled water. For
preparing the sham-operated rats, the left common carotid artery
and external carotid artery were exposed and ligated as above, but
the catheter was not inserted into the vessels. The arteries were
collected on day 14 after balloon injury, and embedded in paraffin
to prepare cross sections. Sections for analysis were taken from
the middle part of the injured segment and cut at equally spaced
intervals of 2 mm. The prepared sections were cleared with xylene,
and hydrated with ethanol. The sections were stained with
hematoxylin and eosin or incubated overnight at 4°C with primary
antibodies (PCNA; 1:200). The incubated sections were treated with
biotinylated pan-specific antibody (Vector Laboratories,
Burlingame, CA, USA) for 1 h and incubated in ABC solution (Elite
ABC kit, Vector Laboratories) prepared according to the
manufacturer's instructions. After 1 h of incubation, the sections
were stained with diaminobenzidine (DAB) reagent (Vector
Laboratories) and with methyl green counterstain. The stained
sections were observed with a BX51 light microscope (Olympus,
Tokyo, Japan). Neointima thickening was assessed using the
intima/media (I/M) thickness ratio measured from haematoxylin and
eosin-stained arterial cross sections with a computer-based
Image-Pro Morphometric System in a double-blind manner. Four
discontinuous sections from each vessel were measured in a
Sprague-Dawley rat, whereas 6 rats were used in each experimental
group [the control (distilled water, the sham-operated rats and the
digoxin-treated rats].
Statistical analysis
Data are presented as the means ± SEM. ANOVA and a
paired or unpaired t-test were performed for statistical analysis
where appropriate. A value of P<0.05 was considered to indicate
a statistically significant difference.
Results
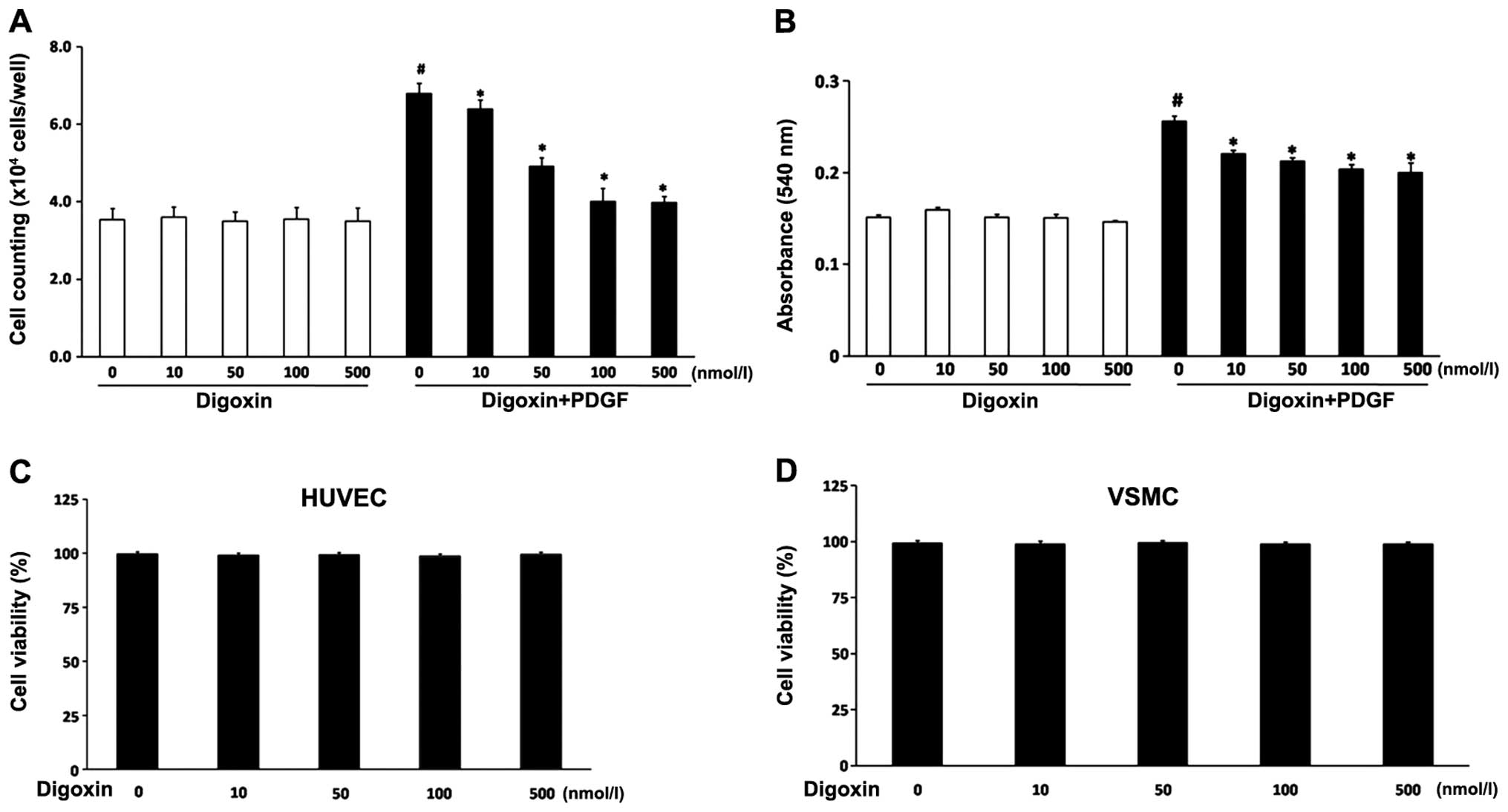
Digoxin inhibits the proliferation of
VSMCs induced by PDGF-BB
The VSMCs were pre-treated with various
concentrations (10–500 nmol/l) of digoxin followed by stimulation
with PDGF-BB (25 µg/l) for 24 h. Digoxin inhibited the
PDGF-BB-induced proliferation of VSMCs in a concentration-dependent
manner (Fig. 1A and B). The
number of cells was significantly increased following treatment
with 25 µg/l PDGF-BB compared to the non-stimulated group.
However, the cell numbers were significantly reduced following
co-culture with 10–500 nM digoxin. In addition, MTT assay revealed
the same inhibitory pattern on VSMC proliferation (Fig. 1B). The cells treated with digoxin
(10–500 nM) for 24 h in the absence of PDGF-BB showed no
significant difference in viability compared with the untreated
cells, suggesting that digoxin is not cytotoxic at the
concentrations tested. Furthermore, re-endothelialization is a
vital process for arterial injury repair. To exclude a cell
cytotoxic effect of digoxin, we determined cell necrosis by trypan
blue exclusion in the absence or presence of digoxin. We examined
the effects of digoxin on endothelial cell viability. The HUVECs
were incubated in the absence or presence of digoxin for 48 h
before their viability was analyzed according to their ability to
exclude trypan blue. Digoxin did not induce the necrosis of HUVECs
at up to 48 h (Fig. 1C), and
digoxin had no negative effect on VSMC viability (Fig. 1D).
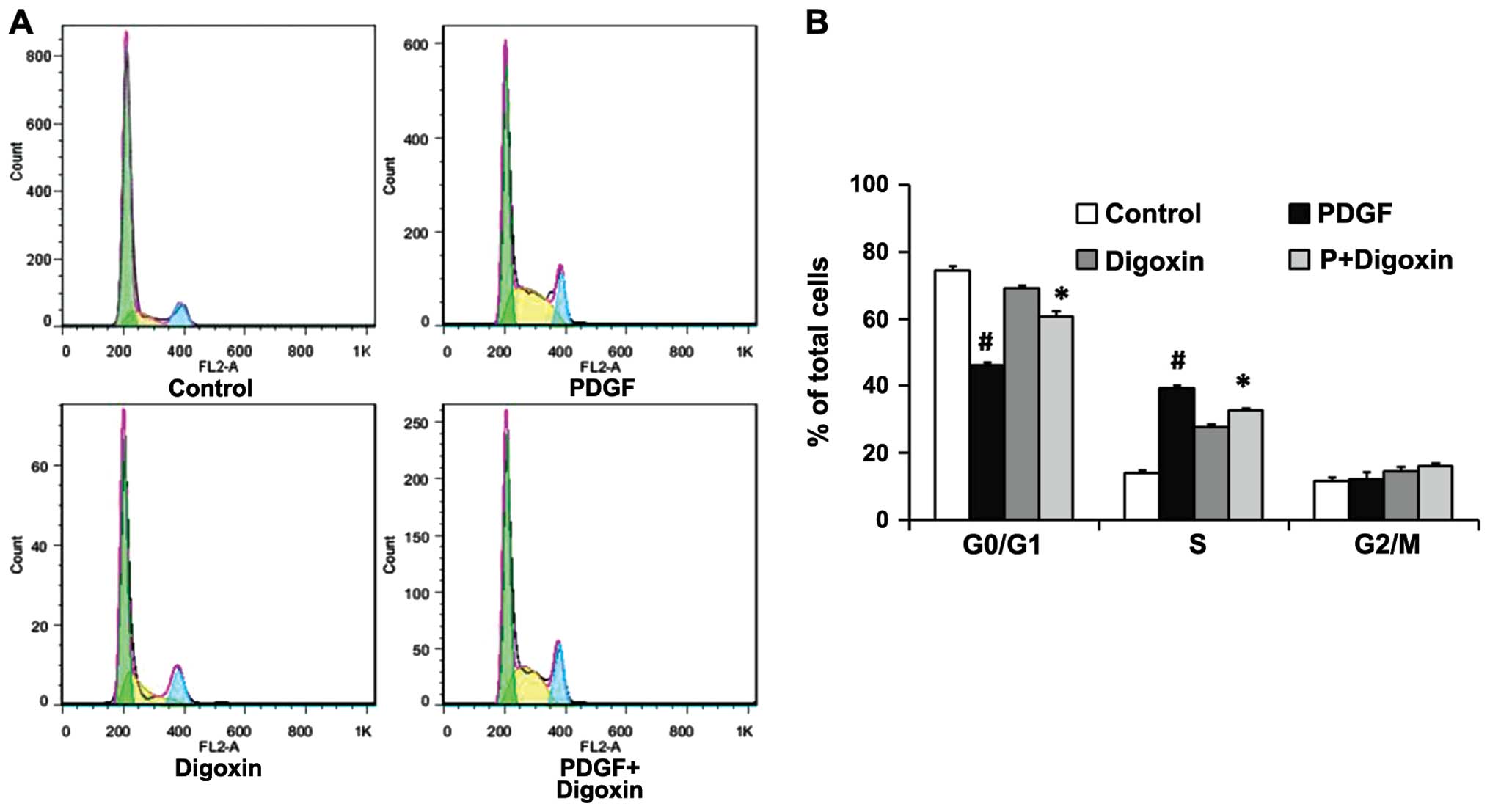
Digoxin inhibits the PDGF-BB-induced cell
cycle progression of VSMCs
To elucidate the mechanisms responsible for the
anti-proliferative effects of digoxin, the effects of digoxin on
cell cycle progression were analyzed. The cells were pre-incubated
in the presence or absence of digoxin in serum-free medium for 24 h
and then stimulated with PDGF-BB (25 µg/l). After 24 h, an
individual nuclear DNA content is reflected as the fluorescence
intensity of incorporated PI. As shown by flow cytometry, treatment
with PDGF-BB alone significantly increased the percentage of cells
in the S phase while decreasing the G0/G1 cell population. By
contrast, digoxin at a concentration of 100 nM significantly
increased the fraction of G0/G1 phase cells and decreased the
number of VSMCs in the S phase, indicating that digoxin prevented
cell cycle entry/progression into the G0/G1 phase (Fig. 2). This result suggests that
digoxin acts at the early stages of cell cycle progression.
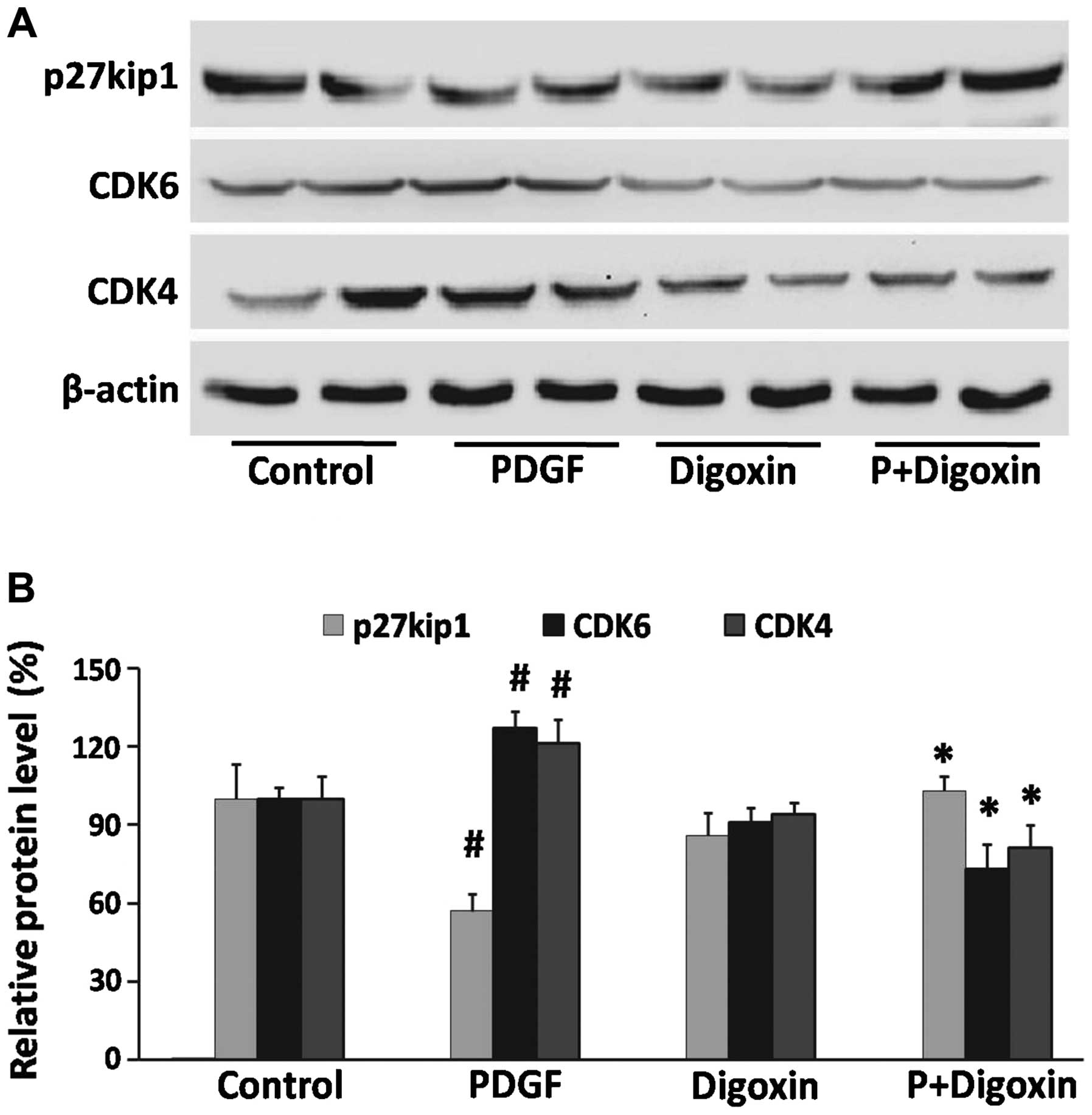
Effects of digoxin on the PDGF-BB-induced
expression of cell cycle-related proteins in VSMCs
Cell cycle progression is tightly regulated through
specific CDK cyclin protein complexes (21). To elucidate the mechanisms
responsible for digoxin-induced cell cycle arrest, the effects of
digoxin on cell cycle events, such as CDK protein expression, were
analyzed by western blot analysis. The expression of CDK4 and CDK6
was induced by PDGF-BB (25 µg/l), whereas treatment with
digoxin (100 nM) significantly decreased the expression of these
molecules (Fig. 3A). Cyclin-CDK
complexes are precisely regulated by cell cycle inhibitors that
block their catalytic activity. One such inhibitor is p27Kip1,
which inactivates the cyclin-CDK complexes in the G1 phase, leading
to cell cycle arrest (22).
Subsequently, we assessed the effects of digoxin on the induction
of p27Kip1 expression. p27Kip1 was constitutively expressed in the
serum-starved quiescent VSMCs and was downregulated by stimulation
with PDGF-BB. By contrast, pre-treatment with digoxin partly
restored p27Kip1 expression to levels that were comparable to those
in the quiescent cells (Fig.
3).
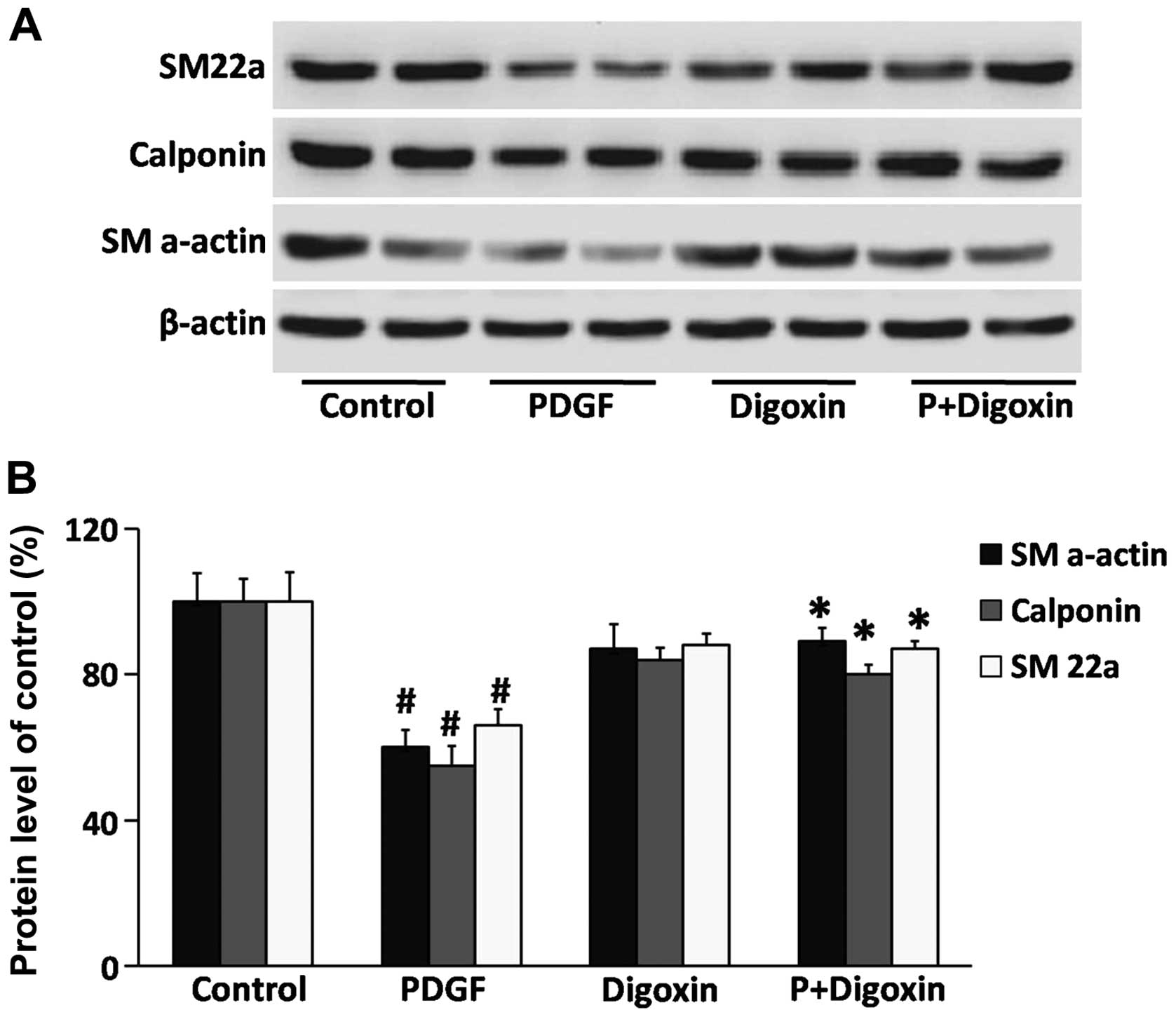
Effects of digoxin on the regulation of
smooth muscle cell contractile gene expression
To evaluate the phenotypic modulation of VSMCs by
digoxin, western blot analysis was used to detect differentiated
phenotype markers. VSMCs were pre-cultured in serum-free medium for
24 h and then treated with digoxin (100 nM) for 24 h followed by
stimulation with or without PDGF-BB (25 µg/l) for 24 h.
PDGF-BB reduced the protein levels of SM22a, calponin and SM
α-actin (Fig. 4). Moreover,
pre-treatment with digoxin partially blocked the suppressive
effects of PDGF-BB, suggesting that digoxin contributes to
maintaining the quiescent (differentiated) state of VSMCs.
Digoxin inhibits the activation of the
GSK-3β/Akt signaling cascade induced by PDGF-BB in VSMCS
To further delineate the cellular and molecular
mechanisms underlying the inhibition of PDGF-BB-induced VSMC growth
by digoxin, the serum-starved VSMCs were stimulated with PDGF-BB
for various periods of time in the absence or presence of didoxin
(100 nM), and the phosphorylation status of ERK1/2, JNK and p38
MAPK, was measured by western blot analysis using antibodies that
identify the active (phosphorylated) forms of these kinases. Our
results revealed that PDGF-BB induced the rapid and sustained
phosphorylation of ERK1/2, JNK and p38, without affecting their
total levels. However, no changes were observed in the
phosphorylated forms of ERK1/2, JNK and p38 when the cells were
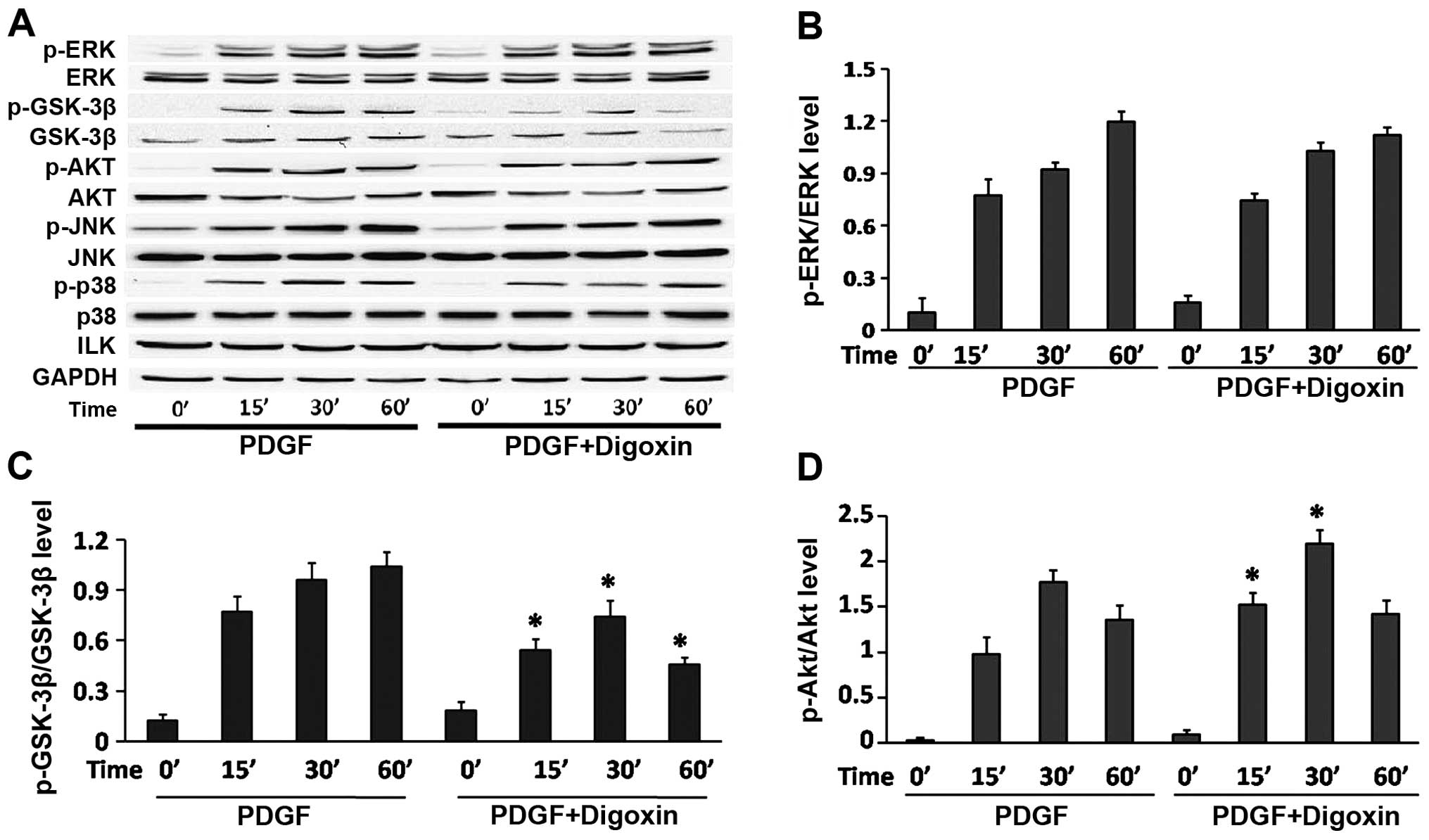
pre-treated with digoxin at the indicated time points (Fig. 5A and B). Previous studies have
also demonstrated that the GSK3β/Akt signal transduction pathway is
critically involved in VSMC proliferation and migration. Therefore,
we evaluated the effects of digoxin on the PDGF-induced activation
of the GSK3β/Akt pathway. Our results revealed that PDGF-BB induced
the rapid and sustained phosphorylation of GSK3β and Akt (Fig. 5A, C and D). However, the
PDGF-BB-induced GSK3β phosphorylation was significantly impaired by
digoxin in a time-dependent manner. However, the increased
phosphorylation of Akt was even observed at 15 min after PDGF-BB
and digoxin co-treatment, and was sustained for up to 30 min
compared to stimulation with PDGF-BB alone as assessed by western
blot analysis. These results suggest that digoxin reduces GSK-3β
and increases Akt activity in PDGF-BB-stimulated VSMCs.
Digoxin activates the ILK signaling
cascade and inhibits the activation of the GSK-3β signaling cascade
induced by PDGF-BB in VSMCs
ILK has been shown to be a critical effector in the
PI3K-dependent signaling pathway that is downstream from both
growth factor and integrin receptor activation (23). The stimulation of ILK results in
the activation of Akt and the inhibition of GSK-3β (23). Thus, we further examined the
effects of digoxin on the PDGF-BB-induced activation of the
PI3K/ILK/GSK-3β signal transduction pathway in VSMCs. The VSMCs
were pre-cultured in serum-free medium for 24 h and then stimulated
with PDGF-BB for 48 h in the absence or presence of digoxin (100
nM). The protein levels of ILK, GSK-3β and Akt were determined by
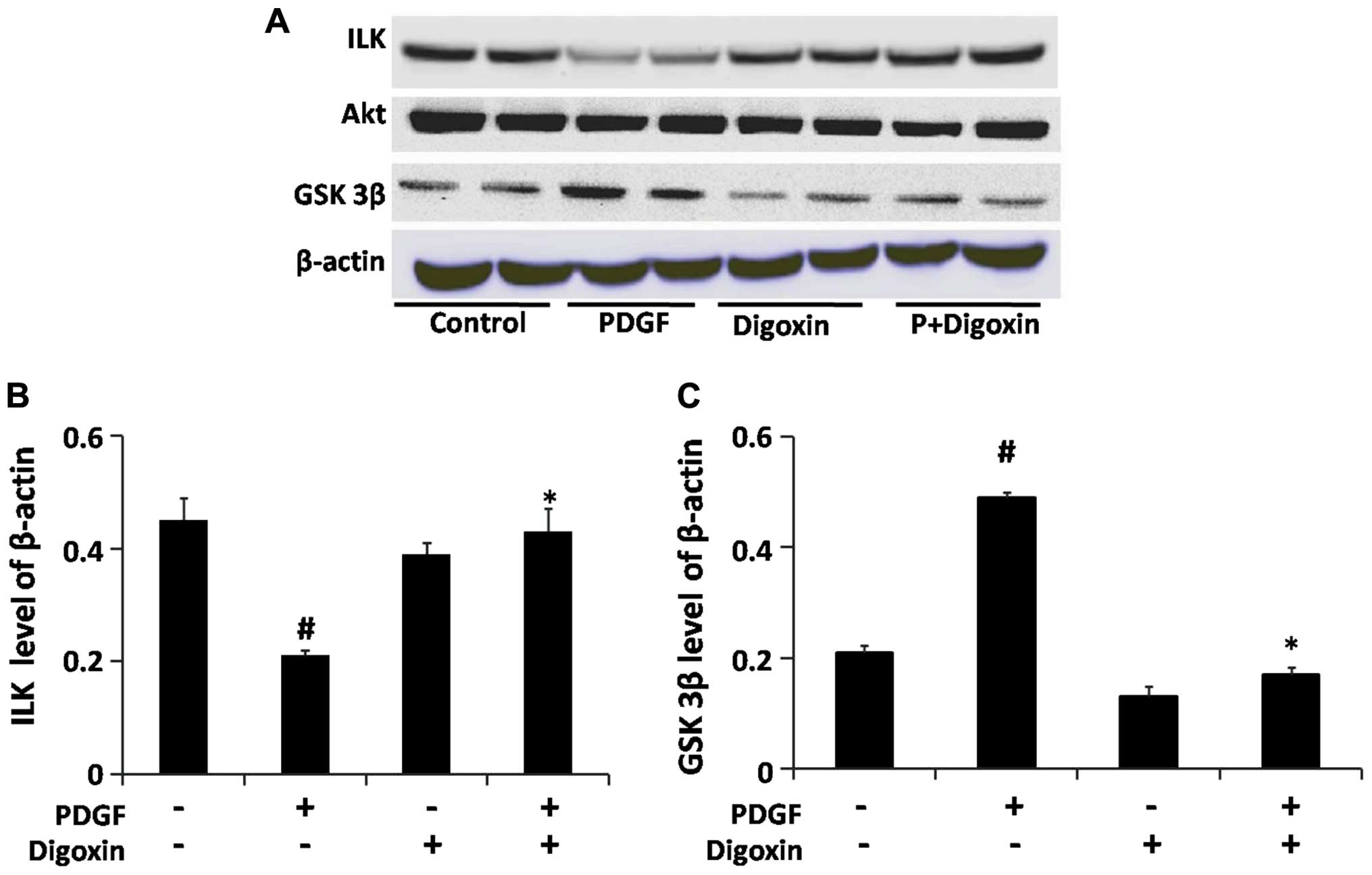
western blot analysis. Our results revealed that digoxin restored
the expression of ILK which was suppressed by PDGF-BB and prevented
the PDGF-BB-induced increase in GSK-3β expression without affecting
the level of Akt in the cells treated with both PDGF-BB and digoxin
(Fig. 6).
Digoxin inhibits PDGF-BB-induced cell
migration through the PI3K/GSK-3β signaling cascade
The migration of smooth muscle cells from the media
to the intimal region is another important component of vascular
lesion formation (24). PDGF-BB
initiates a multitude of biological effects through the activation
of intracellular signal transduction pathways that contribute to
VSMC migration (25). Thus, we
examined the effects of digoxin on PDGF-BB-induced VSMC migration
by Transwell assay which is used to assess chemotaxis. We examined
whether digoxin plays a role in regulating VSMC migration.
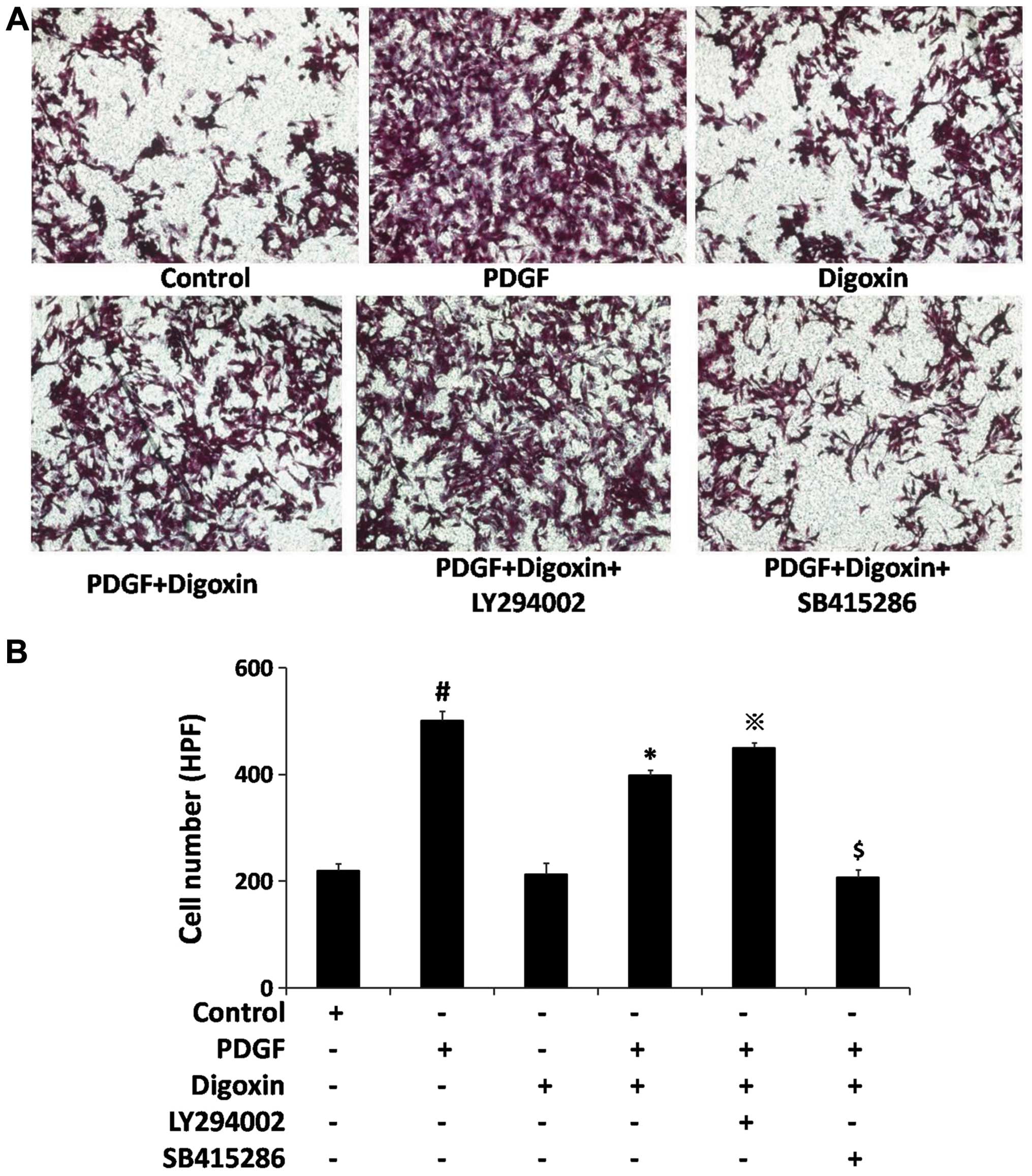
Treatment with PDGF-BB (25 µg/l) for 6 h increased the basal
migration of the VSMCs by >2-fold (Fig. 7); however, compared to stimulation
with PDGF-BB alone, a 30% decrease in cell migration was observed
in the cells treated with both PDGF-BB and digoxin. These results
suggest that digoxin is a potent inhibitor of VSMC migration.
Co-stimulation with LY294002, an inhibitor of PI3K, inactivated
ILK/Akt and prevented the anti-migratory effects of digoxin in the
VSMCs. However, pre-treatment with SB415286, a specific antagonist
of GSK-3β, resulted in a >2-fold decrease in the number of VSMCs
moving across the membrane, compared to co-culture with PDGF-BB and
digoxin. These data demonstrate that digoxin inhibits
PDGF-BB-induced cell migration through the PI3K/GSK-3β signaling
cascade.
Digoxin inhibits the PDGF-BB induced
expression of adhesion molecules and effects the expression of key
proteins in the extracellular matrix (ECM) in VSMCs
To elucidate the molecular mechanisms responsible
for the inhibitory effects of digoxin on VSMC migration, we
examined the effects of digoxin on the migration regulatory
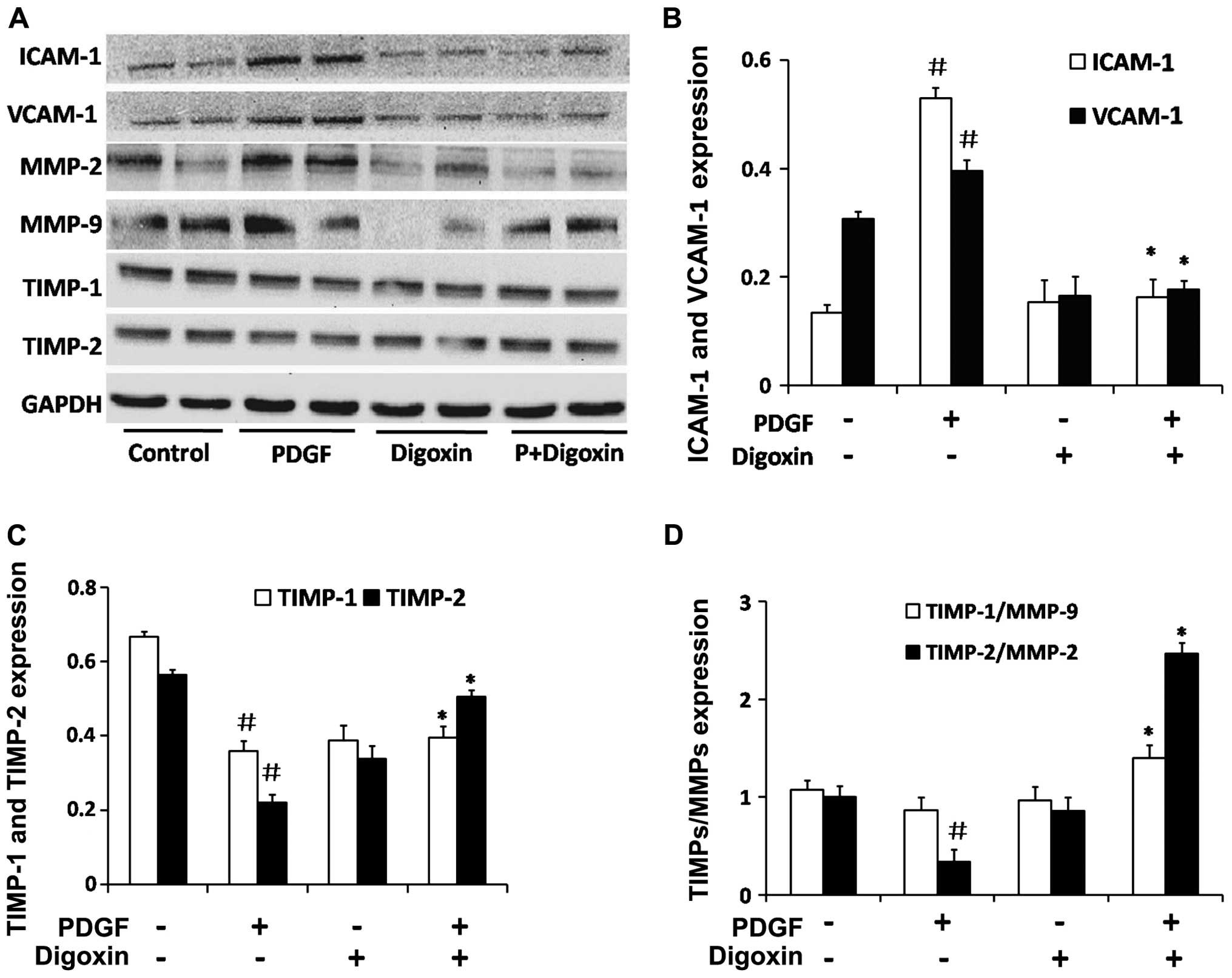
proteins, ICAM-1 and VCAM-1. The stimulation of serum-starved VSMCs
with PDGF-BB for 48 h in the absence of digoxin induced the
upregulation of ICAM-1 and VCAM-1 protein expression (Fig. 8A and B). Treatment with 100 nM
digoxin resulted in an ~60% decrease in the protein levels of
ICAM-1 compared to the levels in the PDGF-BB-treated cells, and a
40% decrease was observed in the VCAM-1 level.
It is well known that MMPs degrade the ECM and
promote VSMC migration from the media to the intimal region. The
balance of MMPs and TIMPs is crucial to maintaining the dynamic
equilibrium of the ECM (26).
Therefore, we detected key factors in the ECM in VSMCs. Digoxin
increased the protein expression of TIMP-1 and TIMP-2 which was
suppressed by PDGF-BB in vitro (Fig. 8C). A similar result was obtained
for the ratio of TIMP-2/MMP-2 and TIMP-1/MMP-9 (Fig. 8D). These findings suggest that
digoxin inhibits the migration of VSMCs induced by PDGF-BB by
suppressing the expression of migration-related proteins in these
cells.
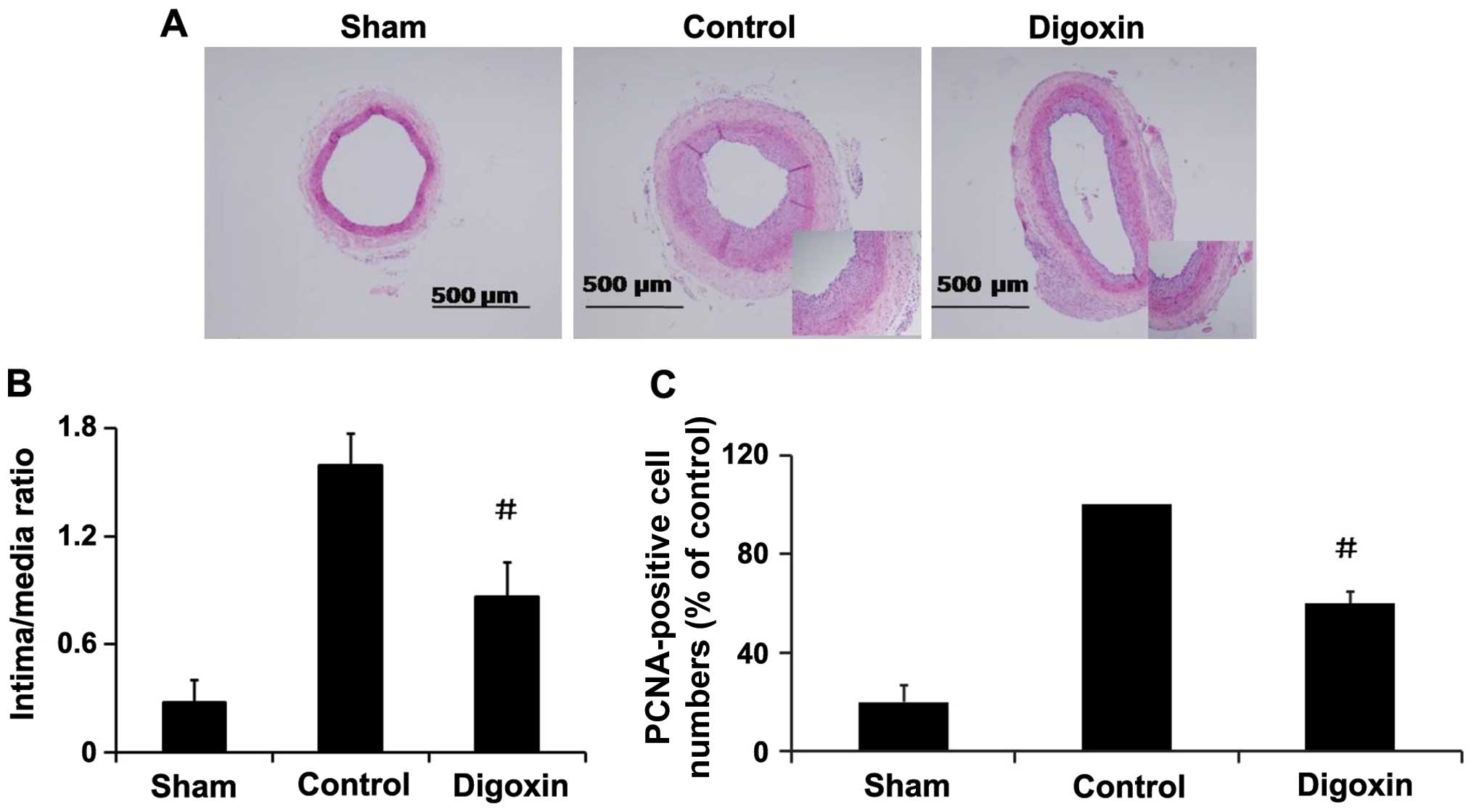
Effects of digoxin on neointima formation
and cell proliferation in vivo
To investigate the role of digoxin in regulating
VSMC proliferation in vivo, rat carotid arteries were
harvested on day 14 following balloon injury, and an increased I/M
thickness ratio of the carotid arteries was observed (Fig. 9A). The administration of digoxin
injection (1.0 mg/kg/day) significantly decreased the I/M thickness
ratio by >45% compared with the injured control rats (Fig. 9B) and inhibited the injury-induced
increase in PCNA expression in the neointima of carotid arteries
(Fig. 9C), and attenuated
neointima formation. These results suggest that digoxin exerts an
inhibitory effect on cell proliferation in vivo, and may be
an effective agent for the prevention of restenosis following
angioplasty.
Discussion
In the present study, to the best of our knowledge,
we demonstrate for the first time that digoxin inhibits the
PDGF-BB-induced phenotypic switching, proliferation and migration
of VSMCs, and prevents neointima formation induced by balloon
injury, at least in part through an increase in ILK/Akt signaling
and a decrease in GSK-3β signaling.
Indeed, a number of studies have described the
effects of cardiac glycosides, such as digoxin and digitoxin, on
the regulation of cell attachment (27), the orientation of polarity
(28), protein trafficking
(29) and the induction of
proliferation (30,31). Overall, it is now clear that the
ultimate response to cardiac glycoside treatment is dependent on
the tissue, exposure time and dose (15). Numerous studies have confirmed the
anti-proliferative effects of cardiac glycosides in several cancer
cell lines, including breast (32), prostate (33), melanoma (34), pancreatic (35), lung (36), leukaemia (37), neuroblastoma (38) and renal adenocarcinoma (39). The exact mechanisms underlying
these effects of cardiac glycosides are not yet fully understood.
Our study demonstrated that the anti-proliferative activity of
digoxin was associated with cell cycle arrest in the G0/G1 phase
(Fig. 2). Cell cycle progression
is a tightly regulated process that involves a complex cascade of
events. A previous study indicated that pharmacological
interventions to regulate the cell cycle inhibit VSMC proliferation
and block neointimal hyperplasia (40). Therefore, the inhibition of VSMC
cycle progression is considered an effective strategy for the
control of VSMC proliferation. In this study, the fact that digoxin
permitted the VSMCs to arrest in the G0/G1 phase suggests that the
modulation of cell cycle regulatory proteins occurs following
digoxin treatment. CDK4 and CDK6 are key mediators during the
progression from the G0/G1 to the S phase of the cell cycle by
forming complexes with cyclin A, E and D1 (41). Our data demonstrated that the
expression of the cell cycle regulatory proteins, CDK4 and CDK6,
decreased after the VSMCs were treated with digoxin, compared to
stimulation with PDGF-BB alone (Fig.
3), indicating that cell cycle arrest in the G0/G1 phase is due
to the downregulation of CDK/cyclin complex expression (4). The results from different research
groups have also demonstrated that p27Kip1 inhibits the
proliferation and migration of VSMCs (42,43). In the present study, the
digoxin-induced upregulation of p27Kip1 expression was consistent
with the inhibitory effects of digoxin on VSMC migration. These
results suggest that the effects of digoxin on p27Kip1 expression
are involved in its inhibitory effects on VSMC proliferation and
migration.
VSMCs can undergo transition from a quiescent,
contractile/differentiated phenotype to a
synthetic/dedifferentiated phenotype (44). To date, compelling evidence has
indicated that PDGF-BB induces a profound suppression of VSMC
marker gene expression through multiple complementary pathways and
plays a pivotal role during restenosis (20,45). In accordance with previous
studies, we observed that PDGF-BB decreased SM α-actin, SM22a and
calponin expression. Our in vitro experiments revealed that
treatment with digoxin partly restored the expression of SM
α-actin, SM22a and calponin (Fig.
4), accompanied by a decrease in cell proliferation and
migration. These results suggest that digoxin halts the change
toward a deleterious VSMC phenotype induced by PDGF-BB, which in
turn contributes to the suppression of neointima formation.
The mechanisms through which digoxin inhibits
PDGF-BB-induced VSMC proliferation, migration and phenotypic
modulation remain largely unclear. ILK is a widely expressed and
evolutionally conserved component of cell-ECM adhesions. Activated
ILK can directly phosphorylate Akt and GSK-3β (46); the phosphorylation of GSK-3β
results in the inhibition of ILK. Yudowski et al (47) found that PI3K-Akt activation was
an important part of the complex and was bound to a proline-rich
region of the catalytic α-subunit of
Na+/K+-ATPase. In a similar manner, ouabain
mediates proliferation through the nitric oxide-induced production
of reactive oxygen species through the activation of the PI3K
cascade (48). Our data
demonstrated that digoxin restored the PDGF-BB induced inhibition
of ILK expression and prevented the PDGF-BB-induced activation of
GSK-3β without affecting the activation of the ERK1/2, JNK and p38
MAPK cascade. Additionally, digoxin prevented the PDGF-BB induced
proliferation and migration of VSMCs accompanied by the activation
of Akt. These findings are consistent with those of a previous
study showing that digoxin prevents TNF-α-induced apoptosis of
endothelial cells accompanied by the activation of Akt through PI3K
signaling (49).
It has previously been demonstrated that MMPs are
rapidly activated following vascular injury in both atherosclerosis
and angioplasty (50). MMPs
degrade the ECM and promote VSMC migration from the media to the
intimal region, and then initial VSMCs undergo proliferation, while
migrating VSMCs secrete vast ECM products, including collagen I and
III, progressively resulting in neointima formation (51). In this study, we found that
digoxin significantly inhibited the expression of MMP-2 and MMP-9
induced by PDGF stimulation, and also had a significant effect on
both TIMP-2 and TIMP-1 (the inhibitors of the MMPs). The ratios of
MMP-2/TIMP-2 and MMP-9/TIMP-1 imply that VSMC migration may be a
disequilibrium process induced by PDGF-BB, and digoxin was able to
balance the ratio to a normal condition. Therefore, the regulatory
effects of digoxin on MMPs and TIMPs which are key factors in the
ECM for VSMC migration, may contribute to its anti-migratory effect
on VSMCs.
In the present study, to determine whether our in
vitro findings have any physiological relevance, we evaluated
the effects of digoxin on neointima formation using an animal model
of arterial balloon-injury. Although the doses of digoxin injection
(1.0 mg/kg/day) administered in this study are higher than those
administered to humans, the comparison of the dosages between
species is complicated by a number of factors. For example, on the
basis of body weight versus surface area measurements, it has been
suggested that a given dosage in humans requires a 12-fold higher
dose in rats (19). Drug
metabolism can also vary considerably owing to differential
mechanisms of uptake, clearance and/or degradation. With these
caveats in mind, plasma digoxin levels measured in this study were
at or below the therapeutic range used in digoxin-treated patients
(0.5–2 ng/ml). Our in vivo data indicated that digoxin
significantly inhibited neointima formation, accompanied by a
decrease in cell proliferation following vascular injury in rats.
These observations indicate the potential therapeutic application
of digoxin in the treatment of cardiovascular diseases, such as
restenosis.
References
|
1
|
Davis-Dusenbery BN, Wu C, Hata A and Sessa
WC: Micromanaging vascular smooth muscle cell differentiation and
phenotypic modulation. Arterioscler Thromb Vasc Biol. 31:2370–2377.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Goel SA, Guo LW, Liu B and Kent KC:
Mechanisms of post-intervention arterial remodelling. Cardiovasc
Res. 96:363–371. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Weintraub WS: The pathophysiology and
burden of restenosis. Am J Cardiol. 100:3K–9K. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Dzau VJ, Braun-Dullaeus RC and Sedding DG:
Vascular proliferation and atherosclerosis: New perspectives and
therapeutic strategies. Nat Med. 8:1249–1256. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Raines EW: PDGF and cardiovascular
disease. Cytokine Growth Factor Rev. 15:237–254. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ahmed A, Pitt B, Rahimtoola SH, Waagstein
F, White M, Love TE and Braunwald E: Effects of digoxin at low
serum concentrations on mortality and hospitalization in heart
failure: A propensity-matched study of the DIG trial. Int J
Cardiol. 123:138–146. 2008. View Article : Google Scholar
|
|
7
|
Pervaiz MH, Dickinson MG and Yamani M: Is
digoxin a drug of the past? Cleve Clin J Med. 73:821–824.
826829–832, passim. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Newman RA, Yang P, Pawlus AD and Block KI:
Cardiac glycosides as novel cancer therapeutic agents. Mol Interv.
8:36–49. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Prassas I and Diamandis EP: Novel
therapeutic applications of cardiac glycosides. Nat Rev Drug
Discov. 7:926–935. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Takara K, Takagi K, Tsujimoto M, Ohnishi N
and Yokoyama T: Digoxin up-regulates multidrug resistance
transporter (MDR1) mRNA and simultaneously down-regulates steroid
xenobiotic receptor mRNA. Biochem Biophys Res Commun. 306:116–120.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Schoner W: Endogenous cardiac glycosides,
a new class of steroid hormones. Eur J Biochem. 269:2440–2448.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Schoner W and Scheiner-Bobis G: Endogenous
and exogenous cardiac glycosides and their mechanisms of action. Am
J Cardiovasc Drugs. 7:173–189. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Schoner W and Scheiner-Bobis G: Endogenous
and exogenous cardiac glycosides: Their roles in hypertension, salt
metabolism, and cell growth. Am J Physiol Cell Physiol.
293:C509–C536. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
López-Lázaro M: Digitoxin as an anticancer
agent with selectivity for cancer cells: Possible mechanisms
involved. Expert Opin Ther Targets. 11:1043–1053. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mijatovic T, Van Quaquebeke E, Delest B,
Debeir O, Darro F and Kiss R: Cardiotonic steroids on the road to
anti-cancer therapy. Biochim Biophys Acta. 1776:32–57.
2007.PubMed/NCBI
|
|
16
|
Winnicka K, Bielawski K and Bielawska A:
Cardiac glycosides in cancer research and cancer therapy. Acta Pol
Pharm. 63:109–115. 2006.
|
|
17
|
Svensson A, Azarbayjani F, Bäckman U,
Matsumoto T and Christofferson R: Digoxin inhibits neuroblastoma
tumor growth in mice. Anticancer Res. 25(1A): 207–212.
2005.PubMed/NCBI
|
|
18
|
Yoshida T, Zhang H, Iwase T, Shen J,
Semenza GL and Campochiaro PA: Digoxin inhibits retinal
ischemia-induced HIF-1alpha expression and ocular
neovascularization. FASEB J. 24:1759–1767. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Abud EM, Maylor J, Undem C, Punjabi A,
Zaiman AL, Myers AC, Sylvester JT, Semenza GL and Shimoda LA:
Digoxin inhibits development of hypoxic pulmonary hypertension in
mice. Proc Natl Acad Sci USA. 109:1239–1244. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang L, Zheng J, Du Y, Huang Y, Li J, Liu
B, Liu CJ, Zhu Y, Gao Y, Xu Q, et al: Cartilage oligomeric matrix
protein maintains the contractile phenotype of vascular smooth
muscle cells by interacting with alpha(7)beta(1) integrin. Circ
Res. 106:514–525. 2010. View Article : Google Scholar
|
|
21
|
Norbury C and Nurse P: Animal cell cycles
and their control. Annu Rev Biochem. 61:441–470. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Malumbres M and Barbacid M: To cycle or
not to cycle: a critical decision in cancer. Nat Rev Cancer.
1:222–231. 2001. View
Article : Google Scholar
|
|
23
|
Qin J and Wu C: ILK: A pseudokinase in the
center stage of cell-matrix adhesion and signaling. Curr Opin Cell
Biol. 24:607–613. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ross R: The pathogenesis of
atherosclerosis: a perspective for the 1990s. Nature. 362:801–809.
1993. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ferns GA, Raines EW, Sprugel KH, Motani
AS, Reidy MA and Ross R: Inhibition of neointimal smooth muscle
accumulation after angioplasty by an antibody to PDGF. Science.
253:1129–1132. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Glass CK and Witztum JL: Atherosclerosis.
the road ahead. Cell. 104:503–516. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Contreras RG, Shoshani L, Flores-Maldonado
C, Lázaro A and Cereijido M: Relationship between Na(+),K(+)-ATPase
and cell attachment. J Cell Sci. 112:4223–4232. 1999.PubMed/NCBI
|
|
28
|
Rajasekaran SA, Palmer LG, Moon SY,
Peralta Soler A, Apodaca GL, Harper JF, Zheng Y and Rajasekaran AK:
Na,K-ATPase activity is required for formation of tight junctions,
desmosomes, and induction of polarity in epithelial cells. Mol Biol
Cell. 12:3717–3732. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang L, Wible BA, Wan X and Ficker E:
Cardiac glycosides as novel inhibitors of human
ether-a-go-go-related gene channel trafficking. J Pharmacol Exp
Ther. 320:525–534. 2007. View Article : Google Scholar
|
|
30
|
Saunders R and Scheiner-Bobis G: Ouabain
stimulates endothelin release and expression in human endothelial
cells without inhibiting the sodium pump. Eur J Biochem.
271:1054–1062. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Abramowitz J, Dai C, Hirschi KK, Dmitrieva
RI, Doris PA, Liu L and Allen JC: Ouabain- and
marinobufagenin-induced proliferation of human umbilical vein
smooth muscle cells and a rat vascular smooth muscle cell line,
A7r5. Circulation. 108:3048–3053. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Bielawski K, Winnicka K and Bielawska A:
Inhibition of DNA topoisomerases I and II, and growth inhibition of
breast cancer MCF-7 cells by ouabain, digoxin and proscillaridin A.
Biol Pharm Bull. 29:1493–1497. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
McConkey DJ, Lin Y, Nutt LK, Ozel HZ and
Newman RA: Cardiac glycosides stimulate Ca2+ increases
and apoptosis in androgen-independent, metastatic human prostate
adenocarcinoma cells. Cancer Res. 60:3807–3812. 2000.PubMed/NCBI
|
|
34
|
Newman RA, Yang P, Hittelman WN, Lu T, Ho
DH, Ni D, Chan D, Vijjeswarapu M, Cartwright C, Dixon S, et al:
Oleandrin-mediated oxidative stress in human melanoma cells. J Exp
Ther Oncol. 5:167–181. 2006.PubMed/NCBI
|
|
35
|
Newman RA, Kondo Y, Yokoyama T, Dixon S,
Cartwright C, Chan D, Johansen M and Yang P: Autophagic cell death
of human pancreatic tumor cells mediated by oleandrin, a
lipid-soluble cardiac glycoside. Integr Cancer Ther. 6:354–364.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Frese S, Frese-Schaper M, Andres AC,
Miescher D, Zumkehr B and Schmid RA: Cardiac glycosides initiate
Apo2L/TRAIL-induced apoptosis in non-small cell lung cancer cells
by up-regulation of death receptors 4 and 5. Cancer Res.
66:5867–5874. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hallböök H, Felth J, Eriksson A, Fryknäs
M, Bohlin L, Larsson R and Gullbo J: Ex vivo activity of cardiac
glycosides in acute leukaemia. PloS one. 6:e157182011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kulikov A, Eva A, Kirch U, Boldyrev A and
Scheiner-Bobis G: Ouabain activates signaling pathways associated
with cell death in human neuroblastoma. Biochim Biophys Acta.
1768:1691–1702. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
López-Lázaro M, Pastor N, Azrak SS, Ayuso
MJ, Austin CA and Cortés F: Digitoxin inhibits the growth of cancer
cell lines at concentrations commonly found in cardiac patients. J
Nat Prod. 68:1642–1645. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Gallo R, Padurean A, Jayaraman T, Marx S,
Roque M, Adelman S, Chesebro J, Fallon J, Fuster V, Marks A, et al:
Inhibition of intimal thickening after balloon angioplasty in
porcine coronary arteries by targeting regulators of the cell
cycle. Circulation. 99:2164–2170. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Sherr CJ and Roberts JM: CDK inhibitors:
Positive and negative regulators of G1-phase progression. Genes
Dev. 13:1501–1512. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wang SH, Liang CJ, Weng YW, Chen YH, Hsu
HY, Chien HF, Tsai JS, Tseng YC, Li CY and Chen YL: Ganoderma
lucidum polysaccharides prevent platelet-derived growth
factor-stimulated smooth muscle cell proliferation in vitro and
neointimal hyperplasia in the endothelial-denuded artery in vivo. J
Cell Physiol. 227:3063–3071. 2012. View Article : Google Scholar
|
|
43
|
Guan H, Gao L, Zhu L, Yan L, Fu M, Chen C,
Dong X, Wang L, Huang K and Jiang H: Apigenin attenuates neointima
formation via suppression of vascular smooth muscle cell phenotypic
transformation. J Cell Biochem. 113:1198–1207. 2012. View Article : Google Scholar
|
|
44
|
Gomez D and Owens GK: Smooth muscle cell
phenotypic switching in atherosclerosis. Cardiovasc Res.
95:156–164. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wang Z, Wang DZ, Hockemeyer D, McAnally J,
Nordheim A and Olson EN: Myocardin and ternary complex factors
compete for SRF to control smooth muscle gene expression. Nature.
428:185–189. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Persad S, Attwell S, Gray V, Mawji N, Deng
JT, Leung D, Yan J, Sanghera J, Walsh MP and Dedhar S: Regulation
of protein kinase B/Akt-serine 473 phosphorylation by
integrin-linked kinase: Critical roles for kinase activity and
amino acids arginine 211 and serine 343. J Biol Chem.
276:27462–27469. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Yudowski GA, Efendiev R, Pedemonte CH,
Katz AI, Berggren PO and Bertorello AM: Phosphoinositide-3 kinase
binds to a proline-rich motif in the Na+,
K+-ATPase alpha subunit and regulates its trafficking.
Proc Natl Acad Sci USA. 97:6556–6561. 2000. View Article : Google Scholar
|
|
48
|
Eva A, Kirch U and Scheiner-Bobis G:
Signaling pathways involving the sodium pump stimulate NO
production in endothelial cells. Biochim Biophys Acta.
1758:1809–1814. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Jagielska J, Salguero G, Schieffer B and
Bavendiek U: Digitoxin elicits anti-inflammatory and vasoprotective
properties in endothelial cells: Therapeutic implications for the
treatment of atherosclerosis? Atherosclerosis. 206:390–396. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Chase AJ and Newby AC: Regulation of
matrix metalloproteinase (matrixin) genes in blood vessels: A
multi-step recruitment model for pathological remodelling. J Vasc
Res. 40:329–343. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Johnson JL, Dwivedi A, Somerville M,
George SJ and Newby AC: Matrix metalloproteinase (MMP)-3 activates
MMP-9 mediated vascular smooth muscle cell migration and neointima
formation in mice. Arterioscler Thromb Vasc Biol. 31:e35–e44. 2011.
View Article : Google Scholar : PubMed/NCBI
|