Introduction
Mitochondria are known as cellular powerhouses due
to their ability to generate adenosine triphosphate (ATP) for use
in metabolic processes. These intracellular organelles of bacterial
origin, are important for bioenergetic function and they retain
their own genome and replication properties. The mitochondria are
also regarded as critical regulators of cell death and are the
major cellular source of reactive oxygen species (ROS) (1), which cause damage to mitochondrial
DNA (mtDNA) in human vascular smooth muscle cells (VSMCs) in a
number of cardiovascular pathologies (2). Thus, mitochondrial dysfunction may
lead to the impairment of various aspects of tissue functioning.
Angiotensin II (Ang II) has been shown to participate in
physiological processes, such as sodium and water homeostasis, and
vascular contraction, as well as in pathophysiological processes,
including hypertrophic cell growth, endothelial dysfunction, and
cardiovascular and renal remodeling (3). Ang II may induce the production of
mitochondrial ROS (mtROS) and several studies have suggested that
this process is dependent on the full enzymatic activity of NADPH
oxidases and the activation of ATP-sensitive potassium channels
(KATP) (3,4). Furthermore, the interplay between
the mitochondria and NADPH oxidase-derived O−2
constitutes a feed-forward cycle (5) and Ang II-induced ROS production may
cause mitochondrial dysfunction in VSMCs (6). Since the mitochondria are both a
target and source of ROS, antioxidant strategies specifically
targeting the mitochondria may prove to be a therapeutically
beneficial approach for the treatment of a number of ROS-induced
cardiovascular diseases (3).
Astragaloside IV (As-IV;
3-O-β-D-xylopyranosyl-6-O-β-D-gluco pyranosyl cycloastragenol), is
a natural saponin purified from Astragalus membranaceus
(Fisch.) Bge., a traditional Chinese herb that has been widely used
in clinical practice for the treatment of cardiovascular diseases
(7). The herb exhibits
antioxidant effects through the inhibition of ROS production, the
reduction of lipid peroxidation and the stimulation of antioxidant
enzymes (8). As-IV has been
reported to possess diverse pharmacological properties, including
anti-inflammatory (9,10), anti-apoptotic (11), anti-infarction (12), anti-hypertensive (13), anti-diabetic (14), anti-heart failure and myocardial
protective properties (15,16). Moreover, As-IV has been shown to
have antioxidant properties in various types of cells, such as
human umbilical endothelial (17), human mesangial (18), SK-N-SH (19) and myocardial cells (20), and plays a role in the treatment
of ischemia and reperfusion injury through energy regulatory
mechanisms (21). Since Ang
II-induced ROS production may cause mitochondrial dysfunction in
VSMCs (6), previous findings have
suggested a possible role for As-IV as a therapeutic agent against
mitochondrial dysfunction (8,
17–20). Thus, the present study was carried
out to investigate the protective effects of As-IV against Ang
II-induced mitochondrial injury in VSMCs and to explore the
potential mechanisms responsible for these effects.
Materials and methods
As-IV preparation
As-IV was purchased from Spring & Autumn
Biological Engineering Co., Ltd., (Nanjing, China) and its purity
was shown to be >98% by high-performance liquid chromatography
(HPLC) analysis and thin layer chromatography (TLC) with a single
dot (data not shown). As-IV was completely dissolved in
hydroxypropyl-beta-cyclodextrin (HPBCD; Sigma, St. Louis, MO, USA)
to obtain a stock solution at a concentration of 6 mg/ml and
subsequently diluted to 50 µg/ml with incubation medium to
prepare a working solution. Preliminary experiments revealed that
As-IV at this concentration demonstrated a more prominent
pharmacodynamic effect (data not shown).
Cell culture and treatments
The VSMCs derived from the thoracic aorta were
obtained from male Sprague-Dawley (SD) rats (weighing 180–200 g).
The rats were purchased from the Animal Center of Nanjing Medical
University (Nanjing, China). The animal experimental procedures
were carried out in accordance with the Experimental Animal Ethics
rules and regulations of the Ethics Committee for Animal
Experimentation of Nanjing Medical University. The vascular media
of the thoracic aortas were isolated from the rats under sterile
conditions. The media were minced and plated into a cell culture
bottle. The tissue was cultured in Dulbecco's modified Eagle's
medium (DMEM), Wisent Inc., Montreal, QC, Canada) with 10% (v/v)
fetal bovine serum (FBS; Gibco, Grand Island, NY, USA), 100 IU/ml
penicillin and 100 g/ml streptomycin at 37°C in a humidified
atmosphere containing 5% CO2. When the newly grown cells
reached 80–90% confluence around the tissue, the cells were
harvested by trypsin (Sigma) enzyme-digestion and were passaged
in vitro for use in later experiments. VSMCs were identified
by immunocytochemistry with anti-α-smooth muscle actin antibody
(1:100; ab5694, Abcam, Cambridge, UK) and by morphological
anlaysis. VSMCs between passages 3 and 5 were used in the present
study. When the cells reached 70–80% confluence, and they were
subjected to 24 h of starvation in serum-free DMEM. The cells were
then incubated in DMEM supplemented with Ang II (Sigma) at a
concentration of 1 µM and 5% FBS for 24 h and subsequently
treated with either a mixture of Ang II (1 µM) and As-IV (50
µg/ml) or Ang II (1 µM) alone for a further 24 h. The
untreated controls were maintained in DMEM only with 5% FBS for 48
h. In addition, this study protocol was approved by the Medical
Ethics Committee of the First Affiliated Hospital of Nanjing
Medical University, Nanjing, China.
Transmission electron microscopy
(TEM)
Following treatment, the VSMCs were collected and
fixed in 2.5% glutaraldehyde overnight, and this was followed by
secondary fixation in 1% osmium tetroxide for 1 h. The fixed
specimens were then dehydrated through a graded series of ethanol
to 100% and embedded in TAAB Epon (Marivac Canada Inc., St.
Laurent, QC, Canada). Ultrathin sections (60 nm) were cut, placed
onto copper grids, stained with uranyl acetate and lead citrate
prior to examination using a transmission electron microscope
(Tecnai G2 Spirit BioTWIN; FEI, Hillsboro, OR, USA) at an
accelerating voltage of 80 kV.
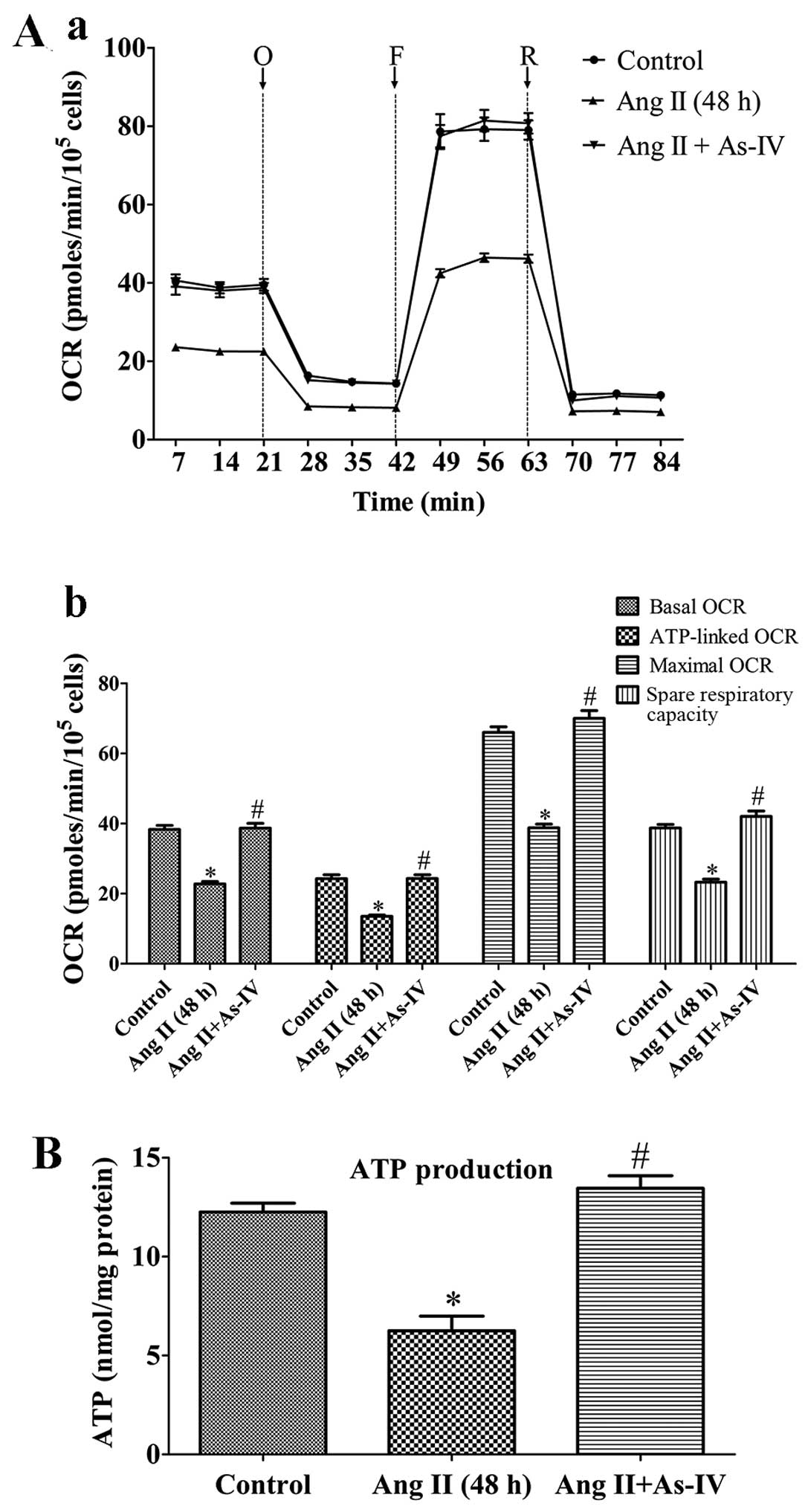
Measurement of mitochondrial bioenergetic
function by extracellular flux (XF) analysis
The XF96 Extracellular Flux Analyzer (Seahorse
Bioscience, Billerica, MA, USA) was used to measure the rate
changes in extracellular flux of dissolved O2 and
protons in the medium immediately surrounding the adherent intact
cells cultured on a XF96-well microplate (Seahorse Bioscience). The
VSMCs were seeded in XF96-well microplates at 8.0×103
cells/well (0.32 cm2) in 80 µl DMEM medium with
10% FBS and incubated at 37°C with 5% CO2 for 24 h. The
VSMCs were then treated as described above. The medium was removed
and replaced with assay medium 1 h prior to the beginning of the
assay and maintained at 37°C. After taking baseline measurements of
the mitochondrial oxygen consumption rates (OCRs), the OCRs were
measured using the XF Cell Mito Stress Test kit (Seahorse
Bioscience), and the following were sequentially added to each
well: oligomycin (1 µM, blocker of the mitochondrial complex
V, inhibiting the electron chain from being coupled to ATP
synthesis), FCCP (2 µM, an uncoupling agent which allows
maximum electron transport) and rotenone (1 µM,
mitochondrial complex I blocker which eliminates mitochondrial
respiration), as prevoiusly described (22). A typical OCR trace on how each
parameter is derived is shown in Fig.
1B (panel a). Values were normalized to the cell number
(105 cells as one unit) per well by cell counting on
completion of the XF assay.
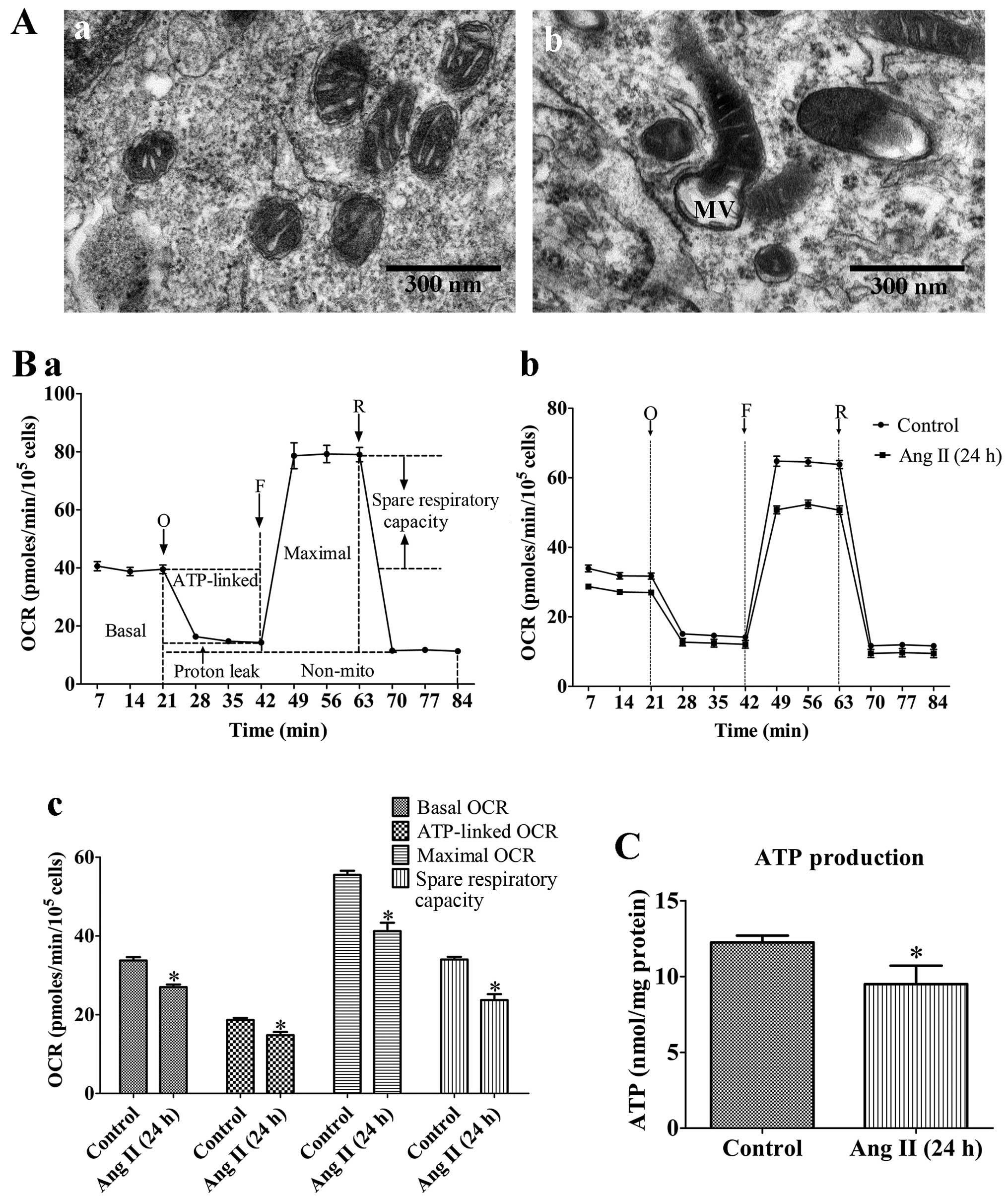 | Figure 1Mitochondrial dysfunction of rat
VSMCs induced by angiotensin II (Ang II; 1 µM) treatment for
24 h. (A) Transmission electron micrographs of mitochondrial
morphology in VSMCs (original magnification, ×80,000). (Panel a)
Untreated VSMCs; (panel b) VSMCs treated with Ang II (1 µM)
for 24 h. The mitochondria in the VMSCs treated with Ang II (1
µM) for 24 h were swollen, and vacuolization had occurred
with almost a complete loss of cristae. MV, mitochondrial
vacuolization; scale bar, 300 nm. (B) Assessment of mitochondrial
function in VSMCs. (Panel a) Schematic representation of the
mitochondrial function assay, including basal oxygen consumption
rate (OCR), ATP-linked OCR, maximal OCR, spare respiratory
capacity, proton leak and non-mito OCR (25). O, oligomycin; F, FCCP; R,
rotenone. (Panel b) Real-time analysis of mitochondrial OCRs.
(Panel c) Quantitative analysis of the effect of Ang II on
mitochondrial OCR readings. Results are the means ± SEM of 3
independent experiments (data were averaged from 12–16 duplicate
wells). (C) Effects of Ang II on cellular ATP production. Control,
untreated VSMCs; Ang II (24 h), VSMCs treated with Ang II (1
µM) for 24 h. Data represents the means ± SEM of 5
independent experiments. *P<0.05 vs. untreated
VSMCs. |
Measurement of mitochondrial ATP
production
Following treatment, the VSMCs were collected and
disrupted in 200 µl of ice-cold lysis buffer from the
luciferase-based luminescence ATP detection kit (Beyotime Institute
of Biotechnlogy, Nantong, China). Following centrifugation at
12,000 × g for 5 min at 4°C, ATP detection working solution (100
µl) was added to each well of a black 96-well culture plate
which was then incubated for 3 min at room temperature. Cell lysate
(40 µl) was then added to the wells of the culture plate,
and luminescence was measured immediately using a luminometer
(Turner BioSystems, Sunnyvale, CA, USA). A standard curve was
generated from the protein concentration of each well using the BCA
Protein assay kit (Thermo Fisher Scientific, Waltham, MA, USA). The
ATP level in each sample was expressed as nmol/mg protein.
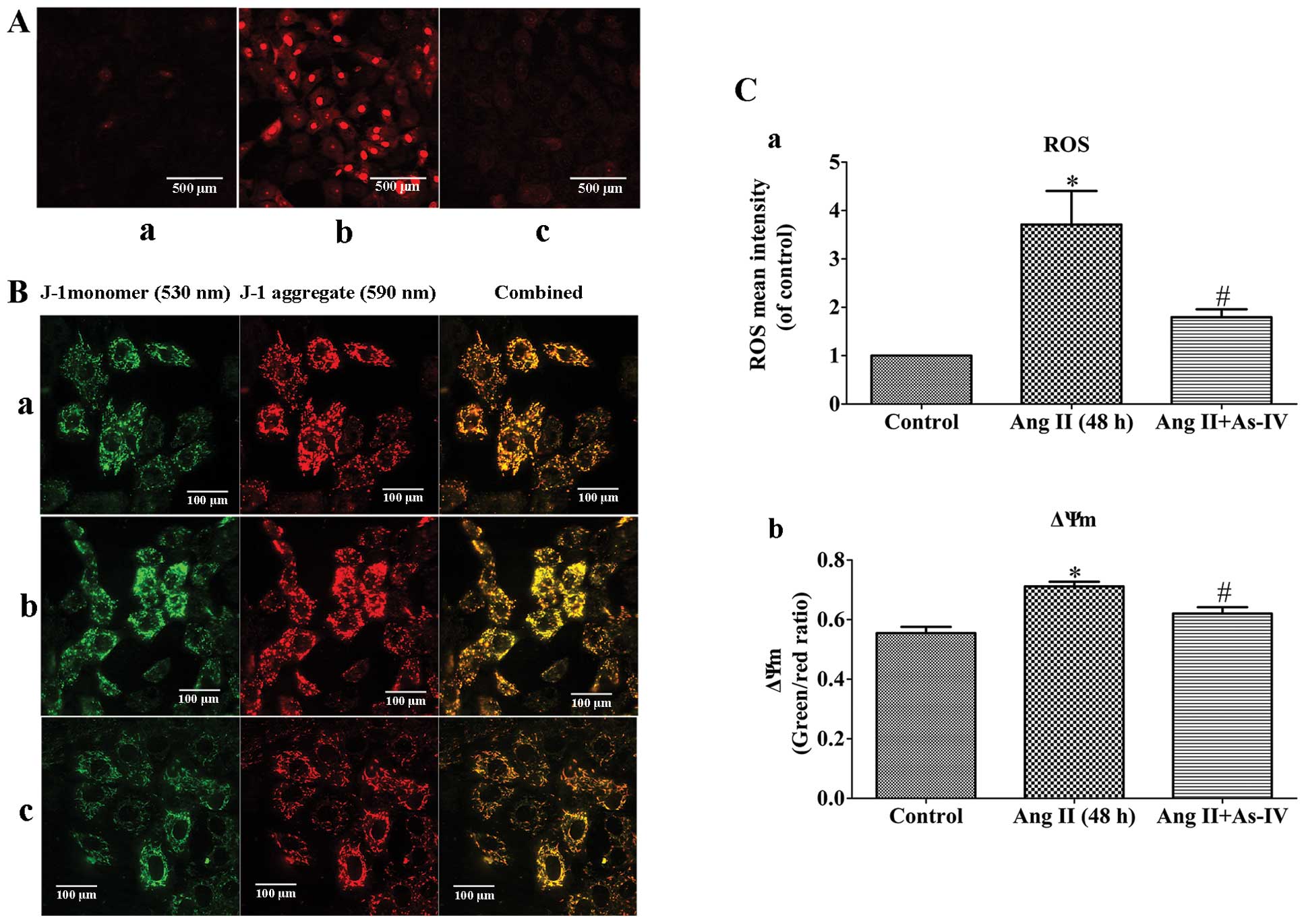
Measurement of ROS production
Following treatment, the VSMCs were incubated with
MitoSOX™ Red mitochondrial superoxide indicator (Molecular Probes,
Inc., Eugene, OR, USA) at 37°C for 20 min and washed gently 3 times
with warm buffer, and then imaged using a confocal microscope
(original magnification, ×200). Mitochondrial ROS generation was
expressed as the mean fluorescence intensity of the red color, as
previously described (23).
Measurement of mitochondrial membrane
potential (ΔΨm)
Following treatment, the VSMCs were stained with 2
µmol/l of JC-1 (Beyotime Institute of Biotechnology) probe
for 30 min and rinsed twice with PBS and then imaged using a
confocal microscope (original magnification, ×400). ΔΨm was
determined by monitoring the dual emissions from the mitochondrial
JC-1 monomers (green) and the aggregates (red) under a confocal
microscope under 488 nm laser excitation. ΔΨm was expressed as the
emission intensity ratio (the ratio of green to red fluorescence)
which represents the relative arbitrary ΔΨm level. A higher ratio
indicates a lower ΔΨm.
Measurement of ROS-related enzyme
activity
Following treatment, the total cellular protein from
the VSMCs was prepared as previously described (24), and the protein concentration was
assessed using a BCA protein assay kit (Thermo Fisher Scientific).
The activities of NADPH oxidase, xanthine oxidase and manganese
superoxide dismutase (Mn-SOD) were measured using an NADPH oxidase
activity quantitative assay kit (Genmed Scientifics Inc.,
Wilmington, DE, USA), a xanthine oxidase assay kit (Nanjing
Jiancheng Bioengineering Institute, Nanjing, China) and a
CuZn/Mn-SOD assay kit (Beyotime Institute of Biotechnology)
according to the manufacturer's instructions, respectively. The
values were calculated according to the manufacturer's instructions
and standardized to the protein concentration of the cells.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Following treatment, total RNA was purified from the
VSMCs using TRIzol reagent (Invitrogen, Carlsbad, CA, USA)
according to the manufacturer's instructions. Total RNA (1
µg) was reverse transcribed into cDNA using the
PrimeScriptTM RT reagent kit (Takara Bio, Dalian, China), and then
the cDNA was amplified with SYBR Premix Ex Taq™ (Taka a Bio) under
the following cycling conditions: 30 sec at 95°C for 1 cycle,
followed by 40 cycles of 5 sec at 95°C and 30 sec at 60°C. The
primer sequences were as follows: peroxisome proliferator-activated
receptor-gamma coactivator-1α (PGC-1α) sense, 5′-CGG AGC AAT CTG
AGT TAT ACG-3′ and antisense, 5′-CAG TCA CAG GAG GCA TCT-3′;
mitochondrial transcription factor A (Tfam) sense, 5′-ATC TCA TCC
GTC GCA GTG-3′ and antisense, 5′-GCA CAG TCT TGA TTC CAG TTC-3′;
and GAPDH sense, 5′-GTG AAG GTC GGT GTG AAC-3′ and antisense,
5′-GGT GAA GAC GCC AGT AGA-3′. The threshold cycle (Ct) value was
set within the exponential phase of the PCR. The relative
quantities of the different mRNAs were calculated by comparing the
cycle times for each target PCR. The target PCR Ct values were
normalized by subtracting the Ct (GAPDH) value, and then the
relative mRNA expression was calculated using the 2−(∆Ct
sample−∆Ct control) method for each sample. Essentially, the
same protocol was used for mtDNA quantification, normalizing the
Rnr2 gene amplification level against the GAPDH gene. The primer
sequences were as follows: Rnr2 sense, 5′-AGC TAT TAA TGG TTC GTT
TGT-3′ and antisense, 5′-AGG AGG CTC CAT TTC TCT TGT-3′; and GAPDH
sense, 5′-GGA CCT CAT GGC CTA CAT GG-3′ and antisense, 5′-ATT CGA
GAG AAG GGA GGG CT-3′. The ratio of mtDNA (Rnr2) to nDNA (GAPDH)
was used as an estimate of the number of mitochondria per cell.
Western blot analysis
Total cellular protein from the treated VSMCs was
extracted following standard protocols (24) and the protein concentration of the
supernatant was determined using the BCA protein assay kit (Thermo
Fisher Scientific). Equal amounts of protein (30 µg) were
electrophoretically separated by 8% SDS-PAGE and then transferred
onto polyvinylidene difluoride membranes. After blocking (non-fat
milk 5%, 2 h), the membranes were incubated at 4°C overnight with
the following primary antibodies: anti-PGC-1α antibody (1:1,000;
ab54481; Abcam), anti-dynamin 1-like protein 1 (Drp1) antibody
(1:1,000; ab56788, Abcam), anti-parkin antibody (1:1,000; Abcam,
ab179812) and anti-GAPDH antibody (1:1,000; 5174P; Cell Signaling
Technology, Inc., Danvers, MA, USA). After washing, the membranes
were incubated with HRP-conjugated secondary antibodies, including
anti-rabbit antibody (1:5,000; ab6721, Abcam) and anti-mouse
antibody (1:2,000; ab97023, Abcam) at room temperature. Signals
were detected with SuperSignal West Pico chemiluminescent substrate
(Thermo Fisher Scientific), and the intensity of the protein band
was quantified using Gel-Pro Analyzer software. Intensity values
were normalized to GAPDH and expressed as the relative protein
expression.
Statistical analysis
Data are expressed as the means ± SEM of a certain
number of independent experiments (the exact number of experiments
performed is indicated in each figure legend). Statistical analysis
was performed by a Student's t-test or by one-way ANOVA followed by
post hoc tests. A value of P<0.05 was considered to indicate a
statisticallydifference.
Results
Damaging effects of Ang II on the
morphology and function of mitochondria in VSMCs
To confirm the effects of As-IV treatment, we
evaluated the extent of Ang II-induced mitochondrial damage by
assessing mitochondrial morphology and bioenergetic function. Our
results revealed that the mitochondria in the VMSCs treated with
Ang II (1 µM) for 24 h were swollen, and vacuolization had
occurred with almost a complete loss of cristae (Fig. 1A). In addition, treatment with Ang
II (1 µM) for 24 h markedly reduced the basal OCR,
ATP-linked OCR, maximal OCR and spare respiratory capacity by 19.6,
16.8, 24.1 and 30.5% compared with the untreated VSMCs,
respectively (Fig. 1B).
Furthermore, treatment with Ang II (1 µM) for 24 h reduced
cellular ATP production compared with the untreated cells (Fig. 1C). These results demonstrated that
the VSMCs treated with Ang II (1 µM) for 24 h exhibited
signs of damage; abnormal mitochondrial morphology and impaired
bioenergetic function.
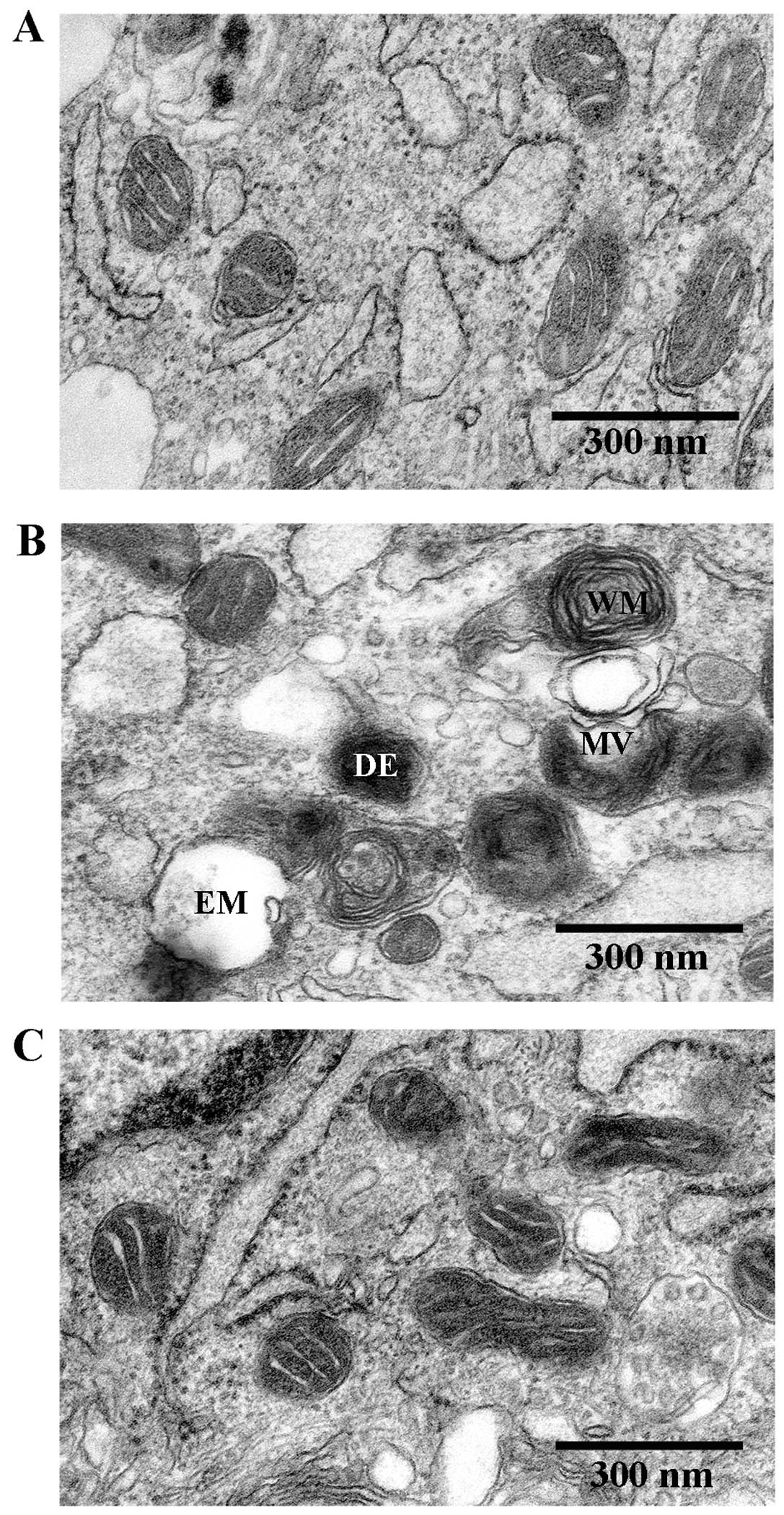
Beneficial effects of As-IV on damaged
mitochondria in VSMCs
To determine the beneficial effects of As-IV on
damaged mitochondria, we used TEM to observe the mitochondrial
ultramicrostructure. TEM revealed that the mitochondrial morphology
remained intact in the untreated VSMCs, whereas the mitochondria
from the VMSCs treated with Ang II (1 µM) for 48 h were
swollen and vacuolization had occurred with almost a complete loss
of cristae. Furthermore, some of the mitochondria contained
electron dense deposits in their matrices and exhibited whorl-like
inner membrane defects. By contrast, the mitochondria from the
VSMCs treated with As-IV (50 µg/ml) for 24 h following
exposure to Ang II for 24 h, exhibited a basically normal
morphology and no significant ultrastructural damage (Fig. 2).
Effects of As-IV on mitochondrial
bioenergetic function
To demonstrate the bioenergetic function of the
mitochondria in VSMCs following treatment with Ang II and the
effects of As-IV on mitochondrial energy metabolism, we exposed the
VSMCs to Ang II for 24 h, and then added As-IV (50 µg/ml)
for a further 24 h. Treatment with Ang II (1 µM) for 48 h
markedly reduced the basal OCR, ATP-linked OCR, maximal OCR and
spare respiratory capacity by 40.6, 44.4, 41.2 and 39.9% compared
with the untreated VSMCs, respectively. By contrast, treatment with
As-IV (50 µg/ml) significantly increased the mitochondrial
respiratory functions in the VSMCs, so that there was almost no
difference with the untreated VSMCs (Fig. 3A). Treatment with Ang II (1
µM) for 48 h also reduced cellular ATP production compared
with the controls, whereas treatment with As-IV (50 µg/ml)
reversed the decrease in ATP production (Fig. 3B).
As-IV prevents the Ang II-induced
excessive generation of mtROS and the decrease in ΔΨm
Ang II-induced mtROS production may be a potential
mechanism responsible for mitochondrial dysfunction (3) and the Ang II-induced depolarization
of ΔΨm has been shown to be dependent on ROS (6). In this study, compared with the
untreated VSMCs, a significant increase in the generation of mtROS
and a decrease in ΔΨm (shown by an increase in the green/red
fluorescence ratio) was observed in the Ang II-treated VSMCs.
However, treatment of the VSMCs with As-IV reversed these effects,
with no significant changes observed in mtROS generation and ΔΨm
compared to the controls (Fig.
4).
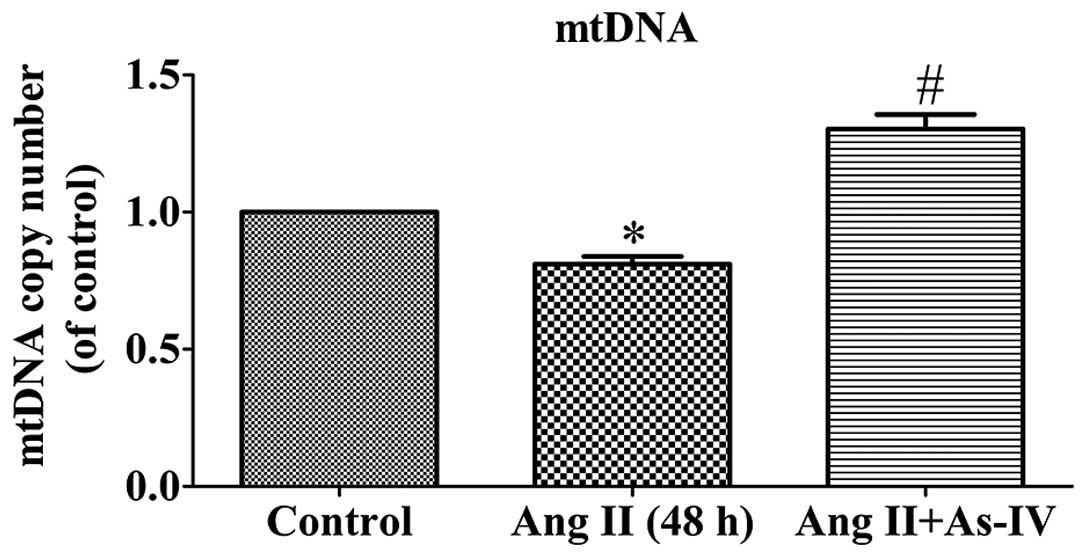
As-IV inhibits the Ang II-induced
decrease in mtDNA copy numbers in VSMCs
mtDNA is particularly susceptible to oxidative
damage resulting from ROS production in the mitochondrial matrix
and the lack of DNA protective histones. Maintaining an adequate
copy number of mtDNA is related to the energy demands that sustain
normal function and it is crucial for cell viability (26). As shown by our results, the mtDNA
copy number of the Ang II-treated VSMCs was lower than that of the
untreated VSMCs. Compared with Ang II treatment alone, the addition
of As-IV to the medium significantly increased the mtDNA copy
number (Fig. 5).
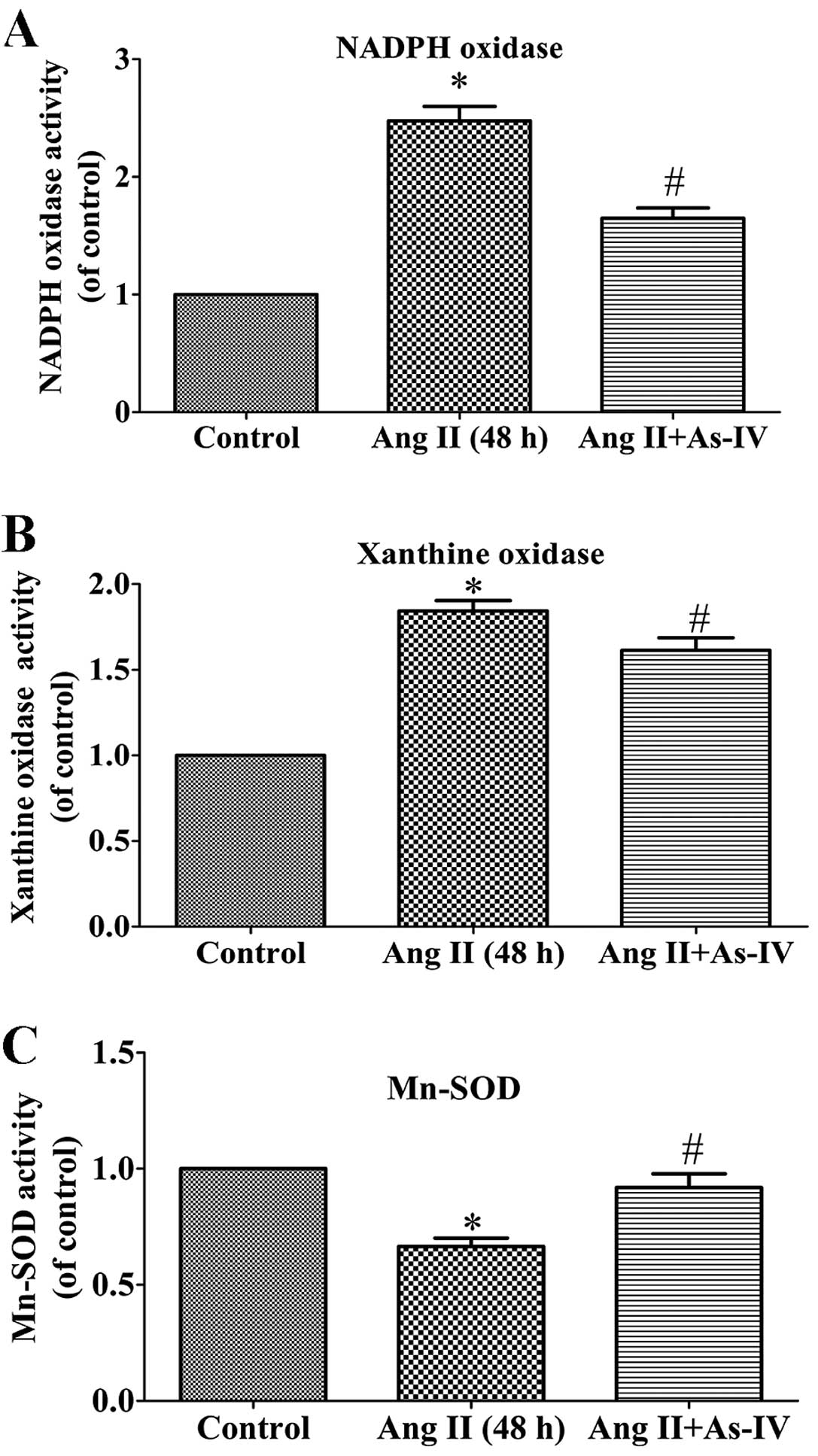
As-IV attenuates the activities of NADPH
oxidase and xanthine oxidase and enhances the activity of
Mn-SOD
ROS are generated from different sources, including
NADPH oxidases and xanthine oxidase, while Mn-SOD
(SOD2), which is located in the mitochondrial matrix, is
an important antioxidant which regulates ROS production (27). The present study demonstrated that
compared with the untreated VSMCs, the activity of NADPH oxidase
(Fig. 6A) and xanthine oxidase
(Fig. 6B) increased significantly
in the Ang II treated VSMCs, whereas this increase in enzyme
activity was alleviated by treatment with As-IV. Furthermore, As-IV
also exhibited the ability to reverse the decrease in Mn-SOD
activity induced by Ang II (Fig.
6C).
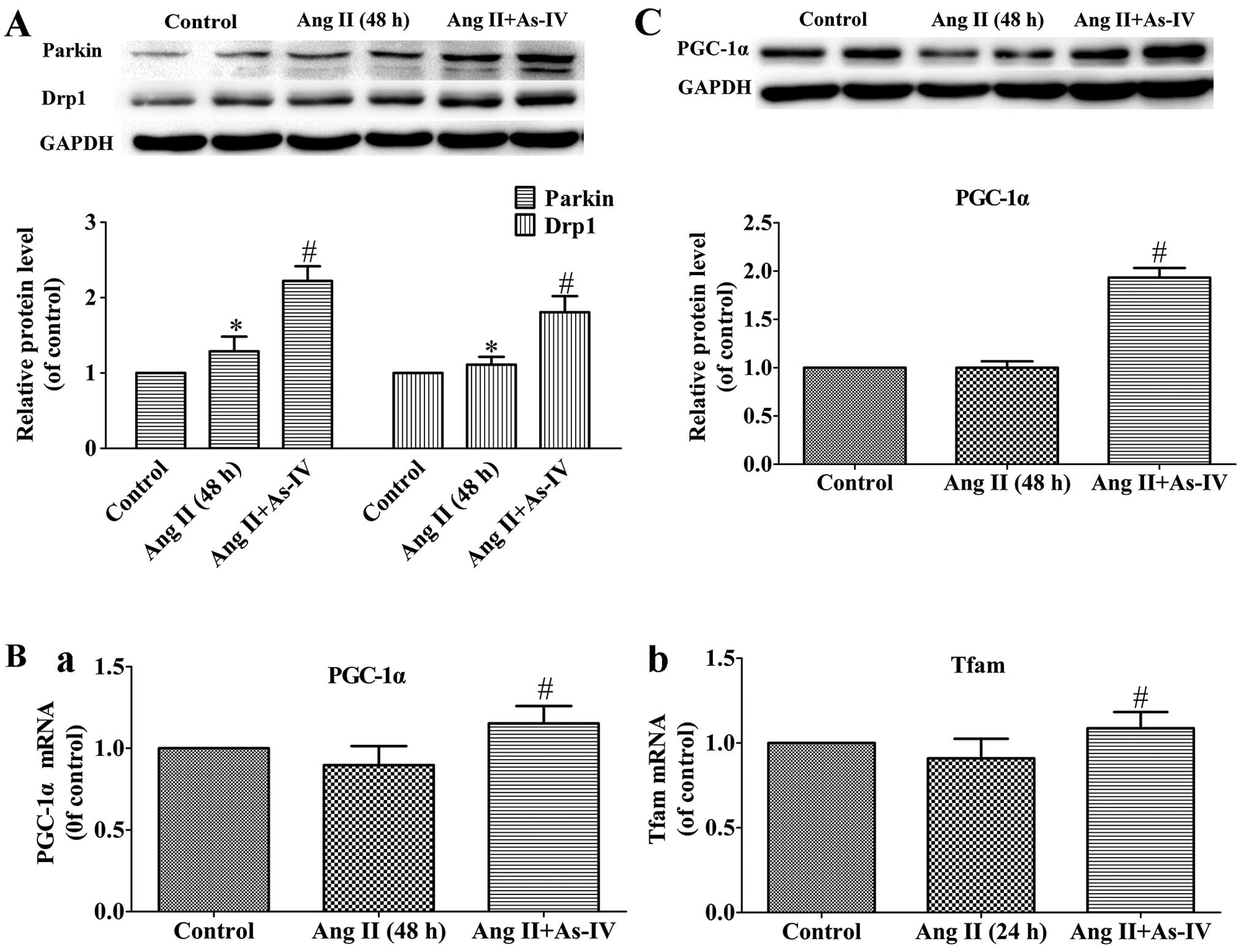
Effects of As-IV on the expression of
genes related to mitochondrial autophagy and biogenesis
Mitochondrial autophagy is mediated by parkin and
Drp1 (43), while mitochondrial
biogenesis is regulated by PGC-1α and Tfam (47). In the present study, our results
revealed that the protein levels of Drp1 and parkin, which are
vital to mitochondrial autophagy, increased in the Ang II-treated
VSMCs, and treatment with As-IV further increased the protein
expression of parkin and Drp1 (Fig.
7A). On the other hand, compared with the Ang II-treated VSMCs,
treatment with As-IV significantly enhanced the mRNA and protein
expression of PGC-1α and the mRNA expression of Tfam. By contrast,
we were unable to detect significant differences in the expression
of PGC-1α and Tfam between the untreated VSMCs and the Ang
II-treated VSMCs (Fig. 7B and
C).
Discussion
The present study demonstrated that As-IV reverses
the Ang II-induced mitochondrial dysfunction in VSMCs by exerting
antioxidant effects against excessive ROS generation and enhancing
mitochondrial autophagy and biogenesis.
In the present study, we demonstrated that Ang II,
which was used at a concentration of 1 µM for 24 h, induced
mitochondrial dysfunction in VSMCs (Fig. 1). These results are in accordance
with those of previous studies (6,28,29). Thus, VSMCs treated with Ang II (1
µM) for 24 h were used as a cell model of mitochondrial
injury prior to treatment with As-IV.
In the present study, the Ang II-treated VSMCs
exhibited mitochondrial morphological abnormalities and
mitochondrial bioenergetic dysfunction, including a reduction in
mitochondrial OCRs and ATP production (Figs. 2 and 3). The Ang II-treated VSMCs also
exhibited a decrease in ΔΨm (Fig. 4B
and C) and mtDNA damage (Fig.
5), which are typical manifestations of mitochondrial damage.
Furthermore, mtROS generation was significantly increased in the
Ang II-treated VSMCs (Fig. 4A and
C) exhibiting mitochondrial damage.
Under normal physiological conditions, the levels of
ROS are generally low and they are controlled by antioxidants, such
as Mn-SOD. However, under pathological conditions, it has been
demonstrated that mtROS generation is triggered by elevated ROS
levels, caused by the Ang II-induced activation of NADPH oxidase
via AT1R-PKC signaling (30) and
the down-regulation of antioxidant expression, resulting in
decreased scavenging capacity (31); and mtROS also plays a critical
role in depressing mitochondrial energy metabolism (32,33). Thus, increased mtROS generation
primarily led to mitochondrial injury in the present study. Our
experiment to visualize mtROS provided evidence that As-IV
diminishes the Ang II-induced excessive generation of mtROS
(Fig. 4A and C). These results
demonstrate that As-IV exerts its antioxidant effects against
mitochondrial injury through the attenuation of the excessive
generation of mtROS.
The physiological generation of ROS occurs through
several mechanisms, including leakage from the electron transport
chain and as a metabolic byproduct of enzymes, such as xanthine
oxidase (34). Ang II-induced
NADPH oxidase activation is an example of a system that generates
ROS not as a byproduct, but rather as the primary function of the
enzyme system (35). On the other
hand, there is growing evidence to suggest that the Ang II-induced
activation of xanthine oxidase may be secondary to ROS generation
from other sources, through the thiol oxidation of the xanthine
dehydrogenase sulfhydryl residues which contribute, to a certain
extent, to Ang II-induced ROS generation (34,36). In this study, we found that Ang II
induced an increase in NADPH oxidase and xanthine oxidase activity,
and this effect was significantly inhibited by treatment with As-IV
(Fig. 6A and B).
In general, the maintenance of ROS production is
normally counteracted and an equilibrium oxidative state is
retained by SODs, which catalyze the dismutation of O−2
into oxygen and H2O2, thereby serving a key
antioxidant function (37). It
has been suggested that Mn-SOD (SOD2), one of the three
isoforms of SOD, is one of the important defenses against oxidative
damage within the mitochondria (27). The beneficial effects of As-IV on
Mn-SOD activity in renal proximal tubular cells (38) and H9c2 cells (39) have been demonstrated. Our
experiment on the activity of Mn-SOD revealed that As-IV possessed
the ability to reverse the reduction in mitochondrial Mn-SOD
activity indcued by Ang II (Fig.
6C). Taken together with the findings that As-IV attenuates the
activity of NADPH oxidase and xanthine oxidase, these results
suggest that As-IV reverses the Ang II-induced excessive production
of mtROS by inhibiting mtROS generation and stimulating the
elimination of mtROS.
In our experiment on the visualization of
mitochondrial morphology, we found that the VSMCs treated with Ang
II exhibited abnormal mitochondrial ultrastructures, while the
morphology of the mitochondria from VSMCs treated with As-IV was
basically normal. Previous studies have suggested that mitochondria
damaged by ROS need to be selectively removed to lysosomes and
degraded in a process known as mitophagy, to protect the cells from
cell death (40,41). They may then be substituted by the
progeny of more bioenergetically active mitochondria (42). Thus, we speculated that treatment
with As-IV may promote mitochondrial biogenesis and autophagy.
Parkin, from the cytosol, eliminates impaired
mitochondria by targeted mitophagy (42). Additionally, parkin-induced
mitochondrial fission, which requires Drp1, appears to be necessary
for mitophagy (42–44). In the present study, treatment
with Ang II increased the protein expression of parkin and Drp1,
and As-IV increased the protein expression of parkin and Drp1
(Fig. 7A). These findings suggest
that As-IV promotes the autophagy of impaired mitochondria. A
previous study suggested that increased ROS levels in the
mitochondrial matrix result in mitochondrial damage and the
subsequent activation of parkin-dependent mitophagy (45). Taken together with the results of
our study, these findings suggest that As-IV promotes the autophagy
of impaired mitochondria.
PGC-1α serves as a central transcriptional control
of mitochondrial biogenesis and respiratory function that
coordinately controls the mitochondrial energy-generating functions
in accordance with the metabolic demands forced by changing
physiological conditions, aging and disease (46). PGC-1α activates the expression of
Tfam, and then the transcription and replication of mtDNA is
directly activated by the translocation of Tfam into the
mitochondria (47). In the
present study, As-IV markedly enhanced the mRNA expression of
PGC-1α and Tfam (Fig. 7B), and
the protein expression of PGC-1α (Fig. 7C). These results may also explain
the stimulative effect of As-IV on the levels of mtDNA (Fig. 5). In this study, we found no
significant difference in the protein expression of PGC-1α and the
mRNA expression of Tfam between the untreated VSMCs and Ang
II-treated VSMCs. A previous study suggested that Ang II is unable
to lead to changes in the mRNA expression of PGC-1α and Tfam
(48). Taken together with the
results of our study, these findings suggest that As-IV enhances
the expression of PGC-1α and Tfam and promotes mitochondrial
biogenesis.
In conclusion, this study clearly demonstrates that
As-IV reverses the structural and biochemical abnormalities caused
by Ang II in VSMCs, by modulating mtROS production and eliminating
mtROS through its antioxidant effects, as well as by promoting
mitochondrial autophagy and mitochondrial biogenesis. These
findings suggest that mitochondria-targeted antioxidants may
represent an attractive novel strategy for the treatment of a
number of pathological conditions caused by the Ang II-mediated
production of mtROS.
Acknowledgments
The present study was funded by grants from the
National Natural Science Foundation of China (nos. 81170220 and
81100156), the Jiangsu Province Health Foundation (RC2011075), the
Priority Academic Program Development of Jiangsu Higher Education
Institutions and the Jiangsu Province Science and Technology
Support Program (BE2011803).
References
|
1
|
Yu E, Calvert PA, Mercer JR, Harrison J,
Baker L, Figg NL, Kumar S, Wang JC, Hurst LA, Obaid DR, et al:
Mitochondrial DNA damage can promote atherosclerosis independently
of reactive oxygen species through effects on smooth muscle cells
and monocytes and correlates with higher-risk plaques in humans.
Circulation. 128:702–712. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mercer JR: Mitochondrial bioenergetics and
therapeutic intervention in cardiovascular disease. Pharmacol Ther.
141:13–20. 2014. View Article : Google Scholar
|
|
3
|
Dikalov SI and Nazarewicz RR: Angiotensin
II-induced production of mitochondrial reactive oxygen species:
potential mechanisms and relevance for cardiovascular disease.
Antioxid Redox Signal. 19:1085–1094. 2013. View Article : Google Scholar :
|
|
4
|
Queliconi BB, Wojtovich AP, Nadtochiy SM,
Kowaltowski AJ and Brookes PS: Redox regulation of the
mitochondrial K(ATP) channel in cardioprotection. Biochim Biophys
Acta. 1813:1309–1315. 2011. View Article : Google Scholar :
|
|
5
|
Dikalov S: Cross talk between mitochondria
and NADPH oxidases. Free Radic Biol Med. 51:1289–1301. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Doughan AK, Harrison DG and Dikalov SI:
Molecular mechanisms of angiotensin II-mediated mitochondrial
dysfunction: linking mitochondrial oxidative damage and vascular
endothelial dysfunction. Circ Res. 102:488–496. 2008. View Article : Google Scholar
|
|
7
|
Wang H, Zhang Y, Xia T, Wei W, Chen F, Guo
X and Li X: Synergistic promotion of blood vessel regeneration by
astragaloside IV and ferulic acid from electrospun fibrous mats.
Mol Pharm. 10:2394–2403. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ko JK, Lam FY and Cheung AP: Amelioration
of experimental colitis by Astragalus membranaceus through
anti-oxidation and inhibition of adhesion molecule synthesis. World
J Gastroenterol. 11:5787–5794. 2005.PubMed/NCBI
|
|
9
|
Yang J, Wang HX, Zhang YJ, Yang YH, Lu ML,
Zhang J, Li ST, Zhang SP and Li G: Astragaloside IV attenuates
inflammatory cytokines by inhibiting TLR4/NF-κB signaling pathway
in isoproterenol-induced myocardial hypertrophy. J Ethnopharmacol.
150:1062–1070. 2013. View Article : Google Scholar
|
|
10
|
Gui D, Huang J, Guo Y, Chen J, Chen Y,
Xiao W, Liu X and Wang N: Astragaloside IV ameliorates renal injury
in streptozotocin-induced diabetic rats through inhibiting
NF-κB-mediated inflammatory genes expression. Cytokine. 61:970–977.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xu W, Shao X, Tian L, Gu L, Zhang M, Wang
Q, Wu B, Wang L, Yao J, Xu X, et al: Astragaloside IV ameliorates
renal fibrosis via the inhibition of mitogen-activated protein
kinases and antiapoptosis in vivo and in vitro. J Pharmacol Exp
Ther. 350:552–562. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wu X, Cao Y, Nie J, Liu H, Lu S, Hu X, Zhu
J, Zhao X, Chen J, Chen X, et al: Genetic and pharmacological
inhibition of Rheb1-mTORC1 signaling exerts cardioprotection
against adverse cardiac remodeling in mice. Am J Pathol.
182:2005–2014. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang WD, Zhang C, Wang XH, Gao PJ, Zhu
DL, Chen H, Liu RH and Li HL: Astragaloside IV dilates aortic
vessels from normal and spontaneously hypertensive rats through
endothelium-dependent and endothelium-independent ways. Planta Med.
72:621–626. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yin Y, Qi F, Song Z, Zhang B and Teng J:
Ferulic acid combined with astragaloside IV protects against
vascular endothelial dysfunction in diabetic rats. Biosci Trends.
8:217–226. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhao Z, Wang W, Wang F, Zhao K, Han Y, Xu
W and Tang L: Effects of Astragaloside IV on heart failure in rats.
Chin Med. 4:62009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Xu XL, Ji H, Gu SY, Shao Q, Huang QJ and
Cheng YP: Modification of alterations in cardiac function and
sarcoplasmic reticulum by astragaloside IV in myocardial injury in
vivo. Eur J Pharmacol. 568:203–212. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Qiu LH, Xie XJ and Zhang BQ: Astragaloside
IV improves homocysteine-induced acute phase endothelial
dysfunction via antioxidation. Biol Pharm Bull. 33:641–646. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sun L, Li W, Li W, Xiong L, Li G and Ma R:
Astragaloside IV prevents damage to human mesangial cells through
the inhibition of the NADPH oxidase/ROS/Akt/NF-κB pathway under
high glucose conditions. Int J Mol Med. 34:167–176. 2014.PubMed/NCBI
|
|
19
|
Sun Q, Jia N, Wang W, Jin H, Xu J and Hu
H: Protective effects of astragaloside IV against amyloid beta1-42
neurotoxicity by inhibiting the mitochondrial permeability
transition pore opening. PLoS One. 9:e988662014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hu JY, Han J, Chu ZG, Song HP, Zhang DX,
Zhang Q and Huang YS: Astragaloside IV attenuates hypoxia-induced
cardiomyocyte damage in rats by upregulating superoxide dismutase-1
levels. Clin Exp Pharmacol Physiol. 36:351–357. 2009. View Article : Google Scholar
|
|
21
|
Tu L, Pan CS, Wei XH, Yan L, Liu YY, Fan
JY, Mu HN, Li Q, Li L, Zhang Y, et al: Astragaloside IV protects
heart from ischemia and reperfusion injury via energy regulation
mechanisms. Microcirculation. 20:736–747. 2013.PubMed/NCBI
|
|
22
|
Shen Y, Tian Y, Yang J, Shi X, Ouyang L,
Gao J and Lu J: Dual effects of carnosine on energy metabolism of
cultured cortical astrocytes under normal and ischemic conditions.
Regul Pept. 192–193:45–52. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mukhopadhyay P, Rajesh M, Haskó G, Hawkins
BJ, Madesh M and Pacher P: Simultaneous detection of apoptosis and
mitochondrial superoxide production in live cells by flow cytometry
and confocal microscopy. Nat Protoc. 2:2295–2301. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Laurindo FR, de Souza HP, Pedro MA and
Janiszewski M: Redox aspects of vascular response to injury.
Methods Enzymol. 352:432–454. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Herst PM and Berridge MV: Cell surface
oxygen consumption: a major contributor to cellular oxygen
consumption in glycolytic cancer cell lines. Biochim Biophys Acta.
1767:170–177. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Xie H, Lev D, Gong Y, Wang S, Pollock RE,
Wu X and Gu J: Reduced mitochondrial DNA copy number in peripheral
blood leukocytes increases the risk of soft tissue sarcoma.
Carcinogenesis. 34:1039–1043. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Elahi MM, Kong YX and Matata BM: Oxidative
stress as a mediator of cardiovascular disease. Oxid Med Cell
Longev. 2:259–269. 2009. View Article : Google Scholar
|
|
28
|
Wei Y, Clark SE, Thyfault JP, Uptergrove
GM, Li W, Whaley-Connell AT, Ferrario CM, Sowers JR and Ibdah JA:
Oxidative stress-mediated mitochondrial dysfunction contributes to
angiotensin II-induced nonalcoholic fatty liver disease in
transgenic Ren2 rats. Am J Pathol. 174:1329–1337. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Prathapan A, Vineetha VP and Raghu KG:
Protective effect of Boerhaavia diffusa L. against mitochondrial
dysfunction in angiotensin II induced hypertrophy in H9c2
cardiomyoblast cells. PLoS One. 9:e962202014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Schulz E, Wenzel P, Munzel T and Daiber A:
Mitochondrial redox signaling: Interaction of mitochondrial
reactive oxygen species with other sources of oxidative stress.
Antioxid Redox Signal. 20:308–324. 2014. View Article : Google Scholar :
|
|
31
|
Lijnen PJ, van Pelt JF and Fagard RH:
Downregulation of manganese superoxide dismutase by angiotensin II
in cardiac fibroblasts of rats: Association with oxidative stress
in myocardium. Am J Hypertens. 23:1128–1135. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Touyz RM: Reactive oxygen species and
angiotensin II signaling in vascular cells - implications in
cardiovascular disease. Braz J Med Biol Res. 37:1263–1273. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kimura S, Zhang GX, Nishiyama A, Shokoji
T, Yao L, Fan YY, Rahman M and Abe Y: Mitochondria-derived reactive
oxygen species and vascular MAP kinases: comparison of angiotensin
II and diazoxide. Hypertension. 45:438–444. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zablocki D and Sadoshima J: Angiotensin II
and oxidative stress in the failing heart. Antioxid Redox Signal.
19:1095–1109. 2013. View Article : Google Scholar :
|
|
35
|
Bedard K and Krause KH: The NOX family of
ROS-generating NADPH oxidases: physiology and pathophysiology.
Physiol Rev. 87:245–313. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Suematsu N, Tsutsui H, Wen J, Kang D,
Ikeuchi M, Ide T, Hayashidani S, Shiomi T, Kubota T, Hamasaki N and
Takeshita A: Oxidative stress mediates tumor necrosis
factor-alpha-induced mitochondrial DNA damage and dysfunction in
cardiac myocytes. Circulation. 107:1418–1423. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Li H, Horke S and Förstermann U: Oxidative
stress in vascular disease and its pharmacological prevention.
Trends Pharmacol Sci. 34:313–319. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Qi W, Niu J, Qin Q, Qiao Z and Gu Y:
Astragaloside IV attenuates glycated albumin-induced
epithelial-to-mesenchymal transition by inhibiting oxidative stress
in renal proximal tubular cells. Cell Stress Chaperones.
19:105–114. 2014. View Article : Google Scholar
|
|
39
|
Wang YY, Peng Y, Zhang Q, Wu YN, Song JQ
and Liu YX: Effect of ERK1/2 signaling pathway on astragaloside IV
protects H9c2 cells against H2O2-induced
oxidative injury. Zhongguo Ying Yong Sheng Li Xue Za Zh.
27:363–367. 2011.In Chinese.
|
|
40
|
Mammucari C and Rizzuto R: Signaling
pathways in mitochondrial dysfunction and aging. Mech Ageing Dev.
131:536–543. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lee J, Giordano S and Zhang J: Autophagy,
mitochondria and oxidative stress: cross-talk and redox signalling.
Biochem J. 441:523–540. 2012. View Article : Google Scholar :
|
|
42
|
Narendra DP and Youle RJ: Targeting
mitochondrial dysfunction: role for PINK1 and Parkin in
mitochondrial quality control. Antioxid Redox Signal. 14:1929–1938.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kageyama Y, Hoshijima M, Seo K, Bedja D,
Sysa-Shah P, Andrabi SA, Chen W, Höke A, Dawson VL, Dawson TM, et
al: Parkin-independent mitophagy requires Drp1 and maintains the
integrity of mammalian heart and brain. EMBO J. 33:2798–2813. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Deng H, Dodson MW, Huang H and Guo M: The
Parkinson's disease genes pink1 and parkin promote mitochondrial
fission and/or inhibit fusion in Drosophila. Proc Natl Acad Sci
USA. 105:14503–14508. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wang Y, Nartiss Y, Steipe B, McQuibban GA
and Kim PK: ROS-induced mitochondrial depolarization initiates
PARK2/PARKIN-dependent mitochondrial degradation by autophagy.
Autophagy. 8:1462–1476. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Scarpulla RC, Vega RB and Kelly DP:
Transcriptional integration of mitochondrial biogenesis. Trends
Endocrinol Metab. 23:459–466. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Wu Z, Puigserver P, Andersson U, Zhang C,
Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC and
Spiegelman BM: Mechanisms controlling mitochondrial biogenesis and
respiration through the thermogenic coactivator PGC-1. Cell.
98:115–124. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Biala A, Tauriainen E, Siltanen A, Shi J,
Merasto S, Louhelainen M, Martonen E, Finckenberg P, Muller DN and
Mervaala E: Resveratrol induces mitochondrial biogenesis and
ameliorates Ang II-induced cardiac remodeling in transgenic rats
harboring human renin and angiotensinogen genes. Blood Press.
19:196–205. 2010. View Article : Google Scholar : PubMed/NCBI
|