Introduction
Melanogenesis is the most important function of
melanocytes, which are located in the basal layer of the epidermis
in human skin (1,2). Melanogenesis has many important
physiological functions, including the photoprotection of human
skin from ultraviolet (UV) irradiation (3). Melanin synthesis is stimulated by
various molecules and conditions, such as α-melanocyte-stimulating
hormone (α-MSH), cyclic AMP (cAMP)-elevating agents [including
forskolin, glycyrrhizin and isobutylmethylxanthine (IBMX)] and UV
radiation (4–6). Various dermatologic disorders result
from the accumulation of excessive levels of epidermal pigmentation
(4–6). Epidermal and dermal
hyperpigmentation may depend on either increased numbers of
melanocytes or melanogenic enzyme activities (7). Three key melanogenic enzymes,
tyrosinase, tyrosinase-related protein-1 (TRP-1), and
tyrosinase-related protein-2 (TRP-2), participate in melanogenesis
(8). Tyrosinase is the
rate-limiting enzyme that catalyzes two critical steps in
melanogenesis: the hydroxylation of L-tyrosine to
3,4-dihydroxyphenylalanine (DOPA) and the oxidation of DOPA to
dopaquinone (9–11). Dopaquinone is converted to
dopachrome, which is in turn converted to dihydroxyindole or
dihydroxyindole-2-carboxylic acid (DHICA) to form melanin.
TRP-1 catalyzes the oxidation of DHICA to
carboxylated indolequinone (12).
Thus, the upregulation or activation of melanogenic enzymes can
increase melanogenesis and hyperpigmentation. Additionally,
stimulators, such as α-MSH, forskolin and IBMX, regulate the
expression of tyrosinase, TRP-1 and TRP-2 by modulating the
activation of transcription factors, such as
microphthalmia-associated transcription factor (MITF) and cAMP
response element-binding protein (CREB) and through protein kinase
A (PKA) signaling pathways (13).
MITF is a master transcription factor for
melanogenesis. With a basic helix-loop-helix-leucine zipper
(bHLHZip) structure, MITF upregulates tyrosinase, TRP-1 and TRP-2
by binding to an M-box motif at the promoter sites (14). MITF regulates melanocyte
pigmentation, proliferation, survival and differentiation (15). Mutation of the MITF gene in humans
can cause abnormal pigmentation of the skin and hair (16,17). Moreover, decreased MITF expression
induces the down-regulation of melanocyte differentiation markers
and inhibits melanogenesis (18).
A key component of MITF induction is the UV-mediated induction of
the proopiomelanocortin (POMC) and MC1 genes encoding α-MSH. α-MSH
binding to MC1R increases the intracellular level of cAMP (19) and activates PKA, which
subsequently phosphorylates CREB protein, an activator of MITF gene
expression. Conversely, the activation of extracellular
signal-regulated kinase (ERK) induces MITF phosphorylation and
degradation, ultimately suppressing melanogenesis (20–22). MITF expression is also regulated
by the Wnt signaling pathway, the activation of which results in
glycogen synthase kinase 3β (GSK3β) inactivation and subsequent
β-catenin accumulation (23).
Furthermore, accumulated β-catenin moves into the nucleus and binds
lymphoid-enhancing factor/T-cell factor transcription factor,
stimulating MITF expression (24). By contrast, it is known that
activated GSK3β leads to the ubiquitination and degradation of
β-catenin (25).
The Wnt/β-catenin pathway, which is activated by the
interactions of Wnt 1, Wnt3a and Wnt8 with Frizzled (Fz) receptors
and low-density lipoprotein receptor-related protein 5/6
co-receptors (26), controls cell
differentiation, cell proliferation and cell motility (27–30). In the absence of a Wnt signal,
N-terminal serine/threonine residues of β-catenin are sequentially
phosphorylated by casein kinase 1 and GSK3β in a multiprotein
complex containing adenomatous polyposis coli and axin (31,32). Phosphorylated β-catenin is then
recognized by F-box beta-transducin repeat-containing protein, a
component of the ubiquitin ligase complex, resulting in the
degradation of β-catenin via an ubiquitin-dependent mechanism
(33). The activation of the
receptor by its Wnt ligands negatively regulates GSK3β, leading to
the accumulation of cytoplasmic β-catenin (34). A link between Wnt/β-catenin
signaling and melanocyte differentiation has been shown by the
finding that β-catenin, which accumulates by the activation of
Wnt/β-catenin signaling, forms a complex with lymphocyte enhancer
factor-1 to upregulate expression of the MITF gene (35). β-catenin also directly interacts
with the MITF protein itself and then activates MITF-specific
target genes (36). In addition
to MITF, Wnt/β-catenin signaling is involved in neural crest
formation, migration, proliferation and differentiation (24).
Several known natural melanin synthesis inhibitors,
including arbutin and kojic acid, have been the focus of previous
studies and are currently being utilized as cosmetic additives
(37). A great deal of attention
has continuously focused on the development of natural products for
future applications in the cosmetics industry (38,39). However, it is necessary to find
safer and more effective skin-whitening compounds due to the
carcinogenic potential and weak whitening effect of kojic acid.
In the present study, we examined the effects of the
novel adamantyl benzylbenzamide derivative, AP736, on melanin
synthesis and tyrosinase activity in B16F10 melanoma cells. In
addition, we examined the effects of AP736 on the expression of
MITF and tyrosinase.
Materials and methods
Materials
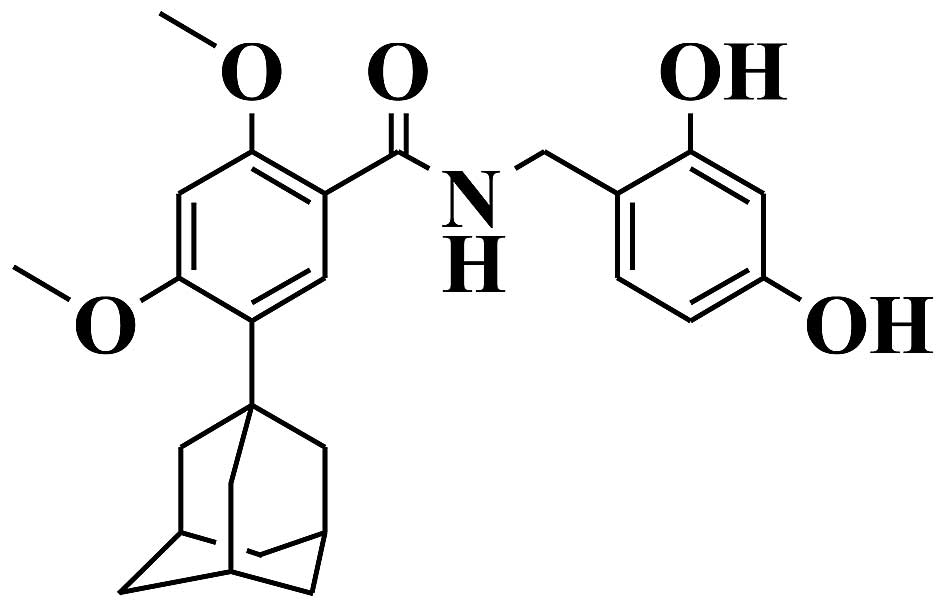
The novel adamantyl benzylbenzamide derivative,
AP736, was synthesized at the AmorePacific R&D Center
(structure shown in Fig. 1).
AP736 was dissolved in dimethyl sulf-oxide (DMSO) and stored at
−20°C as a stock solution (10 mM). α-MSH,
3,4-dihydroxy-L-phenylalanine (L-DOPA), mushroom tyrosinase,
phenylthiourea (PTU), kojic acid and
(2′Z,3′E)-6-bromoindirubin-3′-oxime (BIO) were purchased from
Sigma-Aldrich Co. (St. Louis, MO, USA). The Cell Counting kit-8
(CCK-8) was purchased from Dojindo Molecular Technologies, Inc.
(Kumamoto, Japan). Dulbecco's modified Eagle's medium (DMEM),
phosphate-buffered saline (PBS) and penicillin/streptomycin were
purchased from WelGene Biopharmaceuticals (Daegu, Korea). Fetal
bovine serum (FBS) was purchased from Gibco Life Technologies
(Grand Island, NY, USA). Antibodies specific for tyrosinase (C-19),
TRP-1 and actin were from Santa Cruz Biotechnology, Inc. (Santa
Cruz, CA, USA). Antibodies specific for phosphorylated (p-)GSK3β
(#9336) and total GSK3β (#9315) were purchased from Cell Signaling
Technology (Beverly, MA, USA), and microphthalmia Ab-1 (C5,
MS-771-P0) was obtained from NeoMarkers (Fremont, CA, USA).
β-catenin (610154) was from BD Transduction Laboratories (San
Diego, CA, USA). Secondary antibodies specific for anti-goat IgG
(PI-9500), anti-mouse IgG (PI-2000) and anti-rabbit IgG (PI-1000)
were purchased from Vector Laboratories, Inc. (Burlingame, CA,
USA).
Cell culture
B16F10 mouse melanoma cells (CRL-6475) were
purchased from the American Type Culture Collection (ATCC;
Rockville, MD, USA). The cells were maintained in DMEM supplemented
with 10% (v/v) FBS and 1% penicillin/streptomycin (10,000 U/ml and
10,000 μg/ml, respectively) in 5% CO2 at 37°C The
cells were cultured every 3 days until a maximal passage number of
20 was achieved.
Cell viability assay
The B16F10 melanoma cells were seeded in 96-well
plates (2×104 cells/well). After 24 h of incubation at
37°C, the medium was replaced with medium containing AP736 diluted
to the appropriate concentrations. The control cells were treated
with DMSO at a final concentration of 0.1%. After 24 h, the medium
containing the compounds or DMSO was replaced with medium
containing 10% CCK-8 solution. The cells were then incubated at
37°C for 30 min, and the absorbance was measured at 450 nm using a
spectrophotometer (VersaMax; Molecular Devices, Sunnyvale, CA,
USA). All assays were performed in triplicate. The cytotoxic effect
was expressed as a percentage of cell viability relative to the
untreated control cells. Cell viability was calculated using the
following formula: cell viability (%) =
(Asample/Acontrol) ×100.
Measurement of melanin content
The release of extracellular melanin was measured as
previously described (40), with
a slight modification. The B16F10 melanoma cells were seeded
overnight in 6-well plates (1×105 cells/well). α-MSH (1
μM) was then added or BIO for 30 min prior to the addition
of α-MSH. The cells were treated with increasing concentra tions of
the compound in phenol red-free DMEM for 3 days. Subsequently, 200
ml of the medium were then added to 96-well plates, and the optical
density of each culture well was measured at 405 nm using a
spectrophotometer. The data were normalized to the protein content
of the cell lysates. The cell lysates were subsequently processed
for determination of the protein concentration using a BCA Protein
Assay kit (Pierce Biotechnology, Rockford, IL, USA).
Cell-free enzymatic assay for tyrosinase
activity
Cell-free enzymatic assay for tyrosinase activity
was carried out as previously described (41). Tyrosinase activity was assayed as
a function of DOPA oxidase activity. A total of 70 ml of 0.1 M
phosphate buffer (pH 6.8) containing AP736 and kojic acid in 0.1 M
phosphate buffer were mixed with 20 ml of 10 μg/ml mushroom
tyrosinase in each well of a 96-well plate, and then 10 ml of 10 mM
L-DOPA was added. The absorbance values at 475 nm were measured
every 10 min for at least 1 h using a spectrophotometer at an
incubation temperature of 37°C. The quantity of dopachrome formed
in the reaction mixture was determined against a blank (solution
without enzyme) at 475 nm in the spectrophotometer. The percentage
of tyrosinase activity was calculated as follows: tyrosinase
activity (%) = [(A–B)/(C–D) ×100], where A is
the absorbance of the reaction mixture containing the test sample
and mushroom tyrosinase, B is the absorbance of the blank
sample containing the test sample but no mushroom tyrosinase,
C is the absorbance of the reaction mixture without the test
sample and with mushroom tyrosinase, and D is the absorbance
of the well lacking both the test sample and mushroom tyrosinase
(L-DOPA alone).
Assay of cellular tyrosinase
activity
The assay for cellular tyrosinase activity was
carried out as previously described (41). Tyrosinase activity was estimated
by measuring the rate of L-DOPA oxidation. The B16F10 melanoma
cells were incubated at a density of 1×105 cells in
6-well plates and treated as indicated for 3 days in DMEM. The
cells were then washed in ice-cold PBS and lysed in 100 ml of
phosphate buffer (0.1 M), pH 6.8, containing 1% (w/v) Triton X-100.
The cellular extract was clarified by centrifugation at 15,000 rpm
for 20 min. The tyrosinase substrate, L-DOPA (2 mg/ml), was
prepared in the same lysis buffer. Each extract (200 ml) was then
placed in a 96-well plate, and the enzymatic assay was initiated by
the addition of 2 ml of L-DOPA solution at 37°C. The control wells
contained 200 ml of the lysis buffer. The absorbance at 405 nm was
read every 10 min, for at least 1 h at 37°C, using a
spectrophotometer. Tyrosinase activity was calculated using the
following formula: tyrosinase activity (%) = OD475 of
sample/OD475 of control ×100.
Western blot analysis
The cells were lysed in cell lysis buffer [62.5 mM
Tris-HCl, pH 6.8, 2% sodium dodecyl sulfate (SDS), 5%
β-mercaptoethanol, 2 mM phenylmethylsulfonyl fluoride, protease
inhibitors (Complete™; Roche, Mannheim, Germany), 1 mM
Na3VO4, 50 mM NaF and 10 mM EDTA]. A 20
μg sample of protein per lane was separated by
SDS-polyacrylamide gel electrophoresis and blotted onto
polyvinylidene fluoride membranes (Millipore, Billerica, MA, USA)
which were then saturated with 5% powdered milk in Tris-buffered
saline containing 0.5% Tween-20 (TBST; Sigma-Aldrich Co.). The
blots were then incubated with the appropriate primary antibodies
at a dilution of 1:1,000 or 1:2,000, and further incubated with
horseradish peroxidase-conjugated secondary antibody. Bound
antibodies were detected using enhanced chemiluminescence (Amersham
Pharmacia, Piscataway, UK). Images of the blotted membranes were
captured using an LAS-1000 lumino-image analyzer (Fujifilm, Tokyo,
Japan). Protein levels were compared to those of the appropriate
loading control, such as actin or non-phosphorylated protein.
Immunocytochemistry/immunofluorescence (ICC/IF) assay
The B16F10 melanoma cells (3×104) were
seeded onto glass coverslips that were pre-coated with
poly-L-lysine (0.01; Sigma-Aldrich Co.). Following incubation for
24 h, ICC/IF was performed. The slides were fixed with 4%
paraformaldehyde for 10 min at room temperature. After washing with
PBS, the cells were permeabilized with 0.01% Triton X-100 in PBS or
TBST for 10 min at room temperature and then blocked with 2% bovine
serum albumin in PBS for 60 min at room temperature. The slides
were incubated overnight with tyrosinase antibodies at 4°C. After
washing, the slides were incubated with fluorescein isothiocyanate
(FITC)-conjugated secondary antibody (Santa Cruz Biotechnology,
Inc.), mounted using fluorescent mounting medium with
4′,6-diamidino-2-phenyl-indole (DAPI; Golden Bridge International,
Inc., Bothell, WA, USA). Cell morphology was observed under a DP70
fluorescence microscope with DP Controller software (Olympus
Optical Co., Ltd., Tokyo, Japan).
Statistical analysis
Statistical analyses were performed using SPSS for
Windows, version 18.0 (SPSS Inc., Chicago, IL, USA). The results
are expressed as the means ± standard deviation. Data were analyzed
by one-way ANOVA, followed by Duncan's test for multiple
comparison. A two-tailed P-value <0.05 was considered to
indicate a statistically significant difference.
Results
Effects of AP736 on B16F10 melanoma cell
viability
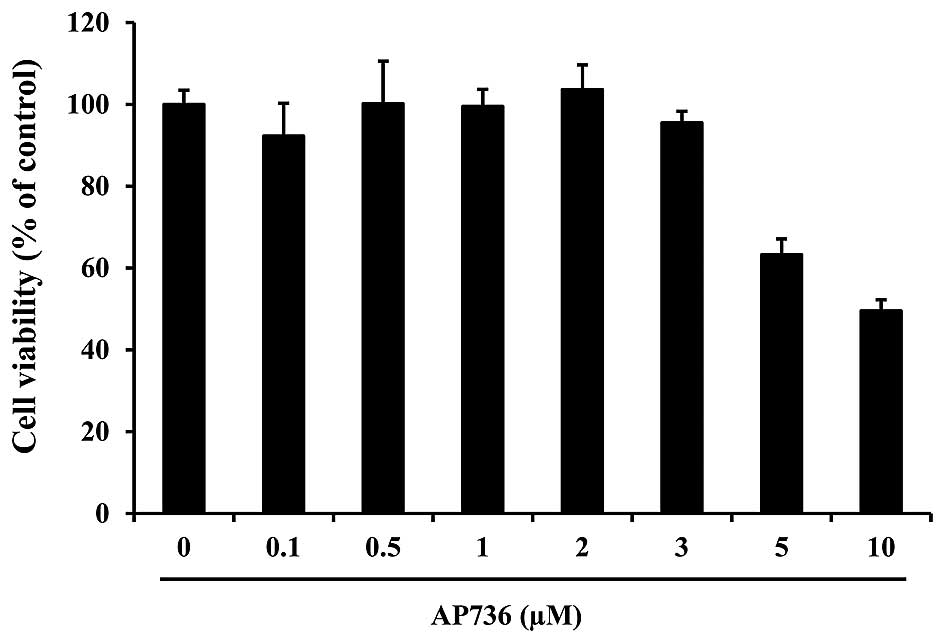
In order to determine whether AP736 inhibits
melanogenesis, the B16F10 melanoma cells were treated with the
extract prior to α-MSH stimulation. However, we first examined the
cytotoxicity of AP736 on B16F10 melanoma cells by CCK-8 assay. The
B16F10 melanoma cells were treated with AP736 at concentrations of
0.1–10 μM for 24 h. As shown in Fig. 2, AP736 was not cytotoxic at
concentrations of 0.1 to 3 μM. Accordingly, we used 0.1–3
μM AP736 in the subsequent experiments.
Effects of AP736 on melanogenesis in
B16F10 melanoma cells
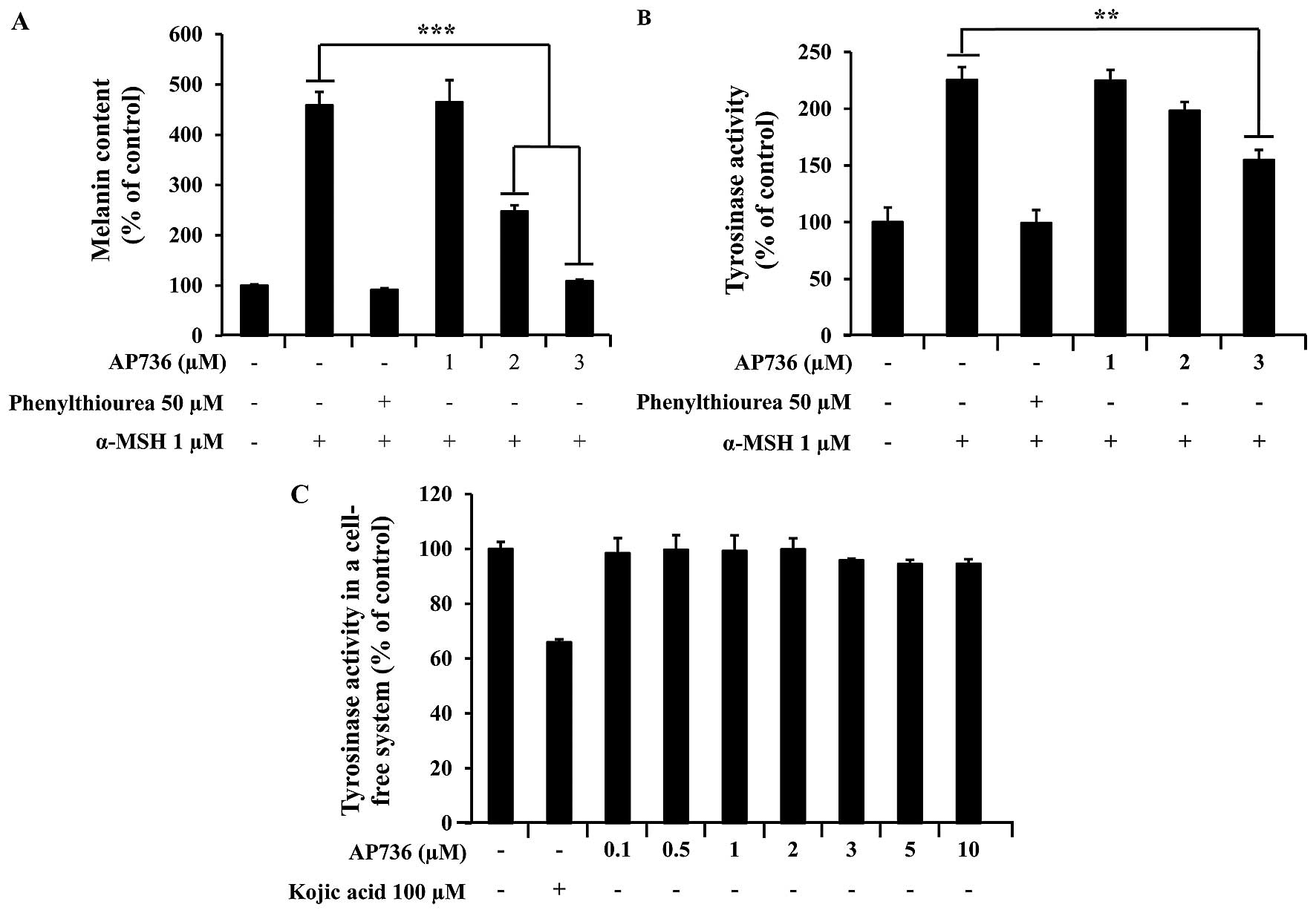
The effects of AP736 on the melanogenesis of B16F10
melanoma cells were examined. To determine whether AP736 inhibits
α-MSH-induced melanin synthesis in B16F10 melanoma cells, we
measured α-MSH-induced melanin production in B16F10 melanoma cells
after 3 days of treatment with AP736 at 1–3 μM. As shown in
Fig. 3A, the addition of α-MSH
increased the amount of melanin in the medium; however, treatment
with AP736 reduced the release of melanin in a dose-dependent
manner. PTU, a well-known tyrosinase inhibitor, was used as a
positive control, as previously described (42).
Melanogenesis is known to be controlled through an
enzymatic cascade that is regulated by tyrosinase (43). Thus, to investigate the mechanisms
of depigmentation by AP736, we measured tyrosinase activity in both
cell-based and cell-free assay systems. As shown in Fig. 3B, the α-MSH-induced tyrosinase
activity was inhibited in a dose-dependent manner by AP736 (1–3
μM). We also found that treatment with 3 μM AP736
reduced tyrosinase activity. Specifically, compared with the
untreated cells, treatment with AP736 at a concentration of 3
μM resulted in an approximately 33% inhibition of
intracellular tyrosinase in the B16F10 melanoma cells. A number of
skin-whitening compounds directly inhibit tyrosinase (44). Thus, to examine the direct effects
of AP736 on tyrosinase, we measured the tyrosinase activity of
mushroom tyrosinase in a cell-free system, as described in the
Materials and methods. AP736 exerted little inhibitory effect on
tyrosinase activity; however, treatment with 100 μM kojic
acid (a direct inhibitor of tyrosinase) exerted a strong inhibitory
effect (Fig. 3C). These results
indicated that the inhibitory effect of AP736 on melanogenesis was
not due to the direct inhibition of tyrosinase.
Effects of AP736 on pathways of
melanogenesis in B16F10 melanoma cells
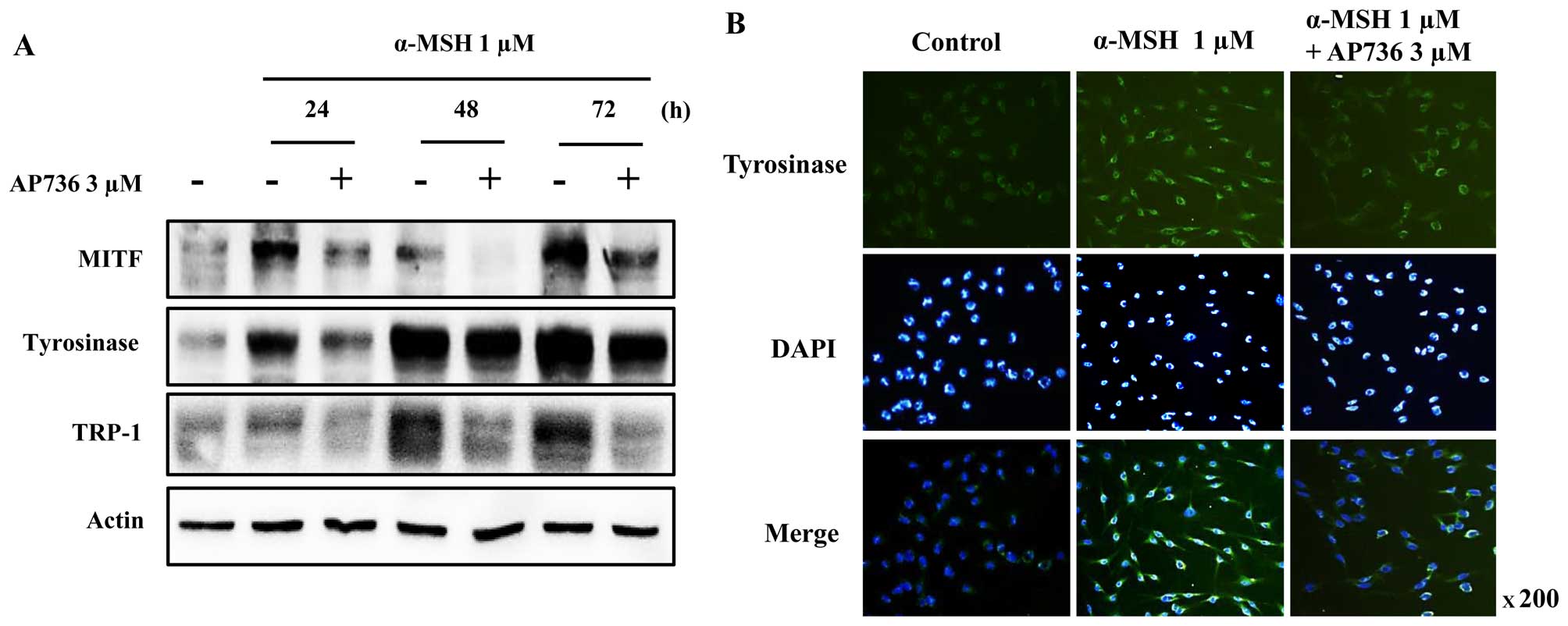
In order to determine whether the inhibitory
activity of AP736 is related to pathways of melanogenesis involving
the expression of tyrosinase, TRP-1 and MITF, the B16F10 mouse
melanoma cells were treated with AP736 prior to stimulation with
α-MSH. The resulting cell lysates were subjected to SDS-PAGE and
western blot analysis. As shown in Fig. 4, the MITF levels were markedly
decreased following treatment with AP736 for 24–72 h, and the
tyrosinase and TRP-1 levels also decreased in a time-dependent
manner. These results suggest that AP736 decreases melanin
synthesis through the downregulation of MITF and tyrosinase.
Furthermore, the effects of AP736 on tyrosinase expression in the
cells was confirmed by ICC/IF assay (Fig. 4B). The results revealed that the
level of tyrosinase in the cells was reduced following treatment
with AP736 for 72 h in the presence of α-MSH.
Effects of AP736 on signal transduction
pathways in B16F10 melanoma cells
To elucidate the mechanisms underlying the
hypopigmentary effects of AP736, changes in melanogenesis-related
signals induced by AP736 were examined by western blot analysis in
a time-course experiment. AP736 has been reported to show
anti-melanogenic activity in melanocytes in vitro and in
artificial skin equivalents through the inhibition of key
melanogenic enzymes and suppression of the cAMP-PKA-CREB signaling
pathway (45). Thus, we
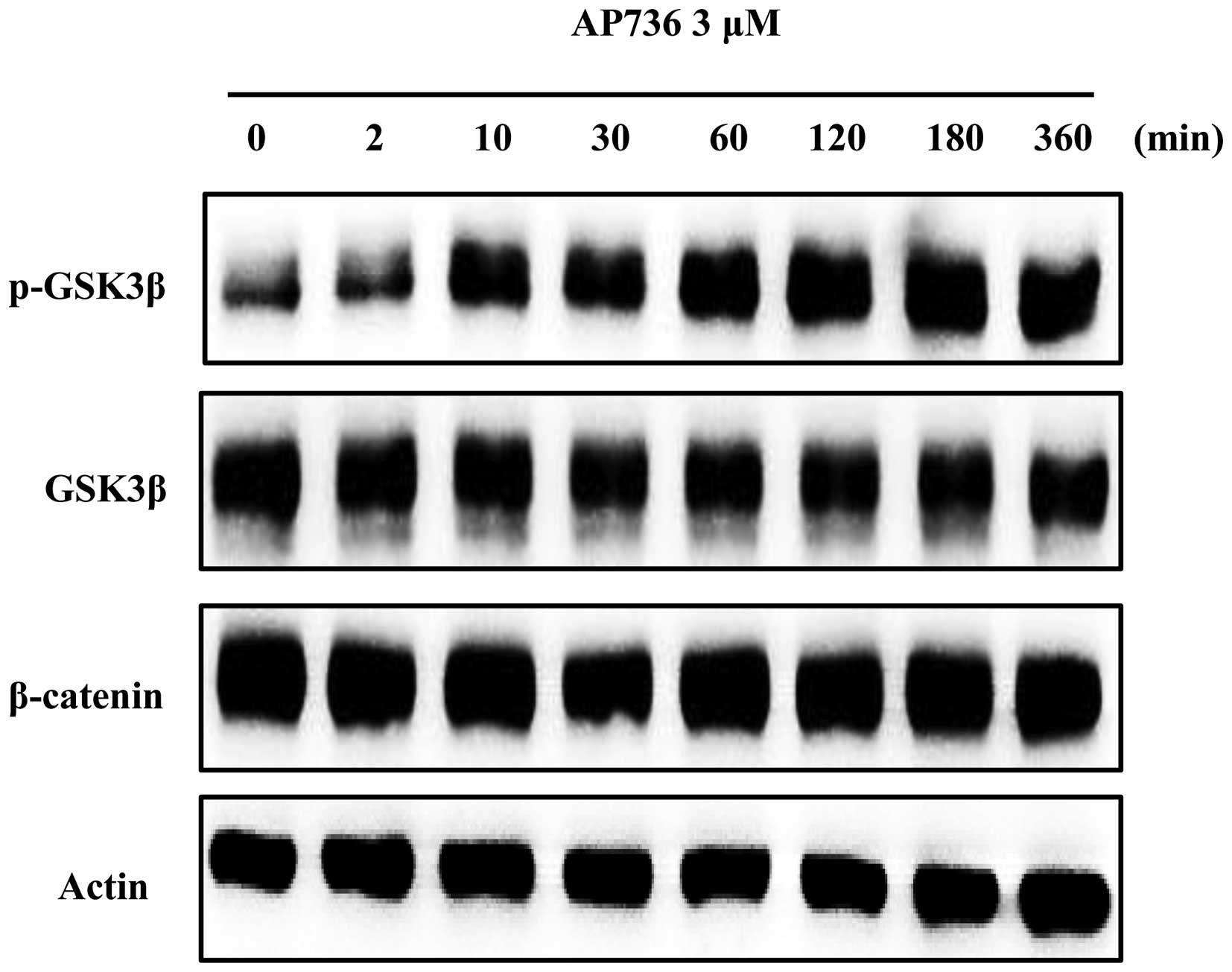
investigated whether AP736 induces the phosphorylation of GSK3β and
the degradation of β-catenin. We confirmed that GSK3β
phosphorylation was clearly induced by treatment with 3 μM
AP736; however, the β-catenin levels were not altered (Fig. 5).
Effects of AP736 on GSK3β phosphorylation
and melanogenesis
The activation of GSK3β has been reported to inhibit
tyrosinase expression (46),
which subsequently decreases cellular melanin synthesis. Therefore,
experiments were carried out to determine whether the AP736-induced
phosphorylation of the GSK3β signaling pathway plays a role in
melanin synthesis in B16F10 melanoma cells treated with AP736 for 6
h in the presence or absence of BIO, a specific GSK3β
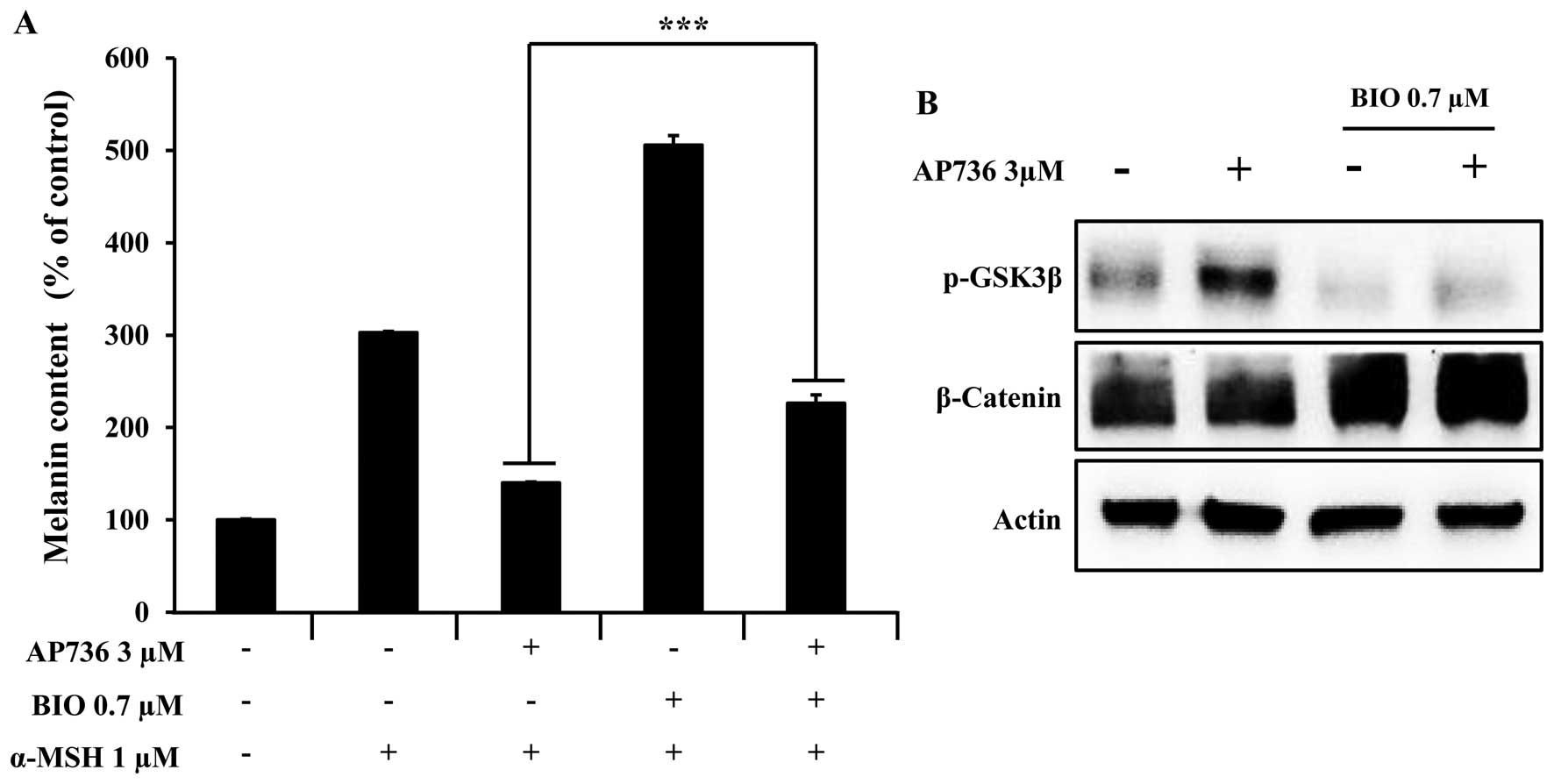
phosphorylation inhibitor. As shown in Fig. 6A, the cellular melanin content of
the cells co-treated with α-MSH and BIO was higher than that in the
cells treated with α-MSH alone. In addition, this synergistic
effect of α-MSH and BIO on the cellular melanin content was offset
by AP736 treatment. We then examined whether BIO inhibits GSK3β
phosphorylation in B16F10 melanoma cells and found that BIO does,
in fact, inhibit the phosphorylation of GSK3β in the AP736-treated
B16F10 melanoma cells (Fig. 6B).
These results clearly indicate that GSK3β phosphorylation is
involved in the inhibition of melanogenesis by AP736.
Discussion
The majority of depigmenting compounds act
specifically to reduce the function of tyrosinase through several
mechanisms which include interference with its transcription and/or
glycosylation, enzyme inhibition, reduction of by-products, and
post-transcriptional control. Some compounds inhibit melanosome
transfer from melanocytes to keratinocytes and accelerate skin
turnover (47,48). The exploration of compounds that
act on the inhibition of melanin synthesis would be helpful in
understanding and studying the precise role of the related signal
transduction pathways and their function in melanogenesis. Thus, in
this study, the effects of AP736 on melanin synthesis and related
signaling pathways were investigated using B16F10 melanoma
cells.
This study demonstrates that AP736 is a
skin-whitening compound that acts via the downregulation of
tyrosinase, TRP-1 and MITF expression. The inhibitory effects of
AP736 were dose-dependent and did not incur significant
cytotoxicity (Fig. 2). The
AP736-induced decrease in melanin production was also accompanied
by a corresponding decrease in tyrosinase activity, suggesting a
possible mechanism of AP736 action (Fig. 3). Tyrosinase has been demonstrated
to catalyze the rate-limiting step of melanin biosynthesis, and it
is the primary target of PTU, which is an agent with efficient
depigmenting effects currently used for cosmetic and medical
purposes (49). AP736 markedly
inhibited intracellular tyrosinase activity in the α-MSH-stimulated
B16F10 melanoma cells, as demonstrated by cellular tyrosinase
(Fig. 3B). However, as shown in
Fig. 3C, AP736 did not directly
inhibit tyrosinase activity, indicating the involvement of
different mechanisms. These results suggest that the decrease in
pigmentation induced by AP736 may be attributed to its effect on
the signaling pathways regulating tyrosinase.
For a better understanding of the depigmenting
effect of AP736 and the way in which tyrosinase synthesis is
targeted, western blot analysis was carried out. Our data confirmed
that the MITF, tyrosinase and TRP-1 protein levels were decreased
by AP736 in a time-dependent manner (Fig. 4A). In addition, the AP736-induced
hypopigmentation correlated with reduced tyrosinase activity
(Fig. 3A and B), which could be
responsible for the hypopigmentation of AP736-treated cells.
Furthermore, the effect of AP736 on tyrosinase expression in B16F10
melanoma cells was confirmed by ICC/IF (Fig. 4B). The results revealed that the
tyrosinase level in B16F10 melanoma cells was reduced by treatment
with AP736 for 72 h in the presence of α-MSH. To elucidate the
mechanisms underlying the hypopigmentary effects of AP736, changes
in melanogenesis-related signals induced by AP736 were examined by
western blot analyiss in a time-course experiment (Fig. 5).
MITF expression is also induced by GSK3β
inactivation (phosphorylation) and subsequent β-catenin
accumulation (50,51). By contrast, it has been reported
that activated (dephosphorylated) GSK3β phosphorylates MITF at
Ser298 in melanoma cells and melanocytes. Furthermore, the
phosphorylation of MITF at Ser298 significantly increases its
ability to bind to the tyrosinase promoter DNA element, resulting
in increased melanin synthesis (23,25,52). Thus, in this study, we
investigated the possible involvement of GSK3β in tyrosinase
expression. In the GSK3β-dependent pathway, GSK3β, which forms a
destruction complex with adenomatous polyposis coli and axin,
phosphorylates the N-terminal Ser/Thr residues of β-catenin
(53), resulting in its
degradation through a ubiquitin-dependent mechanism (54). The N-terminal Ser/Thr residues of
β-catenin are also phosphorylated by protein kinase C (PKC)α,
leading to ubiquitin-dependent β-catenin degradation (55). In the Siah-dependent pathway,
Siah-1 interacts with the carboxyl terminus of adenomatous
polyposis coli, which recruits the ubiquitination complex and
promotes the degradation of β-catenin (56). In this study, we found that
treatment with a specific GSK3β pathway inhibitor, BIO, reversed
the AP736-induced hypopigmentation (Fig. 6). The results of our study also
demonstrated that AP736 induced GSK3β phosphorylation and reduced
the MITF protein level. To the best of our knowledge, this is the
first study to demonstrate the induction of GSK3β phosphorylation
by AP736. To investigate the association between GSK3β
phosphorylation and MITF downregulation, we treated the cells with
BIO prior to treatment with AP736, re-examined the p-GSK3β level,
and found that GSK3β inactivation suppressed melanogenesis in
B16F10 melanoma cells.
In conclusion, in this study, we demonstrated that
treatment with AP736 led to GSK3β phosphorylation, which may have
reduced the binding affinity of MITF and subsequently decreased the
tyrosinase level. Our data suggest that AP736 may prove to be
useful as a novel skin-whitening compound by decreasing the level
of tyrosinase.
References
|
1
|
Eves PC, MacNeil S and Haycock JW:
alpha-Melanocyte stimulating hormone, inflammation and human
melanoma. Peptides. 27:444–452. 2006. View Article : Google Scholar
|
|
2
|
Yokota T, Nishio H, Kubota Y and Mizoguchi
M: The inhibitory effect of glabridin from licorice extracts on
melanogenesis and inflammation. Pigment Cell Res. 11:355–361. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Eller MS, Ostrom K and Gilchrest BA: DNA
damage enhances melanogenesis. Proc Natl Acad Sci USA.
93:1087–1092. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wong G and Pawelek J:
Melanocyte-stimulating hormone promotes activation of pre-existing
tyrosinase molecules in Cloudman S91 melanoma cells. Nature.
255:644–646. 1975. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Halaban R, Pomerantz SH, Marshall S and
Lerner AB: Tyrosinase activity and abundance in Cloudman melanoma
cells. Arch Biochem Biophys. 230:383–387. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hunt G, Todd C, Cresswell JE and Thody AJ:
Alpha-melanocyte stimulating hormone and its analogue Nle4DPhe7
alpha-MSH affect morphology, tyrosinase activity and melanogenesis
in cultured human melanocytes. J Cell Sci. 107:205–211.
1994.PubMed/NCBI
|
|
7
|
Kanwar AJ, Dhar S and Kaur S: Treatment of
melasma with potent topical corticosteroids. Dermatology.
188:1701994. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Körner A and Pawelek J: Mammalian
tyrosinase catalyzes three reactions in the biosynthesis of
melanin. Science. 217:1163–1165. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Boissy RE: Melanosome transfer to and
translocation in the keratinocyte. Exp Dermatol. 12(Suppl 2):
S52003. View Article : Google Scholar
|
|
10
|
Buscà R, Bertolotto C, Ortonne JP and
Ballotti R: Inhibition of the phosphatidylinositol
3-kinase/p70(S6)-kinase pathway induces B16 melanoma cell
differentiation. J Biol Chem. 271:31824–31830. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Costin GE and Hearing VJ: Human skin
pigmentation: melanocytes modulate skin color in response to
stress. FASEB J. 21:976–994. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hearing VJ and Tsukamoto K: Enzymatic
control of pigmentation in mammals. FASEB J. 5:2902–2909.
1991.PubMed/NCBI
|
|
13
|
Jung E, Lee J, Huh S, Lee J, Kim YS, Kim G
and Park D: Phloridzin-induced melanogenesis is mediated by the
cAMP signaling pathway. Food Chem Toxicol. 47:2436–2440. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yasumoto K, Yokoyama K, Takahashi K,
Tomita Y and Shibahara S: Functional analysis of
microphthalmia-associated transcription factor in pigment
cell-specific transcription of the human tyrosinase family genes. J
Biol Chem. 272:503–509. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Steingrímsson E, Moore KJ, Lamoreux ML,
Ferré-D'Amaré AR, Burley SK, Zimring DC, Skow LC, Hodgkinson CA,
Arnheiter H, Copeland NG, et al: Molecular basis of mouse
microphthalmia (mi) mutations helps explain their developmental and
phenotypic consequences. Nat Genet. 8:256–263. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tachibana M: MITF: A stream flowing for
pigment cells. Pigment Cell Res. 13:230–240. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Vakhedi AS: Biological sutures in
ophthalmological microsurgery. Vestn Oftalmol. 5:27–31. 1976.In
Russian.
|
|
18
|
Jiménez-Cervantes C, Martínez-Esparza M,
Pérez C, Daum N, Solano F and García-Borrón JC: Inhibition of
melanogenesis in response to oxidative stress: transient
downregulation of melanocyte differentiation markers and possible
involvement of microphthalmia transcription factor. J Cell Sci.
114:2335–2344. 2001.PubMed/NCBI
|
|
19
|
Corre S, Primot A, Sviderskaya E, Bennett
DC, Vaulont S, Goding CR and Galibert MD: UV-induced expression of
key component of the tanning process, the POMC and MC1R genes, is
dependent on the p-38-activated upstream stimulating factor-1
(USF-1). J Biol Chem. 279:51226–51233. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wu M, Hemesath TJ, Takemoto CM, Horstmann
MA, Wells AG, Price ER, Fisher DZ and Fisher DE: c-Kit triggers
dual phosphorylations, which couple activation and degradation of
the essential melanocyte factor Mi. Genes Dev. 14:301–312.
2000.PubMed/NCBI
|
|
21
|
Xu W, Gong L, Haddad MM, Bischof O,
Campisi J, Yeh ET and Medrano EE: Regulation of
microphthalmia-associated transcription factor MITF protein levels
by association with the ubiquitin-conjugating enzyme hUBC9. Exp
Cell Res. 255:135–143. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kim DS, Hwang ES, Lee JE, Kim SY, Kwon SB
and Park KC: Sphingosine-1-phosphate decreases melanin synthesis
via sustained ERK activation and subsequent MITF degradation. J
Cell Sci. 116:1699–1706. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Larue L and Delmas V: The WNT/Beta-catenin
pathway in melanoma. Front Biosci. 11:733–742. 2006. View Article : Google Scholar
|
|
24
|
Wu J, Saint-Jeannet JP and Klein PS:
Wnt-frizzled signaling in neural crest formation. Trends Neurosci.
26:40–45. 2003. View Article : Google Scholar
|
|
25
|
Bellei B, Flori E, Izzo E, Maresca V and
Picardo M: GSK3beta inhibition promotes melanogenesis in mouse B16
melanoma cells and normal human melanocytes. Cell Signal.
20:1750–1761. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Giles RH, van Es JH and Clevers H: Caught
up in a Wnt storm: Wnt signaling in cancer. Biochim Biophys Acta.
1653:1–24. 2003.PubMed/NCBI
|
|
27
|
Korinek V, Barker N, Morin PJ, van Wichen
D, de Weger R, Kinzler KW, Vogelstein B and Clevers H: Constitutive
transcriptional activation by a beta-catenin-Tcf complex in APC-/-
colon carcinoma. Science. 275:1784–1787. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Morin PJ, Sparks AB, Korinek V, Barker N,
Clevers H, Vogelstein B and Kinzler KW: Activation of
beta-catenin-Tcf signaling in colon cancer by mutations in
beta-catenin or APC. Science. 275:1787–1790. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wodarz A and Nusse R: Mechanisms of Wnt
signaling in development. Annu Rev Cell Dev Biol. 14:59–88. 1998.
View Article : Google Scholar
|
|
30
|
Polakis P: Wnt signaling and cancer. Genes
Dev. 14:1837–1851. 2000.PubMed/NCBI
|
|
31
|
Liu C, Li Y, Semenov M, Han C, Baeg GH,
Tan Y, Zhang Z, Lin X and He X: Control of beta-catenin
phosphorylation/degradation by a dual-kinase mechanism. Cell.
108:837–847. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Amit S, Hatzubai A, Birman Y, Andersen JS,
Ben-Shushan E, Mann M, Ben-Neriah Y and Alkalay I: Axin-mediated
CKI phosphorylation of beta-catenin at Ser 45: a molecular switch
for the Wnt pathway. Genes Dev. 16:1066–1076. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Latres E, Chiaur DS and Pagano M: The
human F box protein beta-Trcp associates with the Cul1/Skp1 complex
and regulates the stability of beta-catenin. Oncogene. 18:849–854.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lee E, Salic A, Krüger R, Heinrich R and
Kirschner MW: The roles of APC and Axin derived from experimental
and theoretical analysis of the Wnt pathway. PLoS Biol. 1:E102003.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Takeda K, Yasumoto K, Takada R, Takada S,
Watanabe K, Udono T, Saito H, Takahashi K and Shibahara S:
Induction of melanocyte-specific microphthalmia-associated
transcription factor by Wnt-3a. J Biol Chem. 275:14013–14016. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Schepsky A, Bruser K, Gunnarsson GJ,
Goodall J, Hallsson JH, Goding CR, Steingrimsson E and Hecht A: The
microphthalmia-associated transcription factor Mitf interacts with
beta-catenin to determine target gene expression. Mol Cell Biol.
26:8914–8927. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Parvez S, Kang M, Chung HS, Cho C, Hong
MC, Shin MK and Bae H: Survey and mechanism of skin depigmenting
and lightening agents. Phytother Res. 20:921–934. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Shimizu K, Yasutake S and Kondo R: A new
stilbene with tyrosinase inhibitory activity from Chlorophora
excelsa. Chem Pharm Bull (Tokyo). 51:318–319. 2003. View Article : Google Scholar
|
|
39
|
Park SH, Kim DS, Kim WG, Ryoo IJ, Lee DH,
Huh CH, Youn SW, Yoo ID and Park KC: Terrein: A new melanogenesis
inhibitor and its mechanism. Cell Mol Life Sci. 61:2878–2885. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kim DS, Jeong YM, Park IK, Hahn HG, Lee
HK, Kwon SB, Jeong JH, Yang SJ, Sohn UD and Park KC: A new
2-imino-1,3-thiazoline derivative, KHG22394, inhibits melanin
synthesis in mouse B16 melanoma cells. Biol Pharm Bull. 30:180–183.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lee do H, Kim DH, Oh IY, Kim SY, Lim YY,
Kim HM, Kim YH, Choi YM, Kim SE, Kim BJ and Kim MN: Inhibitory
effects of Saururi chinensis extracts on melanin biosynthesis in
B16F10 melanoma cells. Biol Pharm Bull. 36:772–779. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Poma A, Bianchini S and Miranda M:
Inhibition of L-tyrosine-induced micronuclei production by
phenylthiourea in human melanoma cells. Mutat Res. 446:143–148.
1999. View Article : Google Scholar
|
|
43
|
Casanola-Martin GM, Le-Thi-Thu H,
Marrero-Ponce Y, Castillo-Garit JA, Torrens F, Rescigno A, Abad C
and Khan MT: Tyrosinase enzyme: 1. An overview on a pharmacological
target. Curr Top Med Chem. 14:1494–1501. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Smit N, Vicanova J and Pavel S: The hunt
for natural skin whitening agents. Int J Mol Sci. 10:5326–5349.
2009. View Article : Google Scholar
|
|
45
|
Lee CS, Jang WH, Park M, Jung K, Baek HS,
Joo YH, Park YH and Lim KM: A novel adamantyl benzylbenzamide
derivative, AP736, suppresses melanogenesis through the inhibition
of cAMP-PKA-CREB-activated microphthalmia-associated transcription
factor and tyrosinase expression. Exp Dermatol. 22:762–764. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Lin YS, Chuang MT, Chen CH, Chien MY and
Hou WC: Nicotinic acid hydroxamate downregulated the melanin
synthesis and tyrosinase activity through activating the MEK/ERK
and AKT/GSK3β signaling pathways. J Agric Food Chem. 60:4859–4864.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Gillbro JM and Olsson MJ: The
melanogenesis and mechanisms of skin-lightening agents - existing
and new approaches. Int J Cosmet Sci. 33:210–221. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Son KH and Heo MY: The evaluation of
depigmenting efficacy in the skin for the development of new
whitening agents in Korea. Int J Cosmet Sci. 35:9–18. 2013.
View Article : Google Scholar
|
|
49
|
Land EJ, Ramsden CA, Riley PA and
Stratford MR: Evidence consistent with the requirement of cresolase
activity for suicide inactivation of tyrosinase. Tohoku J Exp Med.
216:231–238. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Grabacka M, Placha W, Urbanska K, Laidler
P, Płonka PM and Reiss K: PPAR gamma regulates MITF and
beta-catenin expression and promotes a differentiated phenotype in
mouse melanoma S91. Pigment Cell Melanoma Res. 21:388–396. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Grabacka M, Plonka PM, Urbanska K and
Reiss K: Peroxisome proliferator-activated receptor alpha
activation decreases metastatic potential of melanoma cells in
vitro via down-regulation of Akt. Clin Cancer Res. 12:3028–3036.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Takeda K, Takemoto C, Kobayashi I,
Watanabe A, Nobukuni Y, Fisher DE and Tachibana M: Ser298 of MITF,
a mutation site in Waardenburg syndrome type 2, is a
phosphorylation site with functional significance. Hum Mol Genet.
9:125–132. 2000. View Article : Google Scholar
|
|
53
|
Hart MJ, de los Santos R, Albert IN,
Rubinfeld B and Polakis P: Downregulation of beta-catenin by human
Axin and its association with the APC tumor suppressor,
beta-catenin and GSK3 beta. Curr Biol. 8:573–581. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Aberle H, Bauer A, Stappert J, Kispert A
and Kemler R: beta-catenin is a target for the ubiquitin-proteasome
pathway. EMBO J. 16:3797–3804. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Gwak J, Cho M, Gong SJ, Won J, Kim DE, Kim
EY, Lee SS, Kim M, Kim TK, Shin JG and Oh S:
Protein-kinase-C-mediated beta-catenin phosphorylation negatively
regulates the Wnt/beta-catenin pathway. J Cell Sci. 119:4702–4709.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Liu J, Stevens J, Rote CA, Yost HJ, Hu Y,
Neufeld KL, White RL and Matsunami N: Siah-1 mediates a novel
beta-catenin degradation pathway linking p53 to the adenomatous
polyposis coli protein. Mol Cell. 7:927–936. 2001. View Article : Google Scholar : PubMed/NCBI
|