Introduction
CD81, also known as target of antiproliferative
antibody 1 (TAPA-1), is a component of the B cell co-stimulatory
complex. This complex consists of CD19, CD21 and
interferon-inducible Leu-13 (CD225) (1). The simultaneous stimulation of this
complex and the B cell receptor (BCR) by antigens magnifies signal
transduction and increases B cell proliferation (2,3).
CD19 and CD21 are expressed exclusively on B cells, whereas CD81
and CD225 are widely expressed on immune cells (T, B and NK
lymphocytes, monocytes and eosinophils), hepatocytes and on most
stromal and epithelial cells (4).
Genetic defects in CD81 disrupt CD19 complex formation and lead to
antibody deficiency syndromes (5). CD19 expression and B cell activation
by T cell-dependent antigens are also impaired in CD81-knockout
mouse models (6,7).
Hepatitis C virus (HCV) is a positive-stranded RNA
Hepacivirus in the Flaviviridae family (8). HCV infection is associated with
chronic liver diseases, such as chronic hepatitis, cirrhosis and
hepatocellular carcinoma (HCC). HCV infection is also an important
cause of autoimmune disease, type II mixed cryoglobulinemia (MC)
and non-Hodgkin lymphoma (NHL) (9–11).
Viral envelope proteins are composed of the heavily glycosylated
envelope proteins, E1 and E2 (12). The large extracellular loop (LEL)
of CD81 binds to the E2 dimer of HCV (13). The E2 glycoprotein of HCV is,
therefore, the target of neutralizing antibodies as the N-terminal
ectodomain of E2 possesses the entry determinants for infection of
the host cell (14). However,
neutralizing antibodies against E2 are strain-specific and are
modulated by a complex interplay between hypervariable regions
(HVR)1 and 2 (15). Although
certain epidemiological and experimental studies have suggested an
etiopathogenetic role for HCV and Epstein-Barr virus (EBV)
infection in B cell NHL pathogenesis, the distinct contribution of
these two viruses to the progression of B cell NHL remains unclear
and controversial (16,17).
Lymphocytes from HCV-positive patients have been
shown to express CD81 at significantly higher levels than
lymphocytes from controls (18).
CD81 has also been shown to play a role in the infection of primary
human hepatocytes by serum-derived HCV (19). CD81 expression in B cells has been
suggested to be involved in chronic antigenic stimulation related
to HCV infection (20). B cells
have been shown to be susceptible to HCV, and direct HCV infection
through CD81 on B cells has been proposed as a possible cause of
NHL (21,22). However, the binding of the E2
protein of HCV alone is insufficient to explain the function of
CD81 expressed by mature B cells. CD81 engagement in B cells
triggers the Raf/MEK/ERK signaling pathway that appears to be
important for cell proliferation and survival (23). Furthermore, E2-CD81 engagement
protects primary human B lymphocytes (PHB) from apoptosis through
the phosphorylation of IκBα and the increase in the expression of
anti-apoptotic Bcl-2 family proteins (24).
Although previous studies have demonstrated the
proliferative effects of the CD81-HCV E2 interaction on resting B
cells, the role of this interatction in EBV infection and
transformation remains unclear. The effects of CD81 overexpression
on B cells also remain controversial. Previously, we reported that
EBV has the unique ability to transform resting B cells into
lymphoblastoid cell lines (LCLs) in vitro (25,26). In the present study, we aimed to
elucidate the effects of CD81 on resting and activated B cells. For
this purpose, we upregulated the expression of CD81 in B cells by
EBV infection and stimulated the cells with anti-CD81 monoclonal
antibodies (mAbs) or HCV E2 protein, leading to a change in the
effects of CD81 on B cells during the transformation process.
Materials and methods
Ethics statement
Informed consent for the present study was obtained
from all participants and the study was approved by the
Institutional Bioethics Review Board of the Medical College of Inje
University, Busan, Korea (#12-238).
Cells, antibodies and reagents
To establish EBV-transformed B cells, peripheral
blood mononuclear cells (PBMCs) were obtained from the blood of 7
healthy human volunteers and 7 patients with chronic HCV by
Ficoll-Paque density gradient centrifugation (GE Healthcare
Biosciences, Pittsburgh, PA, USA). B cells were purified from the
PBMCs using the MACS B cell isolation kit and the MACS separator
(both from Miltenyi Biotec, Bergisch Gladbach, Germany). Mouse
anti-human CD81 mAb (5A6) for stimulation experiments was purchased
from BD Biosciences (San Jose, CA, USA). FITC-conjugated anti-CD81
mAb (#551108), PE-conjugated anti-CD20 antibody (#346595), Fas
(CD95) antibody (#555674), Fas ligand (CD178) antibody
(#12-9919-42) and PE-conjugated anti-latent membrane protein
(LMP)-1 antibody (#550018) for FACS analysis were purchased from BD
Biosciences. Recombinant purified HCV E2 protein was produced using
the pCMVdhfr-E2 plasmid (a gift from Dr Chang-Yuil Kang, Seoul
National University, Seoul, Korea) according to the protocol
descrbied in the study by Heo et al (27). 2′,7′-Dichlorofluorescin diacetate
(DCFH-DA) and 3,3′-dihexyloxacarbocyanine iodide (DiOC6)
were purchased from Molecular Probes (Eugene, OR, USA).
N-acetylcysteine (NAC),
N-benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone
(Z-VAD-fmk, 20 µM in DMSO, a broad spectrum caspase
inhibitor),
N-benzyloxycarbonyl-Asp-Glu-Val-Asp-fluoromethylketone
(Z-DEVD-fmk, 20 µM in DMSO, a caspase-3 inhibitor), and
N-benzyloxycarbonyl-Ile-Glu-Thr-Asp-fluoromethylketone
(Z-IETD-fmk, 20 µM in DMSO, a caspase-8 inhibitor) were
purchased from Calbiochem (San Diego, CA, USA).
Generation of EBV-transformed B cells and
analysis of phenotype
EBV stock was prepared from an EBV-transformed B95-8
marmoset cell line (a gift from Dr B.G. Han, National Genome
Research Institute, National Institute of Health, Seoul, Korea). To
establish an EBV-infected B cell line from the PBMCs, 10 ml of
peripheral blood were collected from 7 healthy human donors and 7
patients with chronic HCV. The characteristics of the patients with
HCV are presented in Table I.
Qualitative and quantitative PCR analyses for HCV RNA were
evaluated with Amplicor HCV version 2.0 (Roche Molecular System,
Pleasanton, CA, USA). HCV genotypes were determined by reverse
hybridization (INNO-LiPA, Innogenetics, Ghent, Belgium). The B
cells were purified from the PBMCs using the MACS B cell isolation
kit and a MACS separator (Miltenyi Biotec). The purified B cells
were added to EBV stock supernatant in a culture flask, and
following 2 h of incubation at 37°C, an equal volume of RPMI-1640
medium (HyClone, Logan, UT, USA) containing 10% fetal bovine serum
(HyClone) and 1 µg/ml of cyclosporine A (Sigma-Aldrich, St.
Louis, MO, USA) were added (1×106 cells/ml). The
cultures were incubated for 2–4 weeks until clumps of EBV-infected
B cells were visible and the medium had turned yellow, as
previously descrbied (25,26).
The phenotype of EBV-transformed B cells was monitored each week by
flow cytometric analysis and a confocal laser-scanning microscope
(510 META; Carl Zeiss, Oberkochen, Germany) at x400 original
magnification using FITC-conjugated anti-CD81 and PE-conjugated
anti-CD20 mAbs (BD Biosciences).
 | Table ICharacteristics of patients with
chronic HCV. |
Table I
Characteristics of patients with
chronic HCV.
| Characteristic | A | B | C | D | E | F | G |
|---|
| Gender/age
(years) | M/25 | M/58 | F/42 | M/54 | F/45 | M/38 | M/50 |
| ALT (U/l) | 77 | 62 | 73 | 64 | 91 | 99 | 47 |
| PCR for HCV | Positive | Positive | Positive | Positive | Positive | Positive | Positive |
| Genotype | 1b | 1b | 1b | 1b | 1b | 2a/2c | 2b |
| HCV RNA
(×106 IU/ml) | 4.2 | 4.4 | 2.6 | 2.6 | 1.7 | 1.3 | 3.6 |
| Biopsy results | F0 | F3 | F2 | F3 | Not checked | Not checked | Not checked |
| Sampling date | 01.10.04 | 21.12.04 | 04.02.04 | 16.03.05 | 04.07.05 | 09.08.05 | 02.09.05 |
| Date of antiviral
treatment | 28.03.05 | 05.01.05 | 14.03.05 | 25.03.05 | – | 02.09.05 | 02.09.05 |
| Type of
response | SVR | SVR | SVR | relapse | Not treated | Follow-up loss | SVR |
Monitoring the changes in the morphology
of EBV-transformed B cells pre-stimulated with CD81
For CD81 pre-stimulation with antibody, the purified
resting B cells were suspended in 100 µl PBS and incubated
with anti-CD81 mAb (BD Biosciences; 5A6, 10 µg/ml) or an
isotype control antibody (MOPC21; IgG1κ; 10 µg/ml;
Sigma-Aldrich) at 37°C for 30 min. The incubated cells were washed
in PBS and resuspended in 100 µl PBS and then incubated with
goat anti-mouse IgG (2 µg/ml; Sigma-Aldrich) for 15 min at
37°C. The purified resting B cells were also cultured in RPMI-1640
medium supplemented with HCV E2 protein (2 µg/ml) or serum
obtained from patients with HCV (diluted 1/20). CD81-pre-stimulated
B cells were added to EBV stock supernatant in a culture flask,
followed by 2 h of incubation at 37°C. We then generated
EBV-transformed B cells as described above. Cell aggregation and
clumping of the B cells following EBV infection were measured at
the indicated time points (days 2 and 4, and weeks 1, 2, 3 and 4)
under a microscope.
Measurement of the proliferation of
EBV-transformed B cells
B cells from healthy donors or patients with HCV
were stimulated with anti-CD81 antibody (10 µg/ml) or HCV E2
protein (2 µg/ ml) prior to EBV infection. At 2 weeks after
EBV infection, the EBV-transformed B cells from healthy donors or
patients with HCV (2×104 cells/well) were harvested and
cultured in RPMI-1640 medium containing 10% FBS in 96-well flat
bottom plates. After 48 h, cell proliferation was measured using
the alamar-Blue assay (AbD Serotec, Raleigh, NC, USA). The
alamarBlue reagent was added (10% by volume) to each well and
relative fluorescence was determined 7 h later using a fluorometer
(Synergy HT; Bio-Tek Instruments, Inc., Winooski, VT, USA;
excitation, 530 nm; emission, 590 nm). The purified naïve B cells
and B cells stimulated with anti-CD81 mAb were also cultured under
the same conditions as the controls (untreated cells). All
experiments were performed in triplicate, and the relative
fluorescence number used was the mean fluorescence determined for
each culture. Data are expressed as the means ± standard deviation
(SD) and analyzed using the Student's t-test. A p-value <0.05
was considered to indicate a statistically significant
difference.
Detection of EBV-induced protein and
interleukin (IL)-10 mRNA and protein expression
To measure the intracellular expression levels of
LMP-1, LMP-2A, Epstein-Barr virus nuclear antigen (EBNA)-1, EBNA-2,
EBNA-3A and IL-10 in B cells during EBV transformation, the cells
and culture media were harvested each week to measure the LMP-1 and
IL-10 expression levels for 4 weeks. The cells were then
permeabilized with permeabilization buffer (0.1% saponin;
Sigma-Aldrich, in PBS) for 5 min. The cells were washed twice in
PBS following centrifugation (400 x g, at 4°C for 5 min), and cell
pellets were stained with PE-conjugated anti-LMP-1 or IL-10
antibodies for 30 min at 4°C. Following 3 washes, the cells were
fixed in 4% paraformaldehyde solution (Sigma-Aldrich) for 10 min.
Expression levels were determined using a FACSCalibur flow
cytometer (BD Biosciences). To detect EBV proteins by western blot
analysis, B cells were harvested at 2 and 4 weeks following
transformation with EBV. The cells (2×106 cells/sample)
were pelleted and lysed in RIPA buffer (Elpis-Biotech, Inc.,
Daejeon, Korea). Proteins (20 µg/sample) were immediately
heated for 5 min at 100°C. Total cell lysates were subjected to
SDS-PAGE on gels containing 15% (wt/vol) acrylamide under reducing
conditions. Anti-LMP-1 antibody for western blot analysis was
purchased from Abnova (Taipei, Taiwan). Anti-EBNA-1 antibody was
purchased from Thermo Fisher Scientific, Inc., (Pittsburg, PA,
USA). Anti-EBNA-2 and anti-LMP-2A antibodies were purchased from
Santa Cruz Biotechnology, Inc., (Santa Cruz, CA, USA). To measure
the mRNA and protein expression levels of IL-10, B cells isolated
from healthy donors were stimulated with anti-CD81 mAb prior to EBV
infection. Mouse anti-human CD19 mAb (BD Biosciences) was used as a
positive control. The EBV-transformed B cells were harvested each
week to determine the intracellular IL-10 levels. Total RNA from
the EBV-transformed B cells was isolated at the indicated time
points using an RNeasy Mini kit (Qiagen, Hilden, Germany). The RNA
was reverse transcribed into cDNA using oligo(dT) primers (Bioneer
Corp., Daejeon, Korea) and reverse transcriptase. PCR amplification
was performed using specific primer sets (Bioneer Corp.) for human
IL-10 (upstream primer, 5′-CTG AGA ACC AAG ACC CAG ACA TCA AGG and
downstream primer, 5′-GTC AGC TAT CCC AGA GCC CCA GAT CCG; 327-bp
product). As a control, a primer set specific for β-actin (upstream
primer, 5′-ATC CAC GAA ACT ACC TTC AA and downstream primer, 5′-ATC
CAC ACG GAG TAC TTG C) was used. PCR (25 cycles; 20 sec at 94°C, 10
sec at 60°C, 30 secs at 72°C) was performed using AccuPower PCR
PreMix (Bioneer Corp.). PCR products were analyzed by agarose gel
electrophoresis and visualized with ethidium bromide under UV light
using the Multiple Gel DOC system (Fujifilm, Tokyo, Japan).
Densitometry was performed using Multi Gauge version 2.3 software
(Fujifilm). The culture supernatant from EBV-transformed B cells
stimulated with anti-CD81 mAb or serum derived from HCV patients
(diluted 1/20) was harvested at 1, 2 and 4 weeks to detect IL-10
level. IL-10 protein was detected and quantified using the
Single-Analyte ELISArray kit (Qiagen, Hilden, Germany). MOPC, the
isotype control, and healthy donor serum were used as controls.
Induction and detection of CD81-mediated
apoptosis
Four weeks following infection, the EBV-transformed
B cells (1×106 cells/ml) were harvested and washed twice
in cold PBS. The cells were resuspended in 100 µl PBS and
incubated with anti-CD81 mAb (10 µg/ml; BD Biosciences) or
the isotype control antibody (MOPC, Sigma-Aldrich, 10 µg/ml)
at 37°C for 30 min. The cells were washed in PBS and resuspended in
100 µl PBS and then incubated with goat anti-mouse IgG
(Sigma-Aldrich; 2 µg/ml) for 15 min at 37°C. Following
stimulation with antibody, the cells were washed and then cultured
in RPMI-1640 medium at 37°C. For stimulation with recombinant HCV
E2 protein, the EBV-transformed B cells (4 weeks following
infection, 1×106 cells/ml) were resuspended in 100
µl PBS and incubated with HCV E2 protein (1 µg/ml) at
37°C for 1 h. Following incubation, the cells were harvested and
washed twice with PBS and resuspended in 100 µl of 1X
binding buffer (10 mM HEPES/NaOH, pH 7.4; 140 mM NaCl; 2.5 mM
CaCl2). Apoptosis was then determined using an FITC
Apoptosis Detection kit (BD Biosciences) according to the
manufacturer's instructions. To detect the subG1 peak, the cells
were harvested following treatment, washed twice in PBS (2% FBS),
fixed with 70% cold aqueous ethanol, and stored at 4°C for at least
24 h. The cells were washed twice in PBS following centrifugation
(400 x g, at 4°C for 5 min), and cell pellets were stained with
propidium iodide (PI; Sigma-Aldrich) staining solution containing
RNase A (10 µg/ml) and PI (10 µg/ml) in PBS. The cell
suspension was then incubated in the dark at room temperature for
30 min, and the DNA content was determined using a FACSCalibur flow
cytometer (BD Biosciences).
Measurement of mitochondrial membrane
potential (∆ψm) and reactive oxygen species (ROS) levels
To determine whether CD81-induced apoptosis is
related to the disruption of ∆ψm and ROS production, we measured
the ∆ψm and ROS levels in the EBV-transformed B cells following
CD81 cross-linking. The EBV-transformed B cells (1×106
cells/well) were pre-treated with 10 µM DCFH-DA for 30 min
to measure the ROS levels. To measure ∆ψm, the cells were collected
and pre-incubated in 100 µl PBS containing 20 µM
DiOC6 at 37°C for 15 min, and the cells were then washed
twice with cold PBS and treated with mouse-anti-human CD81 mAb or
HCV E2 protein for 3 h at 37°C. The cells were then harvested at
various time points (0, 10, 20, 30 min, 1, 2 and 3 h) and washed 3
times with cold PBS. The ROS levels and ∆ψm were determined using a
FACSCalibur flow cytometry (BD Biosciences). To determine whether
CD81-induced apoptosis is related to mitochondrial dysfunction, the
EBV-transformed B cells (1×106 cells/ml) were
pre-incubated for 30 min with 300 µM antimycin A
(Sigma-Aldrich), a mitochondrial complex III inhibitor. The cells
were then washed with PBS and treated with mouse anti-human CD81
mAb (10 µg/ml) or MOPC (10 µg/ml) for 1 h at 37°C.
The cells were then washed twice with PBS and incubated for 1 h
(5×105 cells/well). The apoptotic rate, ROS production
and ∆ψm were analyzed using a FACSCalibur flow cytometer (BD
Biosciences).
Inhibition of apoptosis
To investigate the effects of caspase inhibitors and
ROS, the EBV-transformed B cells were treated with Z-VAD-fmk,
Z-IETD-fmk, Z-DEVD-fmk (20 µM in DMSO; Calbiochem), or NAC
for 2 h prior to stimulation with the antibodies. The cells were
incubated with anti-CD81 mAb (10 g/ml; BD Biosciences) at 37°C for
40 min followed by cross-linking with goat anti-mouse IgG (2
µg/ml; Sigma-Aldrich) for 15 min at 37°C. The inhibitory
effects of caspase inhibitors or NAC on the apoptosis of
EBV-transformed B cells were detected by Annexin V, DCFH-DA and
DiOC6 staining as described above. To block the Fas-FasL
interaction, antagonistic anti-Fas antibody (ZB4; Abcam, Cambridge,
MA, USA, 0.5 µg/ml) was added 1 h prior to treatment with
anti-CD81 mAb. ZB4 was removed from the cell cultures prior to
stimulation with anti-CD81 mAb. Apoptosis was determined by flow
cytometry following staining with Annexin V (BD Biosciences).
Confocal microscopy to detect
apoptosis-related intracellular molecules
The EBV-transformed B cells were first cross-linked
with anti-CD81 mAb and then secondary antibodies to induce
apoptosis. To detect intracellular apoptosis-related molecules, teh
cells were permeabilized with permeabilization buffer (0.1% saponin
in PBS). The cells were incubated with primary antibodies against
cytochrome c (mouse IgG2b; Santa Cruz Biotechnology, Inc.,
Dallas, TX, USA), apoptosis-inducing factor (AIF; mouse IgG2b;
Santa Cruz Biotechnology, Inc.) and then FITC-conjugated goat
anti-mouse IgG (Sigma-Aldrich) for 30 min. Nuclei were stained with
PI for 10 min at room temperature. Following 3 washes with PBS, the
cells were mounted on microscopic slides under coverslips using
fluorescence mounting medium (DakoCytomation, Glostrup, Denmark).
Fluorescence-stained cells were examined under a confocal
laser-scanning microscope (510 META; Carl Zeiss) at x400 original
magnification and images were acquired using confocal microscopy
software release 3.0 (Carl Zeiss).
Western blot analysis of
apoptosis-related proteins
Following stimulation as described above, the
EBV-transformed B cells were pelleted and lysed in RIPA buffer
(Elpis-Biotech, Inc.). Proteins (10 µg/sample) were
immediately heated for 5 min at 100°C. Total cell lysates
(5×106 cells/sample) were subjected to SDS-PAGE on gels
containing 15% (wt/vol) acrylamide (Sigma-Aldrich) under reducing
conditions. Separated proteins were transferred onto nitrocellulose
membranes (Millipore, Billerica, MA, USA), and membranes were
blocked with 5% skim milk followed by commercial western blot
analysis. Chemiluminescence was detected using an ECL kit (Amersham
Biosciences, Pittsburgh, PA, USA) and the Multiple Gel DOC system
(Fujifilm). The following primary antibodies were used: caspase-8,
caspase-3, caspase-9, β-actin, phosphorylated (p-)ERK1/2, ERK1/2,
p-Akt (Ser473), Akt, p-JNK (Thr183/Tyr185), JNK and
poly(ADP-ribose) polymerase (PARP) from Cell Signaling Technology,
Inc., (Danvers, MA, USA) and p-c-Jun and c-Jun from Santa Cruz
Biotechnology, Inc.
RT-PCR to detect apoptosis and
anti-apoptosis-related molecules
Following stimulation as described above, total RNA
was isolated using the RNeasy Mini kit (Qiagen). RNA was reverse
transcribed into cDNA using oligo(dT) primers (Bioneer Corp.) and
reverse transcriptase. PCR amplification was performed using
specific primer sets (Bioneer Corp.) for Bcl-2 (upstream primer,
5′-GGA TTG TGG CCT TCT TTG AG-3′ and downstream primer, 5′-CAG CCA
GGA GAA ATC AAA CAG-3′; 209-bp product), Bax (upstream primer,
5′-CCA AGA AGC TGA GCG AGT GT-3′ and downstream primer, 5′-CAG CCC
ATG ATG GTT CTG AT-3′; 250-bp product), and Bad (upstream primer,
5′-CGA GTG AGC AGG AAG ACT CC-3′ and downstream primer, 5′-CTG TGC
TGC CCA GAG CTT-3′; 299-bp product). As a control, a specific
primer set for β-actin (upstream primer, 5′-ATC CAC GAA ACT ACC TTC
AA-3, downstream primer, 5′-ATC CAC ACG GAG TAC TTG C-3′) was used.
PCR (25 cycles; 20 sec at 94°C, 10 sec at 60°C, 30 sec at 72°C) was
performed using AccuPower PCR PreMix (Bioneer Corp.). PCR products
were analyzed as described above.
Statistical analysis
The cell proliferation or mean fluorescence
intensity (MFI) data are expressed as the means ± standard error of
the mean (SEM) from representative experiments. All data shown were
confirmed by at least 3 independent experiments and are
representative of at least 3 different experiments. The data were
compared by one-way analysis of variance (ANOVA) using SPSS
software for Windows, version 20.0 (SPSS Inc., Chicago, IL, USA). A
p-value <0.05 was considered to indicate a statistically
significant difference.
Results
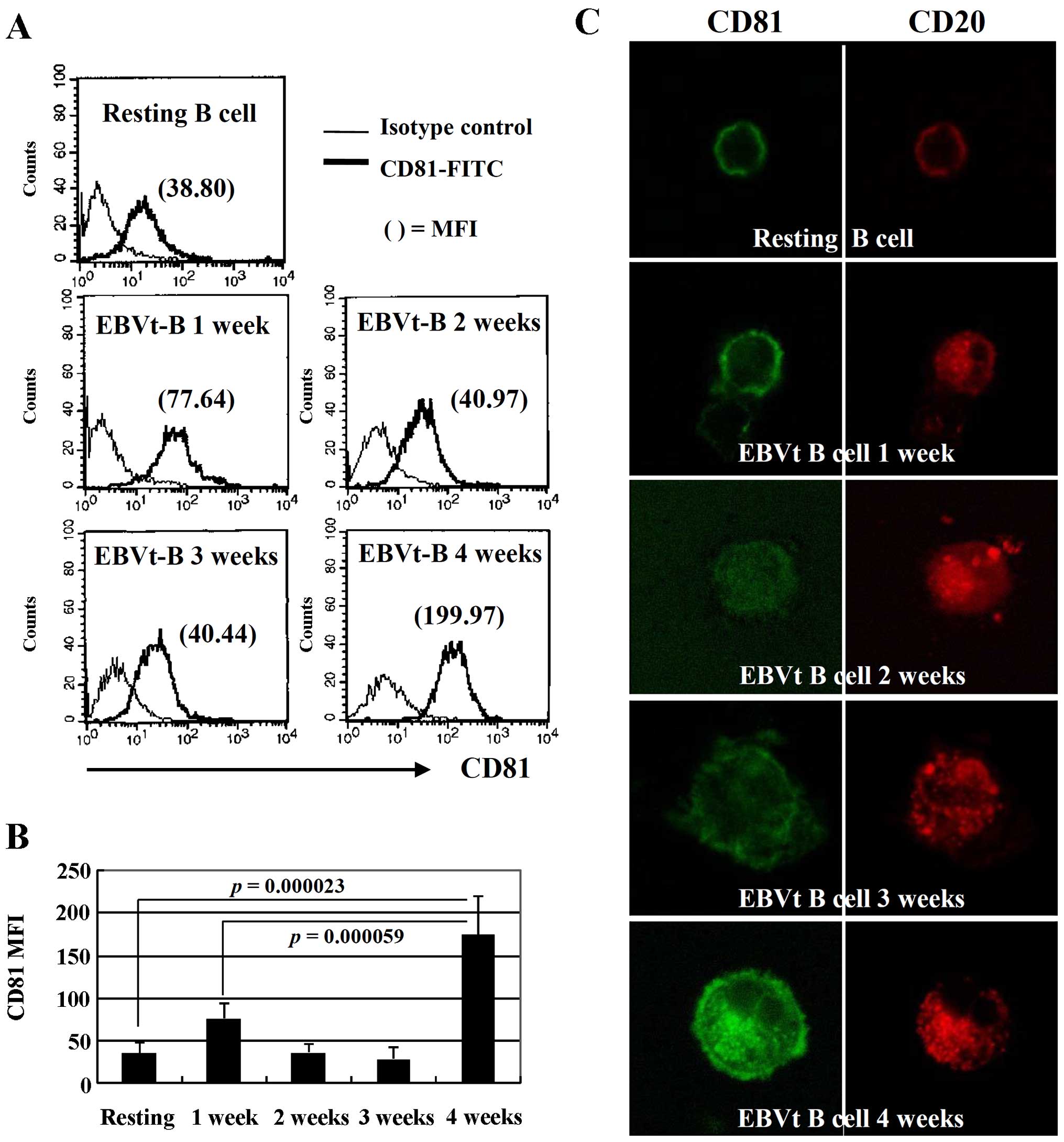
EBV infection enhances CD81 expression in
resting B cells
CD81 (TAPA-1) is expressed on the surface of most
cells in the body, including B cells throughout their cellular
differentiation (1). Resting B
lymphocytes from peripheral blood were harvested to evaluate the
changes in CD81 expression by flow cytometry and confocal
microscopy each week for 4 weeks following EBV infection. The
expression of CD81 was slightly increased at 1 week following
infection; however, the expression of CD81 was not altered in the
second and third weeks compared with resting B cells. Of note, the
MFI of CD81 in EBV-transformed B cells was increased by
approximately 5-fold compared to the resting B cells at 4 weeks
following EBV infection (Fig. 1A and
B). These changes in the expression pattern of CD81 in the
EBV-infected B cells during the transformation process was also
detected by confocal microscopy (Fig.
1C). These results suggest that the expression of CD81, which
is expressed constitutively on mature B cells, may be increased
during the late stages of EBV transformation.
CD81 pre-stimulation by cross-linking or
treatment with HCV E2 protein increases the proliferation of
EBV-infected B cells
HCV E2 induces the proliferation of resting B cells
through interaction with CD81 (28). CD81 expression is also
significantly higher in lymphocytes from HCV-positive patients than
those from controls (18). In
this study, to investigate the effect of stimulation with CD81 on
the proliferation of EBV-transformed B cells, B cells isolated from
normal healthy donors or HCV-positive patients were infected with
EBV. A total of 7 healthy donors and 7 patients with chronic HCV
infection were selected and examined. The clinical and virological
characteristics of the patients with chronic HCV are presented in
Table I. Early signs of
EBV-induced transformation (cell aggregation, increased cell size
and a sudden increase in growth) were observed in the B cells
isolated from HCV-positive patients 4 or 5 days following EBV
infection. In addition, there was an increase in the number and
size of B cell aggregations in the cells isolated from HCV-infected
patients compared with the B cells isolated from the normal
controls (Fig. 2A). To determine
whether HCV infection influences EBV-induced B cell transformation,
we stimulated the B cells from healthy donors with anti-CD81 mAb,
HCV E2 protein, or serum from HCV-positive patients prior to EBV
infection. Notably, all the EBV-induced transformation processes in
the B cells pre-stimulated with anti-CD81 mAb, HCV E2 protein, or
serum from HCV-positive patients occurred much more rapidly than
those in the control cells (Fig.
2B–D). The alamarBlue proliferation assay revealed that the
proliferation rate of the B cells isolated from HCV-positive
patients (p=0.00037, healthy donor PBMCs vs. HCV patient PBMCs), or
CD81-pre-stimulated B cells from healthy donors (p=0.00089, healthy
donor B cells stimulated with isotype control antibodies and EBV
vs. healthy donor B cells stimulated with HCV E2 protein and EBV)
was significantly enhanced at 2 weeks following EBV infection
(Fig. 2E and F). These results
suggest that HCV infection affects the transformation and
proliferation of B cells following EBV infection.
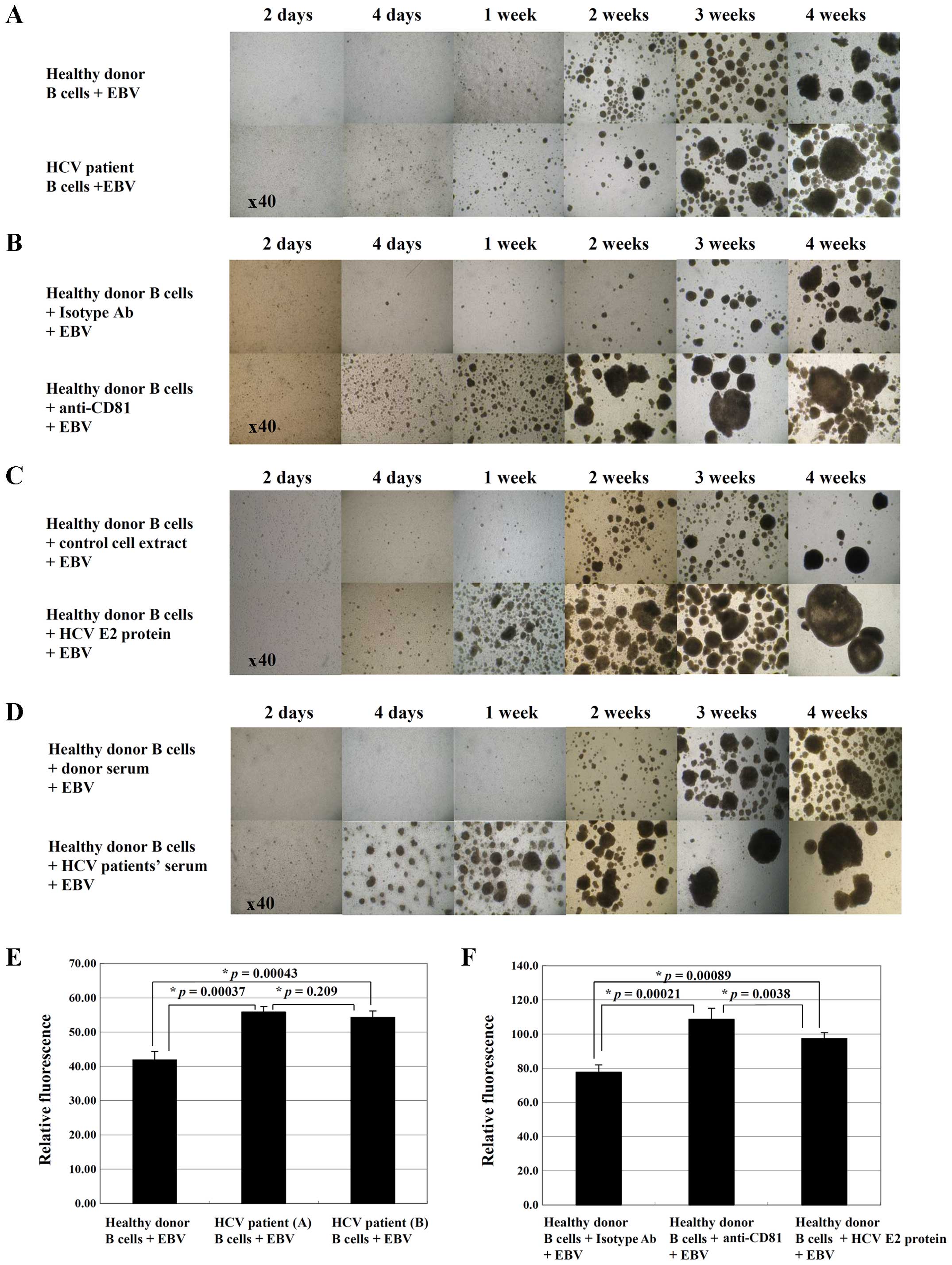 | Figure 2Clumping and proliferation of CD81
pre-stimulated B cells during Epstein-Barr virus (EBV)
transformation. Following the treatment of PBMCs, the cells were
infected with EBV. Cell clumping formation was monitored daily and
measured at the indicated time periods (days 2 and 4, and weeks 1,
2, 3 and 4) using an inverted microscope. (A) EBV-transformed B
cells from HCV-positive patients in comparison with EBV-infected B
cells from normal controls. (B–D) B cells from healthy donors
treated with several CD81 stimuli, including (B) anti-CD81 mAb (10
µg/ml), (C) hepatitis C virus (HCV) E2 protein (2 µg/
ml), (D) serum from HCV-positive patients prior to EBV infection.
MOPCs were used as isotype control antibodies (10 µg/ml).
(E) At 2 weeks following EBV infection, EBV-transformed B cells
from healthy donors or HCV patients were transferred and cultured
for 48 h. To analyze the proliferation rate, alamarBlue dye was
added to each well (10% by volume) and relative fluorescence was
measured 7 h later using a fluorometer. Data represent the means ±
SEM of triplicate cultures. (F) At 2 weeks following EBV infection,
CD81 pre-stimulated EBV-transformed B cells from healthy donors
were transferred and cultured for 48 h, and then the proliferation
rate was analyzed as described above. Data represent the means ±
SEM of triplicate cultures. Comparisons between all individual data
were made using one-way ANOVA. Results are representative of 5
separate experiments. |
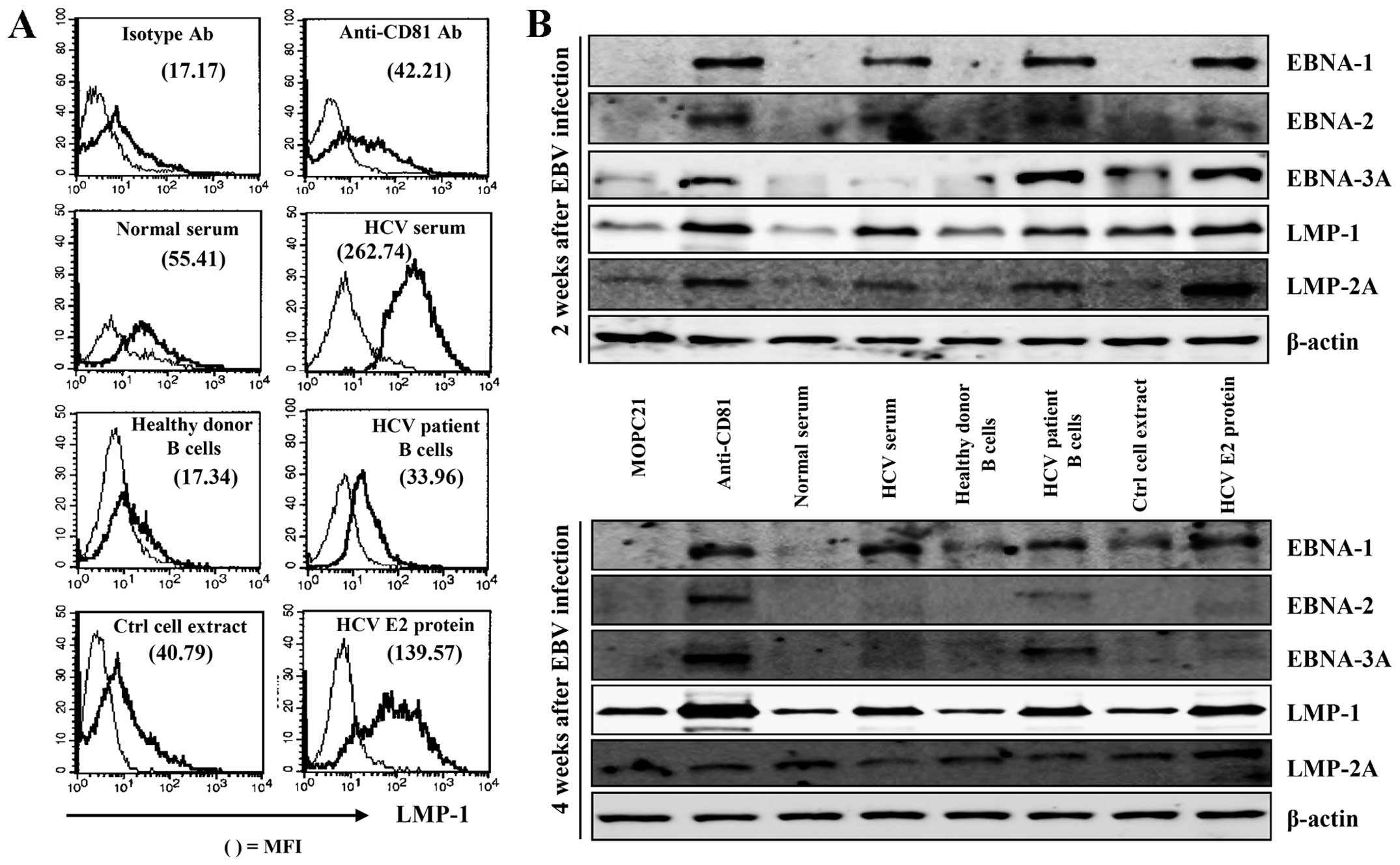
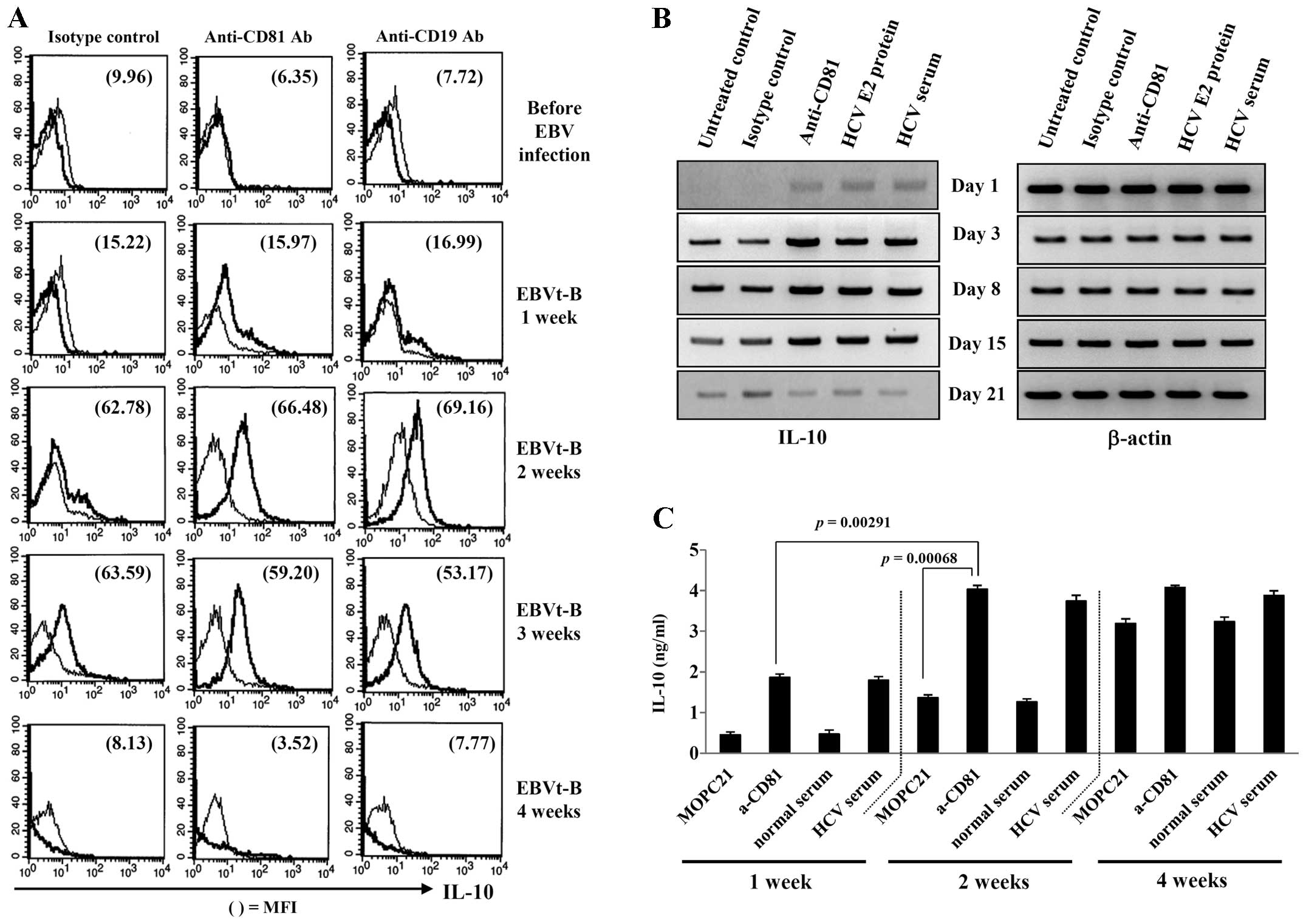
CD81 pre-stimulation promotes the
proliferation of B cells following EBV infection through the early
induction of EBV-induced protein and IL-10 expression
EBV-induced protein expression is upregulated
following EBV infection. LMP-1 of EBV has been shown to induce the
expression of a number of important cellular genes that have
profound effects on cellular growth (29). In this study, to investigate the
effects of CD81 stimulation on the proliferation of EBV-transformed
B cells, we examined intracellular EBNA-1, EBNA-2, EBNA-3A, LMP-1
and LMP-2A expression in the EBV-transformed B cells. LMP-1
expression in EBV-infected-cells from HCV-positive patients was
significantly higher than that in healthy donor B cells (Fig. 3A). Pre-stimulation of CD81 with
serum from obtained HCV-positive patients, HCV E2 protein, or
anti-CD81 mAb accelerated the expression of LMP-1 in comparison
with the respective controls (Fig.
3A). We also performed western blot analyses to determine the
expression of EBV-induced proteins, which are involved in
immortalization and growth. CD81 stimulation with anti-CD81 mAb,
HCV E2 protein, or serum obtained from HCV-positive patients prior
to EBV infection induced the early and high expression of
EBV-induced EBNA-1, EBNA-2, EBNA-3A, LMP-1 and LMP-2A compared with
that of the control group at 2 weeks. EBNA-1, LMP-1 and LMP-2A
expression was maintained at significantly high levels following
CD81 stimulation at 4 weeks (Fig.
3B).
The expression of LMP-1 induces the expression of
human IL-10 in transfected sublines of the EBV-negative DG75 and
BL41 cell lines, and IL-10 can also induce the expression of LMP-1
in B lymphocytes and in Burkitt's lymphoma (30,31). Next, we examined the effects of
CD81 stimulation on IL-10 production by EBV-transformed B cells.
The IL-10 level was measured following CD19 stimulation with
anti-CD19 mAb in EBV-transformed B cells. Pre-stimulation of CD81
with anti-CD81 mAb promoted the intracellular IL-10 expression at
an earlier time point in the EBV-transformed B cells than in the
unstimulated cells (Fig. 4A). We
also observed that human IL-10 mRNA expression and protein
secretion increased rapidly in the EBV-transformed B cells
following pre-stimulation of CD81 (Fig. 4B and C). These results suggest
that the stimuli associated with CD81 signaling induces the early
expression of EBV-induced proteins, and that the increased IL-10
protein expression facilitates the survival and proliferation of
EBV-transformed B cells.
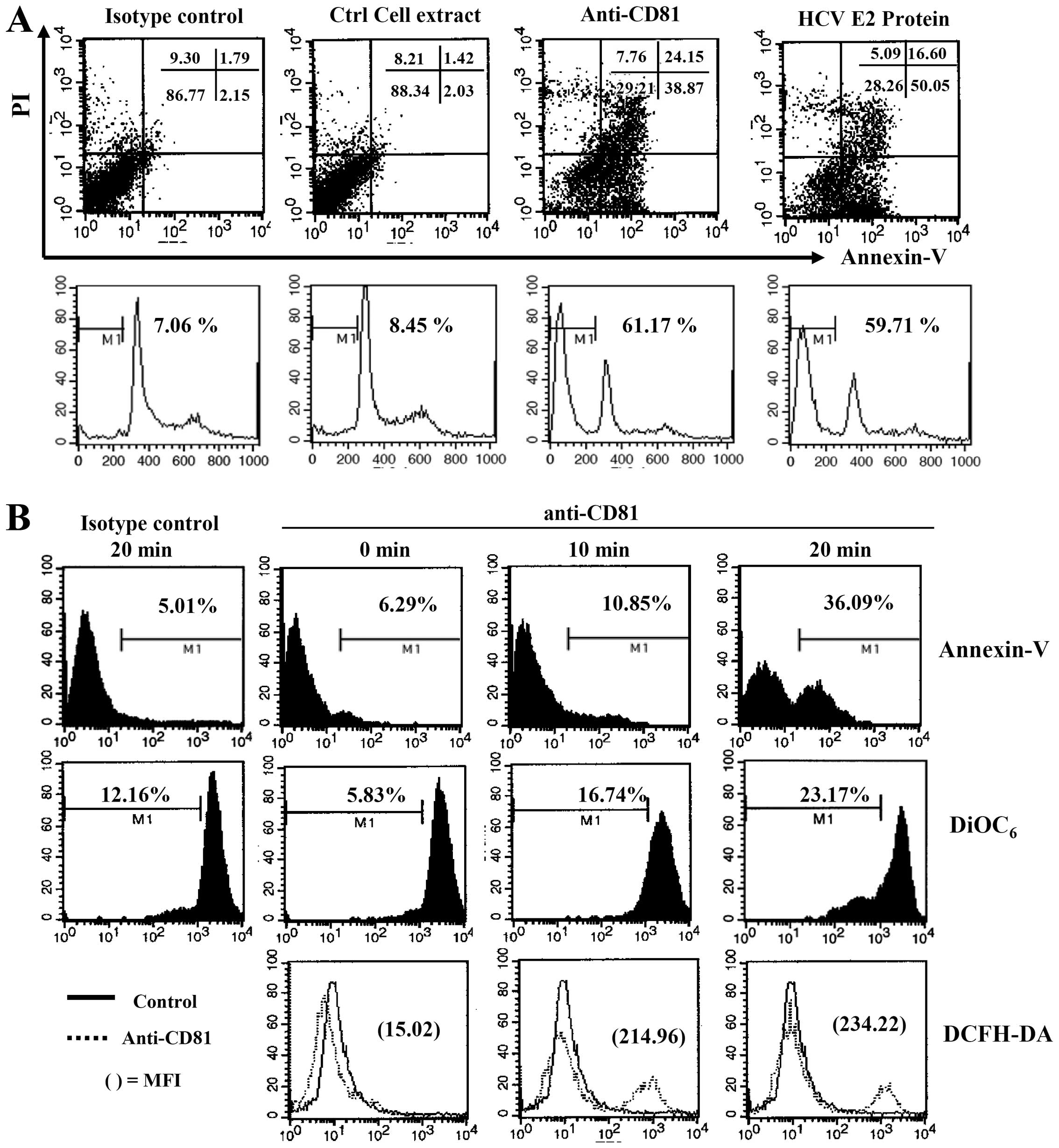
ROS and caspases contribute to the
apoptosis of EBV-transformed B cells following the induction of the
overexpression of CD81
The above-mentioned results indicated that CD81 was
upregulated by EBV infection. In addition, B cells pre-stimulated
with various CD81 stimuli proliferated more rapidly following EBV
infection than the healthy donor B cells through the production of
LMP-1 and IL-10. However, the overexpression of CD81 by
EBV-transformed B cells through stimulation with HCV E2 protein or
anti-CD81 mAb inhibited cell proliferation (data not shown). In
addition, we observed that the IL-10 mRNA level decreased abruptly
at 3 or 4 weeks following CD81 stimulation, and the intracellular
IL-10 protein level also dropped (Fig. 4A, lowest panel; Fig 4B, lowest lane). To investigate
whether the overexpression of CD81 affects B cell survival and
apoptosis, B cells transformed with EBV for 4 weeks and
overexpressing CD-81 were stimulated with anti-CD81 mAb or HCV E2
protein. This induced the apoptosis of the EBV-transformed B cells.
In addition, cell cycle analysis revealed that the stimulation with
anti-CD81 mAb or HCV E2 resulted in a significant increase in the
subG1 arrest of EBV-transformed B cells (Fig. 5A).
We then examined whether this CD81-mediated
apoptosis is related to ROS production and the disruption of Δψm,
as caspases and ROS are important mediators of apoptosis following
cross-linking with antibodies (32). The induction of the overexpression
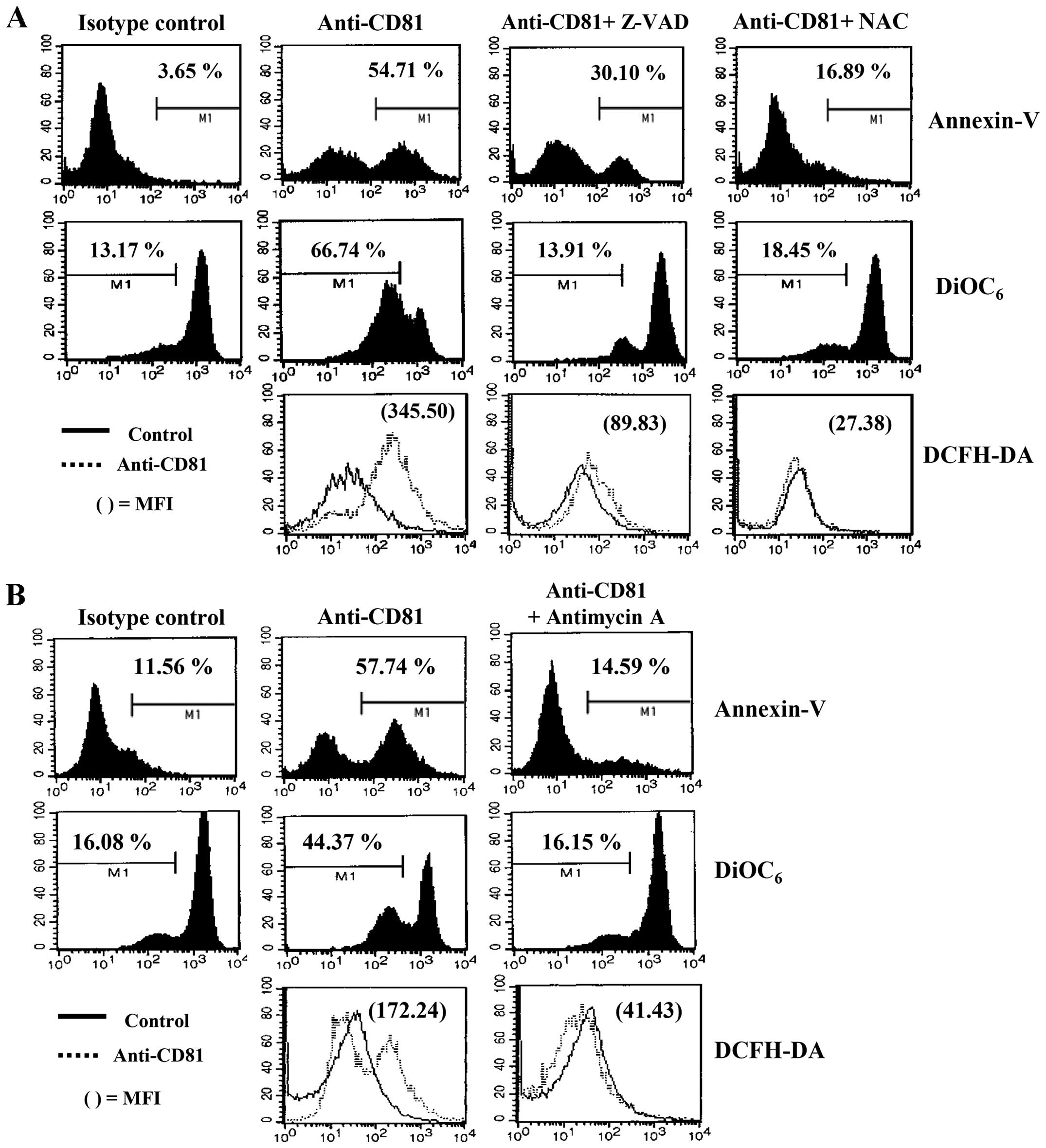
of CD81 on EBV-transformed B cells resulted in the immediate
generation of ROS, it increased the number of Annexin V-positive
apoptotic cells, and disrupted Δψm (Fig. 5B). The EBV-transformed B cells
were pre-incubated with a pan-caspase inhibitor, Z-VAD-fmk, or a
ROS quencher, NAC, for 2 h prior to CD81 stimulation with anti-CD81
mAb. Z-VAD-fmk and NAC significantly decreased CD81-dependent
apoptosis and ROS generation by blocking the disruption of Δψm
(Fig. 6A). In addition, treatment
with antimycin A (300 µM), a mitochondrial complex III
inhibitor, also completely blocked the apoptosis of EBV-transformed
B cells induced by the enhanced expression of CD81 (Fig. 6B). These results suggest that
CD81-mediated apoptosis in EBV-transformed B cells may be related
to ROS generation and may be dependent on the disruption of
Δψm.
CD81-mediated apoptosis of
EBV-transformed B cells is mediated by the translocation of
mitochondrial proteins
Based on our observation that Z-VAD-fmk, a
pan-caspase inhibitor, incompletely blocked CD81-mediated
apoptosis, we investigated whether the apoptosis mediated by the
overexpression of CD81 is related to individual caspase activity
and other apoptotic proteins released from the mitochondria. The
EBV-transformed B cells were incubated with the caspase-8
inhibitor, Z-IETD, or the caspase-3 inhibitor, Z-DEVD, for 2 h
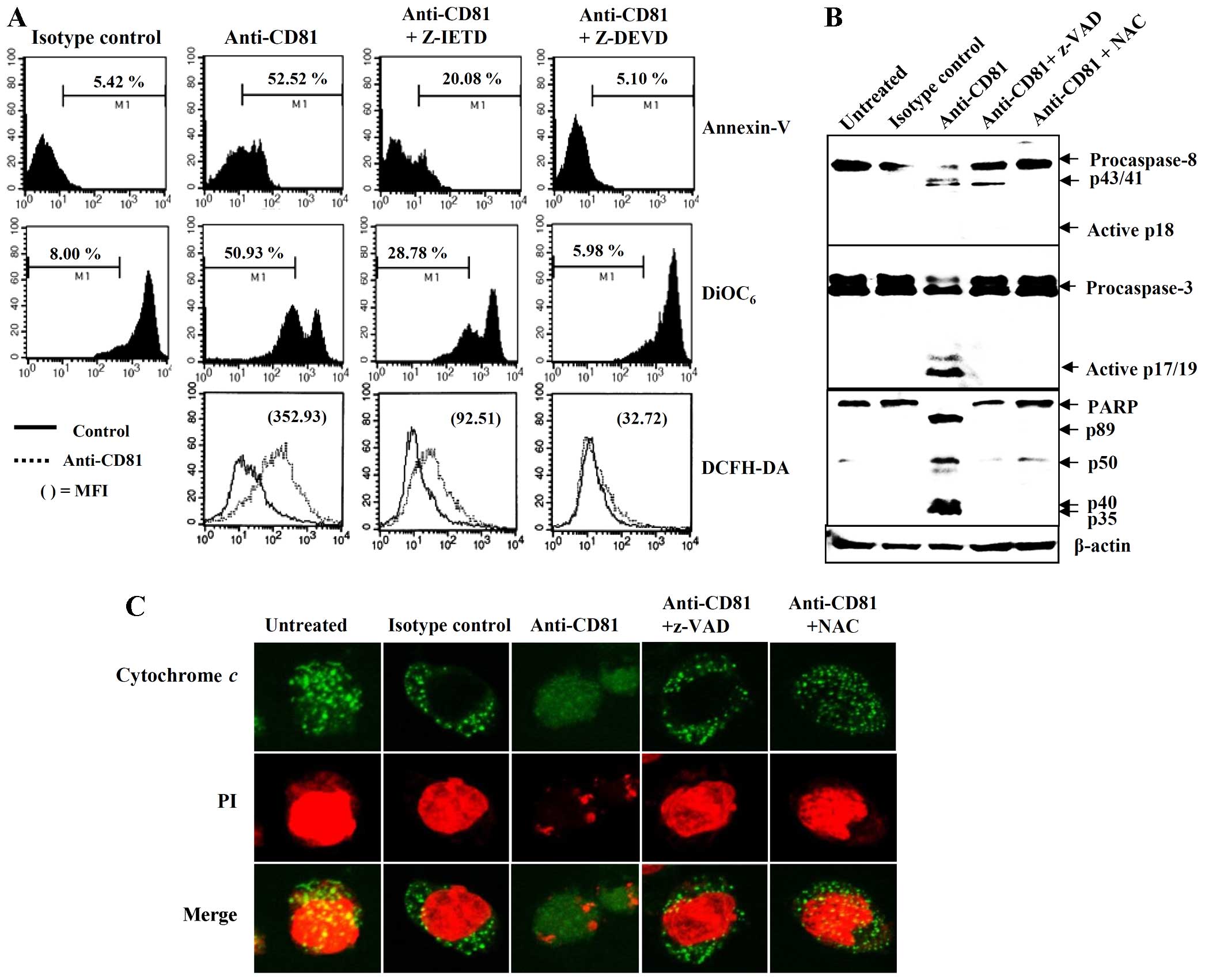
prior to CD81 stimulation. Pre-treatment with Z-IETD partially
reduced CD81-mediated apoptosis; by contrast, pretreatment with
Z-DEVD completely blocked CD81-mediated apoptosis (Fig. 7A). We subsequently examined the
expression of the cleaved forms of caspase-8, caspase-3 and PARP by
western blot anlaysis. The EBV-transformed B cells stimulated with
anti-CD81 mAb exhibited cleaved PARP and generated the active
cleaved forms of caspase-3 (17/19-kDa fragments), indicating the
induction of apoptosis. However, 41/43-kDa fragments corresponding
to the initial form of caspase-8 were observed. The untreated or
NAC pre-treated EBV-transformed B cells only expressed precursor
proteins (Fig. 7B). Thus, we
performed confocal microscopy analysis using anti-cytochrome
c antibody to determine the apoptotic proteins released from
the mitochondria. Stimulation with anti-CD81 mAb caused the
translocation of cytochrome c from the mitochondria to the
cytosol in the EBV-transformed B cells. By contrast, treatment with
Z-VAD-fmk and NAC prior to stimulation with anti-CD81 mAb almost
completely blocked the release of cytochrome c from the
mitochondria (Fig. 7C). These
results suggest that the CD81-mediated signal results in the
release of apoptotic molecules, mainly from the mitochondria and
the activation of the caspase-3 pathway.
JNK-associated Bcl-2 pathway is involved
in CD81-mediated apoptosis
To investigate the mechanisms responsible for the
apoptosis mediated by the overexpression of CD81, we examined
certain candidate signaling molecules by RT-PCR for the detection
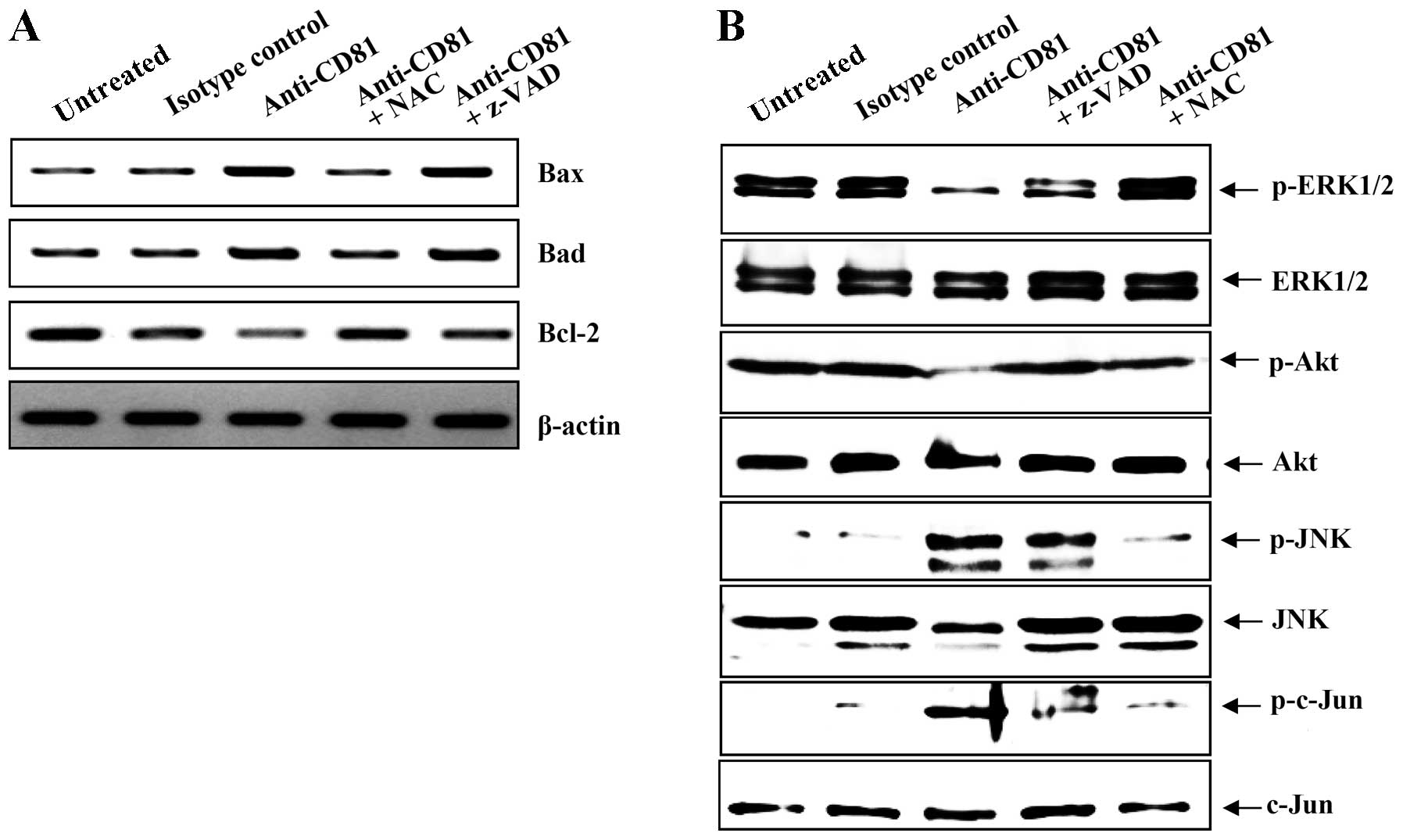
of RNA derived from EBV-transformed B cells. Bax, Bad and Bcl-2
mRNA was constitutively expressed in the EBV-transformed B cells,
but CD81 cross-linking increased the mRNA expression of Bax and
Bad, which are known pro-apoptotic genes (Fig. 8A, 1st and 2nd panels from the
top). By contrast, the expression of Bcl-2, an anti-apoptotic gene,
was decreased by CD81 stimulus (Fig.
8A, 3rd panel from the top). NAC almost completely blocked the
CD81-mediated changes in the expression levels of
apoptosis-associated genes (Fig.
8A, lane 4). However, the broad-spectrum caspase inhibitor,
Z-VAD-fmk did not inhibit the CD81-mediated upregulation of Bax and
Bad in the EBV-transformed B cells. To elucidate the identity of
the upstream proteins in the CD81-mediated mitochondrial apoptotic
pathway, we examined the expression levels of JNK, c-Jun, ERK1/2
and Akt, which mediate signaling to members of the Bcl-2 family
(33,34). CD81 stimulation upregulated the
expression of the phosphorylated form of JNK and its major
substrate, the phosphorylated form of c-Jun (Fig. 8B, panels 5 and 7 from the top),
whereas it decreased the expression of the phosphorylated forms of
ERK1/2 and Akt relative to the control cells (Fig. 8B, panels 1 and 3 from the top).
NAC almost completely blocked the CD81-mediated changes in the
phosphorylation of the kinases (Fig.
8B, lane 5). These results suggest that the
apoptosis-associated genes of EBV-transformed B cells, stimulated
by CD81, are regulated by MAPK signaling.
Discussion
Although previous studies have confirmed the
specificity of binding between E2 and CD81, these studies were not
able to fully explain the effect of overexpressed CD81 on
EBV-transformed B cells (35,36). In this study, the pattern of CD81
expression in EBV-infected B cells during transformation remained
unaltered for the first 3 weeks; however, there was a rapid
increase in CD81 expression at the fourth week following EBV
infection. Resting peripheral blood B cells stimulated with HCV
envelope protein E2, as well as anti-CD81 mAb exhibited an enhanced
proliferation and transformation following EBV infection. By
contrast, the induction of the overexpressin of CD81 on
EBV-transformed B cells induced apoptosis and increased the subG1
peak in cell cycle analysis. Previous studies have demonstrated
that the multimeric engagement of CD81 is required for the
activation of resting B cells (28) and for the protection of primary
human B lymphocytes from Fas-mediated apoptosis (24). However, the results of our study
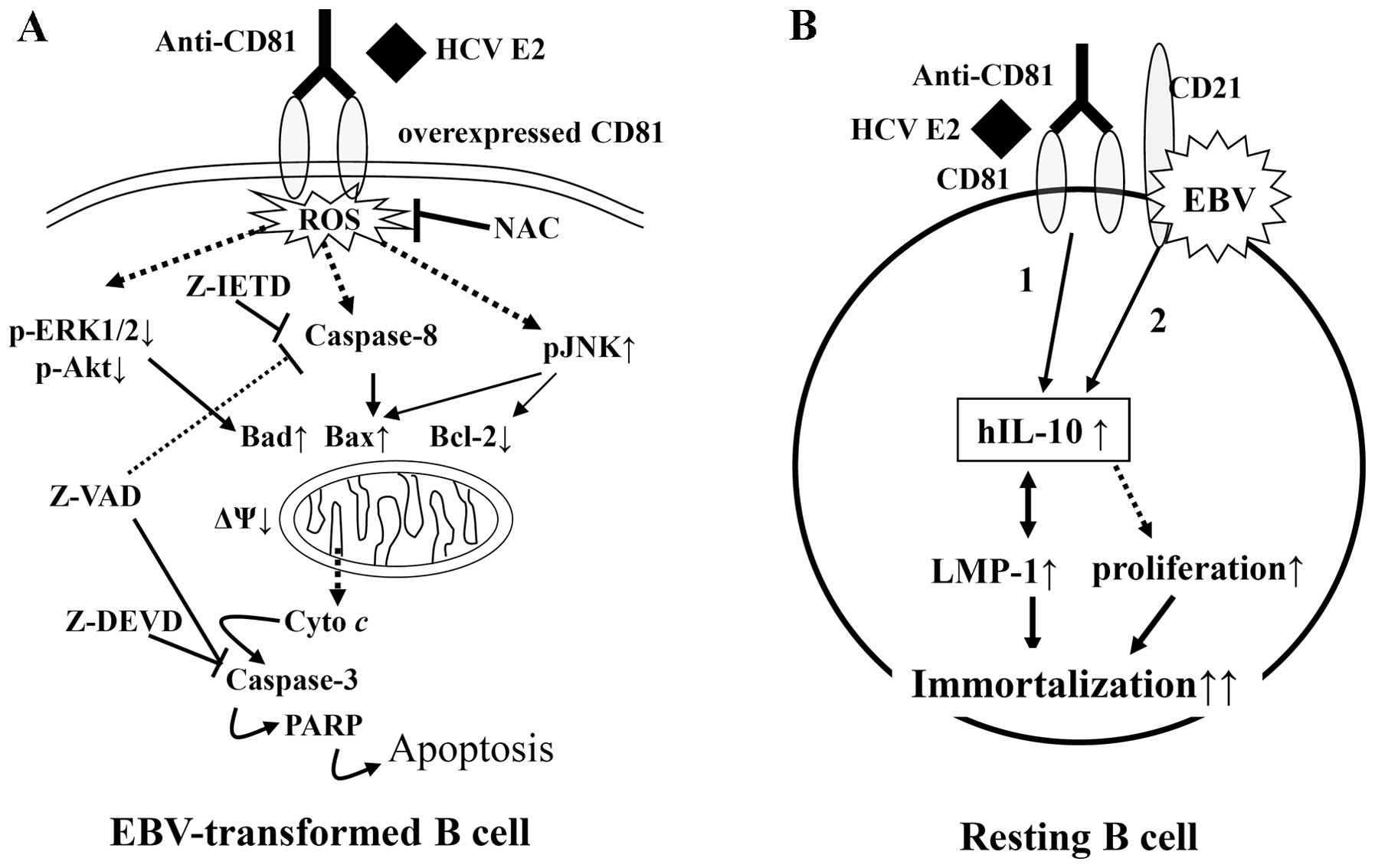
suggest that CD81 signaling is transmitted through different
mechanisms in resting B cells and EBV-transformed B cells (Fig. 9).
CD81 is a cellular ligand for E2 that is expressed
by most human cell types, but not by red blood cells or platelets
(13). It is known that this
complex reduces the threshold for B cell activation via the BCR by
bridging Ag-specific recognition and CD21-mediated complement
recognition (2,37). A statistically significant
increase in CD81 expression has been observed on lymphocytes and
monocytes from HCV-positive patients as compared with healthy
controls (18). The engagement of
CD81 has been shown to result in an increased percentage of B cells
expressing the early activation marker, CD69, the transferrin
receptor, CD71, and the co-stimulatory molecule, CD86 (28). For these reasons, in this study, B
cells derived from patients with HCV were pre-activated chronically
compared with the cells from normal healthy donors. We also
observed that the EBV transformation process of B cells isolated
from HCV-positive patients occurred more rapidly than that of B
cells isolated from normal controls in this study. In addition, the
proliferative activity of the EBV-transformed B cells isolated from
HCV-positive patients surpassed that of the normal controls. This
acceleration of the EBV transformation process also occurred in
healthy donor B cells that were stimulated with serum obtained from
HCV-positive patients, HCV E2 protein, or anti-CD81 mAb prior to
EBV infection. However, we could not exclude the possibility that
contamination with other blood components had occurred in the
stimulation with serum obtained from HCV-positive patients. Thus,
anti-CD81 mAb was used for CD81 stimulation in the experiment shown
in Fig. 5 and thereafter.
In previous studies, it has been observed that the
time required for EBV transformation is shortened by CD19
cross-linking through earlier intracellular LMP-1, EBNA-1 and
EBNA-2 expression than in the controls (38), and that EBV infection induces the
expression of LMP-1, which promotes B cell immortalization
(39). The upregulation of
constitutive nuclear factor NF-κB, as well as CD30, CD40, tumor
necrosis factor (TNF)-α, and Notch1 interactions contributes to
these processes (30). CD81
signaling of resting B cells was accelerated during transformation
into EBV-induced lymphoblastoid B cells in this study. We observed
that EBV-specific proteins (LMP-1, LMP-2A, EBNA-1, EBNA-2 and
EBNA-3A) and human IL-10 expression increased during the early
period of the EBV transformation of CD81 pre-stimulated B cells
compared with the unstimulated controls. EBNA-1 and EBNA-2 are
required for B cell immortalization. LMP-1 is a direct target of
EBNA-2 (40). CD19 and BCR
ligation has been demonstrated to increase the mRNA expression of
EBNA-1 and EBNA-2 in the early stages of infection (38). For these reasons, we hypothesized
that EBNA-2 may be a more important factor than EBNA-1. However,
CD81 stimulation significantly increased and maintained EBNA-1
expression levels compared with EBNA-2 and EBNA-3A in
EBV-transformed B cells. These results suggest that proliferation
and EBV transformation are accelerated through different mechanisms
by CD81 signaling prior to EBV infection and that CD81 signaling is
initiated and maintained by HCV infection in vivo.
Both EBV and HCV bind to the same cell surface
protein complex to deliver stimulatory signals to B cells. This
complex contains CD21 which binds EBV, and CD81 which binds E2
(13). In a previous study, EBV
was detected in 37% of HCC cases in Japan and EBV DNA is also
frequently found in cases with HCV (41). However, another group reported
that of 31 HCC cases, none were positive for EBV while 10 (32%)
tested positive for HCV and 12 (38%) tested positive for hepatitis
B virus (HBV) (42). These data
suggest that EBV infection may cooperate with HCV infection;
however, the association between these two viruses needs to be
investigated further.
In patients chronically infected with HCV, the cell
surface tetraspanin CD81 is overexpressed by different PBMC
subtypes compared with healthy controls. CD81 expression by PBMCs,
including CD4+, CD8+, CD19+ and
CD56+ cells, has been shown to be downregulated during
interferon α-based antiviral therapy (43). In a prevoius study, patients with
splenic lymphoma with villous lymphocytes and HCV infection who
were treated with interferon α-2b went into remission after the
loss of detectable HCV RNA (44).
In another study, cross-linking of CD81 on the cell surface of the
EBV-positive lymphoblastoid cell line JY induced the release of
TNF-α and homotypic aggregation and inhibited JY cell proliferation
(45). However, the differential
effects of normal or elevated CD81 on B cells are not yet fully
understood due to the lack of a suitable model. For these reasons,
in this study, we examined the effects of CD81 overexpression on
the survival or apoptosis of EBV-transformed B cells (normal
expression vs. overexpression). EBV-infected B cells are known to
resist Fas-mediated apoptosis due to defects in the proximal Fas
signaling pathway (46) or the
expression of the FLICE-inhibitory protein (FLIP) (47). We also observed that Fas and Fas
ligand expression by EBV-transformed B cells was not altered
following CD81 stimulation, and that apoptosis induced by CD81
cross-linking was not recovered by treatment of the cells with ZB4
antibody, which blocks Fas-mediated apoptosis (data not shown).
Thus, we focused primarily on the mitochondria and ROS to elucidate
the mechanisms respon-bisle for CD81-mediated apoptosis.
In the present study, we found that cross-linking
CD81 on EBV-transformed B cells resulted in the immediate
generation of ROS, which subsequently disrupted Δψm. However, the
precise mechanism of ROS generation remains unclear. ROS modulate
cytoskeletal metabolism, intracellular Ca2+ levels, and
influence the mitochondrial membrane directly or indirectly
(48). It has been reported that
ROS induce caspase activity, JNK phosphorylation, and even
participate in ERK1/2 signaling (33,49). These findings are supported by our
observations that the ROS quencher, NAC, completely blocked
apoptosis and ROS generation and that Z-VAD-fmk, a pan-caspase
inhibitor, effectively blocked CD81-mediated apoptosis. In
addition, blocking experiments using individual caspase inhibitors
(Z-IETD and Z-DEVD) and the results from western blot analysis
revealed that CD81 cross-linking induced apoptosis mainly via
caspase-3 and partly via caspase-8. These results indicate that
CD81 signaling induces the apoptosis of EBV-transformed B cells
through a caspase-dependent pathway. A previous study demonstrated
that upstream regulators such as JNK, c-Jun, ERK1/2 and Akt
transduce signals to members of the Bcl-2 family (33). However, the engagement of CD81 on
human B cells by a combination of HCV E2 and an anti-CD81 mAb
triggers the JNK pathway and leads to the preferential
proliferation of the naïve B cell subset (28). Notably, stimulation through the
overexpressio of CD81 also induced the phosphorylation of JNK and
c-Jun, but repressed the phosphorylation of ERK1/2 and Akt to
induce the apoptosis of EBV-transformed B cells. The expression of
Bcl-2, regulated by the JNK pathway, was restored in
EBV-transformed B cells treated with NAC prior to CD81 stimulation.
These results suggest that stimulation of overexpressed CD81 on
EBV-transformed B cells may initiate ROS production and activate
the apoptotic signaling cascade through a JNK-mediated
mitochondrial apoptotic pathway, as well as suppress survival
signaling through inhibition of ERK1/2 and Akt phosphorylation.
Taken together, our results suggest that the normal
expression of CD81 vs. the overexpression of CD81 has different
effects on EBV-infected B cells. HCV is a lymphotrophic and a
hepatotrophic virus (50). This
unusual lymphotropism may, at least in part, induce the multiple
immune-mediated extrahepatic manifestations of HCV infection, such
as MC, the presence of serum rheumatoid factor (RF), and B cell
non-Hodgkin lymphoma (9–11). However, the persistence of HCV
infection results in CD81 overexpression and chronic stimulation of
B cells (51). Our results
suggest that CD81 overexpression may induce the eradication of
activated B cells producing immunoglobulin against HCV through
apoptosis and may eventually lead to malignant transformation and
the development of lymphoma by EBV. Our results provide insight
into changes in the expression patterns of CD81 and the high
prevalence of B cell proliferative disorders, including non-Hodgkin
B cell lymphoma in HCV-positive patients. In addition, the results
of the present study indicate potential targets for the treatment
of EBV infection in HCV-positive patients.
Acknowledgments
The present study was supported by the Basic Science
Research Program through the National Research Foundation of Korea
(NRF) funded by the Ministry of Education (grant no.
NRF-2013R1A1A2010668) and the National R&D Program for Cancer
Control, Ministry for Health, Welfare and Family Affairs, Republic
of Korea (grant no. 0920040).
References
|
1
|
Bradbury LE, Kansas GS, Levy S, Evans RL
and Tedder TF: The CD19/CD21 signal transducing complex of human B
lymphocytes includes the target of antiproliferative antibody-1 and
Leu-13 molecules. J Immunol. 149:2841–2850. 1992.PubMed/NCBI
|
|
2
|
Matsumoto AK, Martin DR, Carter RH,
Klickstein LB, Ahearn JM and Fearon DT: Functional dissection of
the CD21/CD19/TAPA-1/Leu-13 complex of B lymphocytes. J Exp Med
Oct. 178:1407–1417. 1993. View Article : Google Scholar
|
|
3
|
Carter RH and Fearon DT: CD19: lowering
the threshold for antigen receptor stimulation of B lymphocytes.
Science. 256:105–107. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Levy S, Todd SC and Maecker HT: CD81
(TAPA-1): a molecule involved in signal transduction and cell
adhesion in the immune system. Annu Rev Immunol. 16:89–109. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
van Zelm MC, Smet J, Adams B, Mascart F,
Schandené L, Janssen F, Ferster A, Kuo CC, Levy S, van Dongen JJ
and van der Burg M: CD81 gene defect in humans disrupts CD19
complex formation and leads to antibody deficiency. J Clin Invest.
120:1265–1274. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Miyazaki T, Müller U and Campbell KS:
Normal development but differentially altered proliferative
responses of lymphocytes in mice lacking CD81. EMBO J.
16:4217–4225. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tsitsikov EN, Gutierrez-Ramos JC and Geha
RS: Impaired CD19 expression and signaling, enhanced antibody
response to type II T independent antigen and reduction of B-1
cells in CD81-deficient mice. Proc Natl Acad Sci USA.
94:10844–10849. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Choo QL, Kuo G, Weiner AJ, Overby LR,
Bradley DW and Houghton M: Isolation of a cDNA clone derived from a
blood-borne non-A, non-B viral hepatitis genome. Science.
244:359–362. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ferri C, La Civita L, Longombardo G,
Zignego AL and Pasero G: Mixed cryoglobulinaemia: a cross-road
between autoimmune and lymphoproliferative disorders. Lupus.
7:275–279. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Dammacco F, Sansonno D, Piccoli C,
Racanelli V, D'Amore FP and Lauletta G: The lymphoid system in
hepatitis C virus infection: autoimmunity, mixed cryoglobulinemia,
and Overt B cell malignancy. Semin Liver Dis. 20:143–157. 2000.
View Article : Google Scholar
|
|
11
|
Quinn ER, Chan CH, Hadlock KG, Foung SK,
Flint M and Levy S: The B cell receptor of a hepatitis C virus
(HCV)-associated non-Hodgkin lymphoma binds the viral E2 envelope
protein, implicating HCV in lymphomagenesis. Blood. 98:3745–3749.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Machida K, Cheng KT, Pavio N, Sung VM and
Lai MM: Hepatitis C virus E2-CD81 interaction induces hypermutation
of the immunoglobulin gene in B cells. J Virol. 79:8079–8089. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Pileri P, Uematsu Y, Campagnoli S, Galli
G, Falugi F, Petracca R, Weiner AJ, Houghton M, Rosa D, Grandi G
and Abrignani S: Binding of hepatitis C virus to CD81. Science.
282:938–941. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Burlone ME and Budkowska A: Hepatitis C
virus cell entry: role of lipoproteins and cellular receptors. J
Gen Virol. 90:1055–1070. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Roccasecca R, Ansuini H, Vitelli A, Meola
A, Scarselli E, Acali S, Pezzanera M, Ercole BB, McKeating J,
Yagnik A, et al: Binding of the hepatitis C virus E2 glycoprotein
to CD81 is strain specific and is modulated by a complex interplay
between hyper-variable regions 1 and 2. J Virol. 77:1856–1867.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Challine D, Buisson M, Cadilhac M,
Germanidis G, Joab I, Eliaszewicz M, Caumes E, Flahault A, Fillet
AM, Pawlotsky JM and Seigneurin JM: Hepatitis C virus-Epstein-Barr
virus interaction in patients with AIDS. J Med Virol. 67:510–515.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ferri C, Lo Jacono F, Monti M, Caracciolo
F, La Civita L, Barsanti LA, Longombardo G, Lombardini F, Careccia
G and Zignego AL: Lymphotropic virus infection of peripheral blood
mononuclear cells in B cell non-Hodgkin's lymphoma. Acta Haematol.
98:89–94. 1997. View Article : Google Scholar
|
|
18
|
Roque-Cuéllar MC, Sánchez B, García-Lozano
JR, Garrido-Serrano A, Sayago M, Praena-Fernández JM, Núñez-Roldán
A and Aguilar-Reina J: Expression of CD81, SR-BI and LDLR in
lymphocytes and monocytes from patients with classic and occult
hepatitis C virus infection. J Med Virol. 84:1727–1736. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Molina S, Castet V, Pichard-Garcia L,
Wychowski C, Meurs E, Pascussi JM, Sureau C, Fabre JM, Sacunha A,
Larrey D, et al: Serum-derived hepatitis C virus infection of
primary human hepatocytes is tetraspanin CD81 dependent. J Virol.
82:569–574. 2008. View Article : Google Scholar :
|
|
20
|
Petracca R, Falugi F, Galli G, Norais N,
Rosa D, Campagnoli S, Burgio V, Di Stasio E, Giardina B, Houghton
M, et al: Structure-function analysis of hepatitis C virus
envelope-CD81 binding. J Virol. 74:4824–4830. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Fornasieri A, Bernasconi P, Ribero ML,
Sinico RA, Fasola M, Zhou J, Portera G, Tagger A, Gibelli A and
D'amico G: Hepatitis C virus (HCV) in lymphocyte subsets and in B
lymphocytes expressing rheumatoid factor cross-reacting idiotype in
type II mixed cryoglobulinaemia. Clin Exp Immunol. 122:400–403.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sung VM, Shimodaira S, Doughty AL, Picchio
GR, Can H, Yen TS, Lindsay KL, Levine AM and Lai MM: Establishment
of B cell lymphoma cell lines persistently infected with hepatitis
C virus in vivo and in vitro: the apoptotic effects of virus
infection. J Virol. 77:2134–2146. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Brazzoli M, Bianchi A, Filippini S, Weiner
A, Zhu Q, Pizza M and Crotta S: CD81 is a central regulator of
cellular events required for hepatitis C virus infection of human
hepatocytes. J Virol. 82:8316–8329. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chen Z, Zhu Y, Ren Y, Tong Y, Hua X, Zhu
F, Huang L, Liu Y, Luo Y, Lu W, et al: Hepatitis C virus protects
human B lymphocytes from Fas-mediated apoptosis via E2-CD81
engagement. PLoS One. 6:e189332011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kim YS, Park GB, Lee HK, Song H, Choi IH,
Lee WJ and Hur DY: Cross-linking of B7-H1 on EBV-transformed B
cells induces apoptosis through reactive oxygen species production,
JNK signaling activation, and fasL expression. J Immunol.
181:6158–6169. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Song H, Park G, Kim YS, Hur I, Kim H, Ryu
JW, Lee HK, Cho DH, Choi IH, Lee WJ and Hur DY: B7-H4 reverse
signaling induces the apoptosis of EBV-transformed B cells through
Fas ligand up-regulation. Cancer Lett. 266:227–237. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Heo TH, Chang JH, Lee JW, Foung SK,
Dubuisson J and Kang CY: Incomplete humoral immunity against
hepatitis C virus is linked with distinct recognition of putative
multiple receptors by E2 envelope glycoprotein. J Immunol.
173:446–455. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Rosa D, Saletti G, De Gregorio E, Zorat F,
Comar C, D'Oro U, Nuti S, Houghton M, Barnaba V, Pozzato G and
Abrignani S: Activation of naïve B lymphocytes via CD81, a
pathogenetic mechanism for hepatitis C virus-associated B
lymphocyte disorders. Proc Natl Acad Sci USA. 102:18544–18549.
2005. View Article : Google Scholar
|
|
29
|
Poppema S: Immunobiology and
pathophysiology of Hodgkin lymphomas. Hematology Am Soc Hematol
Educ Program. 2005:231–238. 2005. View Article : Google Scholar
|
|
30
|
Nakagomi H, Dolcetti R, Bejarano MT, Pisa
P, Kiessling R and Masucci MG: The Epstein-Barr virus latent
membrane protein-1 (LMP1) induces interleukin-10 production in
Burkitt lymphoma lines. Int J Cancer. 57:240–244. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kis LL, Takahara M, Nagy N, Klein G and
Klein E: IL-10 can induce the expression of EBV-encoded latent
membrane protein-1 (LMP-1) in the absence of EBNA-2 in B
lymphocytes and in Burkitt lymphoma- and NK lymphoma-derived cell
lines. Blood. 107:2928–2935. 2006. View Article : Google Scholar
|
|
32
|
Saelens X, Festjens N, Vande Walle L, van
Gurp M, van Loo G and Vandenabeele P: Toxic proteins released from
mitochondria in cell death. Oncogene. 23:2861–2874. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wang M, Zhang L, Han X, Yang J, Qian J,
Hong S, Samaniego F, Romaguera J and Yi Q: Atiprimod inhibits the
growth of mantle cell lymphoma in vitro and in vivo and induces
apoptosis via activating the mitochondrial pathways. Blood.
109:5455–5462. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Spencer JP, Rice-Evans C and Williams RJ:
Modulation of pro-survival Akt/protein kinase B and ERK1/2
signaling cascades by quercetin and its in vivo metabolites
underlie their action on neuronal viability. J Biol Chem.
278:34783–34793. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Allander T, Forns X, Emerson SU, Purcell
RH and Bukh J: Hepatitis C virus envelope protein E2 binds to CD81
of tamarins. Virology. 277:358–367. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Higginbottom A, Quinn ER, Kuo CC, Flint M,
Wilson LH, Bianchi E, Nicosia A, Monk PN, McKeating JA and Levy S:
Identification of amino acid residues in CD81 critical for
interaction with hepatitis C virus envelope glycoprotein E2. J
Virol. 74:3642–3649. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Tedder TF, Zhou LJ and Engel P: The
CD19/CD21 signal transduction complex of B lymphocytes. Immunol
Today. 15:437–442. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hur DY, Lee MH, Kim JW, Kim JH, Shin YK,
Rho JK, Kwack KB, Lee WJ and Han BG: CD19 signalling improves the
Epstein-Barr virus-induced immortalization of human B cell. Cell
Prolif. 38:35–45. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Dolcetti R and Masucci MG: Epstein-Barr
virus: induction and control of cell transformation. J Cell
Physiol. 196:207–218. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Bornkamm GW and Hammerschmidt W: Molecular
virology of Epstein-Barr virus. Philos Trans R Soc Lond B Biol Sci.
356:437–459. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Sugawara Y, Mizugaki Y, Uchida T, Torii T,
Imai S, Makuuchi M and Takada K: Detection of Epstein-Barr virus
(EBV) in hepatocellular carcinoma tissue: a novel EBV latency
characterized by the absence of EBV-encoded small RNA expression.
Virology. 256:196–202. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Akhter S, Liu H, Prabhu R, DeLucca C,
Bastian F, Garry RF, Schwartz M, Thung SN and Dash S: Epstein-Barr
virus and human hepatocellular carcinoma. Cancer Lett. 192:49–57.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Welker MW, Hofmann WP, Lange CM, Herrmann
E, Sarrazin C, Zeuzem S and Kronenberger B: CD81 expression for
discrimination between sustained virologic response and relapse in
patients with chronic hepatitis C. Scand J Gastroenterol.
46:973–980. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Hermine O, Lefrère F, Bronowicki JP,
Mariette X, Jondeau K, Eclache-Saudreau V, Delmas B, Valensi F,
Cacoub P, Brechot C, et al: Regression of splenic lymphoma with
villous lymphocytes after treatment of hepatitis C virus infection.
N Engl J Med. 347:89–94. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Altomonte M, Montagner R, Pucillo C and
Maio M: Triggering of target of an antiproliferative antibody-1
(TAPA-1/CD81) up-regulates the release of tumour necrosis
factor-alpha by the EBV-B lymphoblastoid cell line JY. Scand J
Immunol. 43:367–373. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Snow AL, Chen LJ, Nepomuceno RR, Krams SM,
Esquivel CO and Martinez OM: Resistance to Fas-mediated apoptosis
in EBV-infected B cell lymphomas is due to defects in the proximal
Fas signaling pathway. J Immunol. 167:5404–5411. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Tepper CG and Seldin MF: Modulation of
caspase-8 and FLICE-inhibitory protein expression as a potential
mechanism of Epstein-Barr virus tumorigenesis in Burkitt's
lymphoma. Blood. 94:1727–1737. 1999.PubMed/NCBI
|
|
48
|
Andreyev AY, Kushnareva YE and Starkov AA:
Mitochondrial metabolism of reactive oxygen species. Biochemistry
(Mosc). 70:200–214. 2005. View Article : Google Scholar
|
|
49
|
Filomeni G, Aquilano K, Rotilio G and
Ciriolo MR: Reactive oxygen species-dependent c-Jun NH2-terminal
kinase/c-Jun signaling cascade mediates neuroblastoma cell death
induced by diallyl disulfide. Cancer Res. 63:5940–5949.
2003.PubMed/NCBI
|
|
50
|
Zignego AL, Macchia D, Monti M, Thiers V,
Mazzetti M, Foschi M, Maggi E, Romagnani S, Gentilini P and Bréchot
C: Infection of peripheral mononuclear blood cells by hepatitis C
virus. J Hepatol. 15:382–386. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Zuckerman E, Slobodin G, Kessel A, Sabo E,
Yeshurun D, Halas K and Toubi E: Peripheral B cell CD5 expansion
and CD81 overexpression and their association with disease severity
and autoimmune markers in chronic hepatitis C virus infection. Clin
Exp Immunol. 128:353–358. 2002. View Article : Google Scholar : PubMed/NCBI
|