Introduction
Alzheimer's disease (AD) is a chronic
neurodegenerative disease that is characterized by progressive
learning and memory loss, the incidences of which increase with
age. The classical pathological hallmarks of AD are an
intracellular accumulation of amyloid plaques and neurofibrillary
tangles (NFTs) along with the loss of neurons (1,2).
The β-amyloid peptide (Aβ) is the main component of amyloid
plaques, and it plays a vital role in the formation of both amyloid
plaques and NFTs. In addition, Aβ interactions with receptors
(e.g., ryanodine receptors, the N-methyl-D-aspartate receptor,
N-formyl peptide receptor like-1, CD36 receptor and α7 nicotinic
acetylcholine receptor) which are expressed in brain cells [e.g.,
neuronal cells, endothelial cells and microglia (MG)/microglial
(MG) cells] induce neurotoxicity (3). MG cells are the resident
antigen-presenting cells in the central nervous system (CNS). They
can be activated in response to environmental toxins and are known
to display diverse reactions that are associated with both
protective and deleterious effects. It is widely accepted that Aβ
can accelerate neurodegeneration by activating MG cells (4,5)
and that MG cell activation results in the release of soluble
factors, including reactive oxygen species, various inflammatory
mediators and chemokines. When neurons are exposed to an excess of
soluble factors, neuronal apoptosis increases. Although neural stem
cells (NSCs) are capable of self-renewal and are multipotent cells
that can differentiate into the main cell phenotypes of the CNS in
order to maintain CNS homeostasis (6,7),
the proliferation and differentiation of NSCs are also suppressed
due to the release of numerous soluble factors. Therefore, one
possible treatment strategy for AD would be to protect NSCs against
MG-derived soluble factors. The murine MG cell line, BV-2, has been
used previously as a substitute for primary MG cells, as it
exhibits very similar behavior (8). In addition, the C17.2 NSC line is
capable of self-renewal and differentiation (9) and has been used as a model system
for neurodegenerative diseases (10). Thus, in the present study, we used
BV-2 cells and C17.2 cells as replacements for primary MG cells and
primary NSCs, respectively.
Previous studies have confirmed that the
mitochondrial permeability transition pore (mPTP) plays an
important role in cell apoptosis. The opening of the mPTP results
in potential dissipation of the membrane, mitochondrial swelling,
rupture of the outer membrane, the release of pro-apoptotic
proteins, such as cytochrome c (Cytc) and the induction of
caspase-3-like activity, which eventually initiates apoptosis
(11–13). Indeed, irreversible mitochondrial
impairment caused by mPTP opening is a key step in apoptosis under
Aβ-induced neurotoxic, and other, conditions. These findings
suggest that mPTP activation is a key player in cell death and is a
potential target for cytoprotective intervention.
Heat shock protein 75 (Hsp75) is a member of the
Hsp70 chaperone family and is expressed predominantly in
mitochondria (14) as a marker of
stress. Hsp75 is not heat-inducible, but it has been reported to
respond to other forms of stress, including glucose deprivation,
oxidative injury, focal ischemia and certain drugs (15–20). However, it remains unclear as to
whether Hsp75 can respond to MG-derived soluble factors. Thus, in
this study, we examined Hsp75 expression in NSCs in response to
MG-derived soluble factors. Recent studies have revealed that Hsp75
expression plays a vital role in maintaining mitochondrial function
and cell survival under various pathological conditions, including
AD (21–23). However, further details of the
neuroprotective mechanisms associated with Hsp75 remain to be
elucidated, particularly its anti-apoptotic effects on NSCs. We
hypothesized that the overexpression of Hsp75 would inhibit the
formation of cyclophilin D (CypD)-dependent mPTP opening and reduce
the release of Cytc into the cytosol following treatment with
Aβ1–42 in an NSC-MG cell co-culture system. To examine
this hypothesis, NSCs overex-pressing Hsp75 protein were
constructed and then subjected to the above-mentioned treatment.
Apoptosis was evaluated by flow cytometry. In addition, changes in
the protein expression of related proteins were assessed by western
blot analysis.
The purpose of this study was to investigate changes
in Hsp75 expression following treatment with soluble factors and to
observe whether Hsp75 overexpression provides protection against
MG-derived soluble factor-induced neurotoxicity by regulating mPTP
opening.
Materials and methods
Cell culture and the NSC-MG cell
co-culture system
In the present study, we used BV-2 cells and C17.2
cells as replacements for primary MG cells and primary NSCs,
respectively. The immortalized murine NSC line, C17.2, was
generously provided by Professor Wei Lin Jin (Shanghai Jiao Tong
University, Shanghai, China). The C17.2 cells were cultured in high
glucose Dulbecco's modified Eagle's medium (DMEM) containing 10%
fetal bovine serum (FBS), 5% horse serum, and 2 mM glutamine. The
murine MG cell line, BV-2, was a gift from Professor Ai Min Ji
(Southern Medical University, Guangzhou, China). The BV-2 cells
were propagated in flasks containing DMEM supplemented with 10%
FBS, at 37°C with 5% CO2. Cells in the exponential
growth phase were used for the experiments. The morphology of C17.2
cells was observed under a microscope.
The MG cells were cultured in a Transwell system
(3450; Corning Corp., Corning, NY, USA) that was placed above the
NSC layer. The NSCs and MG cells shared the same medium, but had no
direct cell-cell interactions, as the cells were physically
separated with a polyester membrane. The pore size of the Transwell
(0.4 µm) does not permit cell migration through the
membrane. The MG cells were then stimulated with Aβ1–42
(10 µM). In the presence of inserts containing MG cells, on
top of the NSC layer, we observed the response of the NSCs to the
diffusible factors secreted by the stimulated MG cells. C17.2 cells
in the control group were placed in the lower chamber and the upper
chamber was left empty. C17.2 cells were placed in the lower
chamber in a final concentration of 10 µM Aβ1–42
(group of C17.2 cells directly exposed to Aβ1–42).
Preparation of Aβ1–42
Aβ1–42 (US Biological, Salem, MA, USA)
was dissolved in 35% acetonitrile and then further diluted to 10 mM
with phosphate-buffered saline (PBS). The peptide solution was
subsequently incubated at 37°C for 72 h to promote aggregation and
fibrillization, followed by freezing and storage at −20°C. The
final working concentration was 10 µM Aβ1–42
diluted in culture medium.
Recombinant adenoviral vector for Hsp75
overexpression
A recombinant adenoviral vector overexpressing Hsp75
(Ad-Hsp75-GFP) and a negative control adenoviral vector (Ad-GFP)
were produced by HanBio (Shanghai, China). The vectors encoded the
green fluorescent protein (GFP) sequence, which served as a marker
gene. When the C17.2 cells reached 60% confluence, recombinant
adenovirus was added at a multiplicity of infection of 100 for 36
h. The cells were observed under a fluorescence microscope
(Olympus, Tokyo, Japan) to detect the presence of fluorescent
protein. All the recombinant adenoviruses were tested for Hsp75
protein expression in the C17.2 cells by western blot analysis.
Isolation of mitochondria
Mitochondria were isolated from the NSCs (C17.2
cells) using the Mitochondria Isolation kit for cultured cells
(Thermo Fisher Scientific, Waltham, MA, USA) according to the
manufacturer's instructions. Briefly, the samples were harvested,
and this was followed by the addition of 800 µl mitochondria
isolation reagent A, 10 µl mitochondria isolation reagent B
and 500 µl mitochondrial isolation reagent C (from
Mitochondria Isolation kit for cultured cells; Thermo Fisher
Scientific, Waltham, MA, USA). Cytosolic and mitochondrial proteins
were harvested according to the manufacturer's instructions and
then concentrated using an Amicon® Ultra-0.5 Centrifugal
Filter device (Millipore, Billerica, MA, USA).
Cell viability assays
The apoptotic rates of the NSCs (C17.2 cells) were
examined using an Annexin V-APC/7-AAD apoptosis detection kit (BD
Pharmingen, San Diego, CA, USA) in accordance with the
manufacturer's instructions. Following treatment with
Aβ1–42 for 36 h, the cells were harvested and
resuspended in binding buffer at a density of 1×106
cells/ml. The cells were mixed with 5 µl Annexin V-APC and 5
µl 7-AAD and then incubated for 15 min at room temperature
in the dark. Finally, the cells were analyzed using a flow
cytometer (BD Pharmingen).
Western blot analysis and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
Following treatment, the C17.2 cells were collected
and lysed for protein expression analysis. The protein
concentration was determined using the BCA protein assay kit
(Thermo Fisher Scientific) with bovine serum albumin as a standard.
Total protein equivalents were denatured and separated on a 10–15%
sodium dodecyl sulfate (SDS)-polyacrylamide gel, and the proteins
were then transferred onto Immobilon-P Transfer membranes
(Millipore). The membranes were blocked with 5% non-fat dry milk
for 1 h at room temperature and then incubated with primary
antibodies. The primary antibodies were as follows: Hsp75
(ab151239; Abcam, Cambridge, MA, USA), CypD (A3208; ABclonal,
Cambridge, MA, USA), Cytc (ab133504; Abcam), caspase-3 (WL0146;
Wanleibio, Shenyang, China), COX IV (internal control for
mitochondrial protein; ab140643; Abcam), glyceraldehyde 3-phosphate
dehydrogenase (GAPDH; KC-5G5) and β-actin (KC-5A08; both from
KangChen Bio-tech, Shanghai, China) and α-tubulin (AT819; Beyotime
Institute of Biotechnology, Haimen, China). The secondary
antibodies were either goat anti-rabbit (BA1054; Boster Biotech
Co., Ltd., Wuhan, China) or mouse (bs-0296G-HRP; Bioss Co.,
Beijing, China) immunoglobulin G (IgG). Horseradish-conjugated
secondary antibody labeling was detected using enhanced
chemiluminescence (Thermo Fisher Scientific) according to the
manufacturer's instructions. The optical density of the bands was
analyzed with ImageJ software.
For RT-qPCR, total RNA was extracted from the C17.2
cells using TRIzol® reagent (Invitrogen, Carlsbad, CA,
USA). The RNA (2 µg) was reverse transcribed into cDNA using
ReverTra Ace® qPCR RT Master Mix with the gDNA Remover
kit (Toyobo, Osaka, Japan). Real-time monitoring of the PCR
amplification of cDNA was detected using a real-time PCR detection
system (ABI PRISM® 7500 sequence detection system). PCR
amplification was conducted as follows: 40 cycles at 95°C for 15
sec and an extension at 60°C for 32 sec. The data were quantified
using the 2−ΔΔCt method. The following primers were
used: mouse CypD, 5′-AACTTCAGAGCCCTATGCA-3′ (forward) and
5′-TCCTGTGCCATTGTGGTT-3′ (reverse); mouse caspase-3,
5′-GTTCATCCAGTCCCTTTGC-3′ (forward) and 5′-TGTTAACGCGAGTGAGAATG-3′
(reverse); mouse β-actin, 5′-GCTTCTAGGCGGACTGTTAC-3′ (forward) and
5′-CCATGCCAATGTTGTCTCTT-3′ (reverse). β-actin (a housekeeping gene)
was used as an internal reference.
Immunocytochemistry
Cell cultures in 96-well plates were examined by
fluorescence immunocytochemistry. The cells were washed twice in
PBS and then fixed in 4% paraformaldehyde for 30 min at room
temperature. The cells were then washed twice with PBS and
permeabilized with 0.3% Triton X-100 for 20 min. Non-specific
binding sites were blocked via incubation with 3% goat serum for 40
min. The cells were then incubated with a mouse monoclonal primary
antibody against nestin (a marker of NSCs) (ab6142; Abcam) at a
dilution of 1:200 in 1% BSA overnight at 4°C. On the following day,
the cells were washed 3 times for 5 min each time and incubated
with rhodamine-conjugated goat anti-mouse secondary antibody
(SA00006-1; Proteintech, Chicago, IL, USA) at room temperature for
1.5 h in the dark; this was followed by 2 washes in PBS.
Subsequently, the nuclei were counterstained with
4′,6-diamidino-2-phenylindole (DAPI), and the stained cells were
observed under a fluorescence microscope (Olympus AX80;
Olympus).
Mitochondrial membrane potential
The mitochondrial membrane potential was monitored
using the fluorescent dye, tetramethylrhodamine ethyl ester (TMRE;
Molecular Probes, Eugene, OR, USA). Following treatment, the cells
were washed with PBS and incubated in the dark with 100 nM TMRE at
37°C for 15 min. The cells were then washed 3 times with PBS and
observed under a fluorescence microscope (Olympus AX80;
Olympus).
Statistical analysis
Data are expressed as the means ± standard error of
the mean (SEM). Differences among groups were analyzed by one-way
ANOVA followed by the least significant difference (LSD) or
Dunnett's T3 post-hoc test. A p-value <0.05 was considered to
indicate a statistically significant difference.
Results
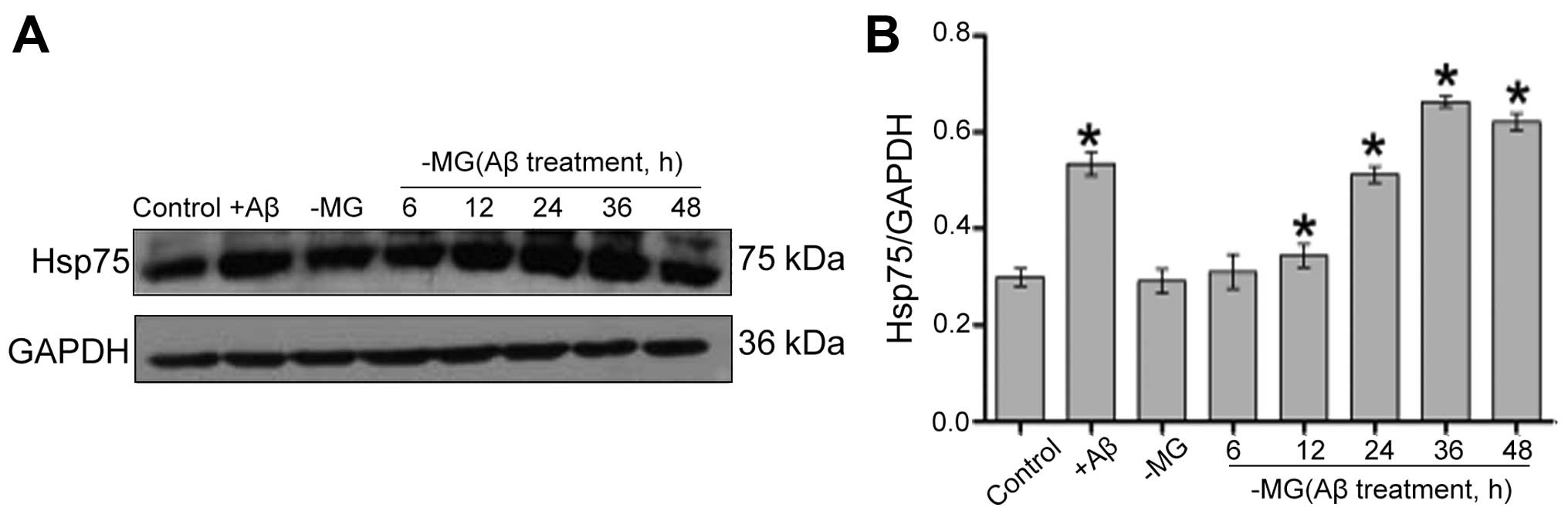
The stimulation of MG cells with
Aβ1–42 increases Hsp75 expression in C17.2 cells
Western blot analysis was used to measured the Hsp75
expression levels following treatment with Aβ1–42 in the
NSC-MG co-culture system. The level of Hsp75 increased in the C17.2
cells following direct exposure to Aβ1–42 (+Aβ group).
Although Hsp75 expression was low in the control group, the levels
of Hsp75 in the C17.2 cells gradually increased from 12 to 36 h
following treatment of the MG cells with Aβ1–42 in the
co-culture system (Fig. 1). Based
on this result, 36 h was used as the optimal treatment time in
subsequent experiments.
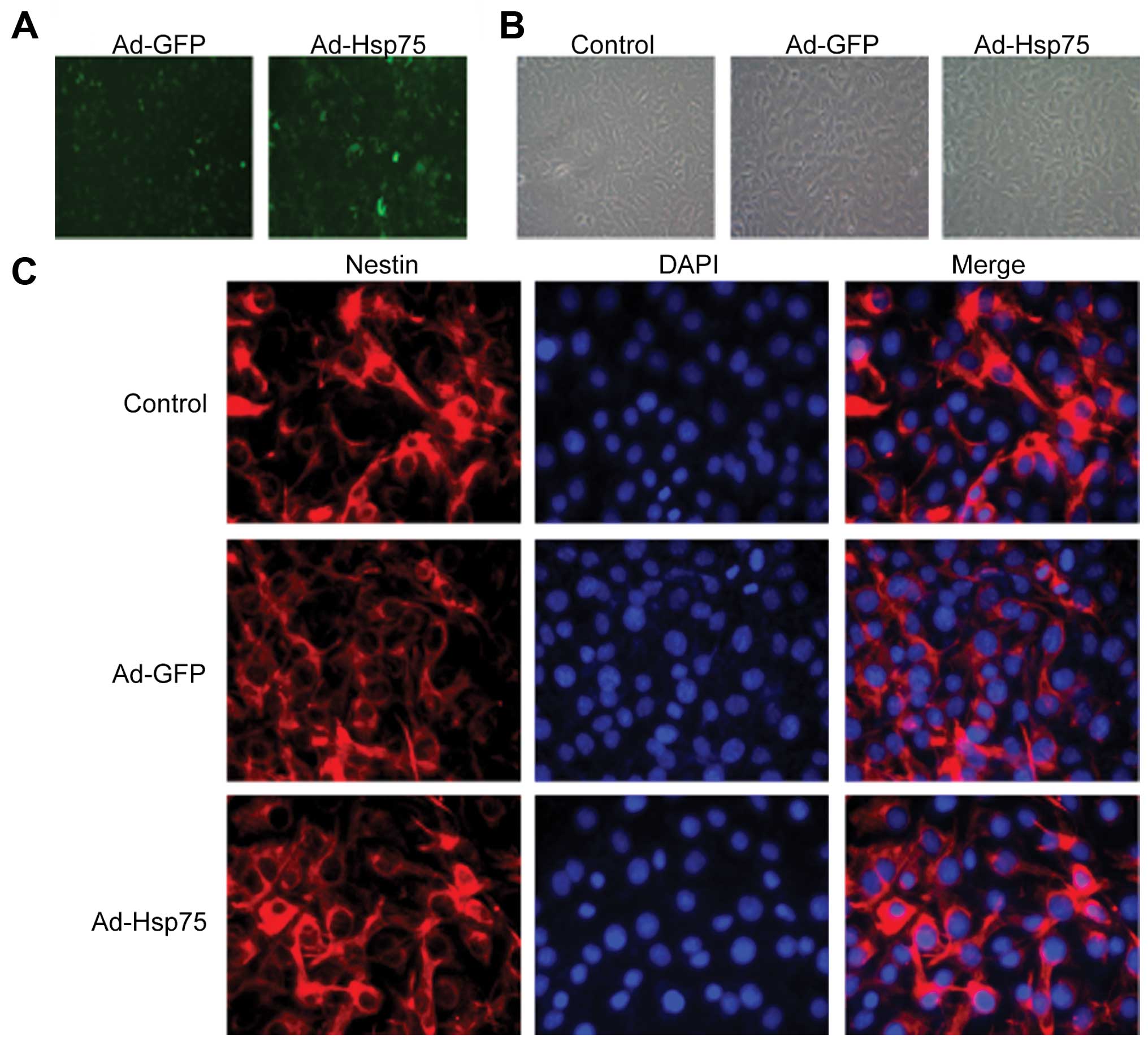
Verification of Ad-GFP and Ad-Hsp75
transfection
We constructed a recombinant adenoviral vector for
transfection using a GFP tag. Following 36 h of infection, the
infection efficiency was visualized by measuring GFP expression
(Fig. 2A). At 36 h following
transfection, C17.2 cells were shuttle-like or irregular shaped and
no obvious changes in the morphology of the C17.2 cells were
observed among these groups (Fig.
2B). In addition, we detected the expression of nestin (a NSC
marker) in 3 groups (control group, Ad-GFP group and Ad-Hsp75
group). As shown in Fig. 2C,
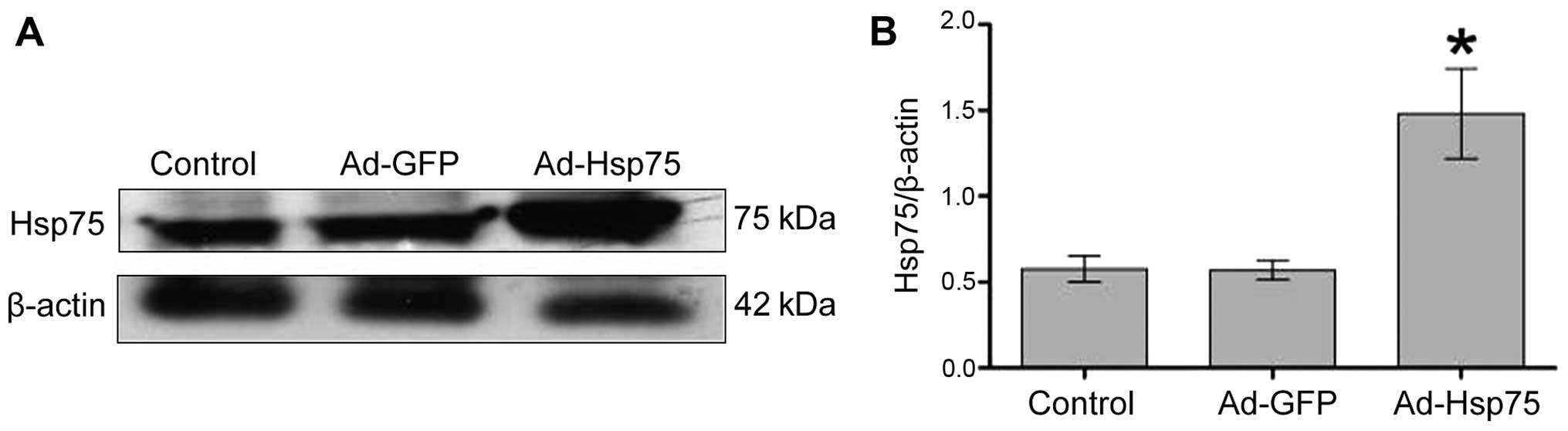
C17.2 NSCs were all nestin-positive. Additionally, Hsp75 expression
increased significantly in the C17.2 cells that were infected with
Ad-Hsp75 compared to those that were infected with the negative
control vector (Ad-GFP), and to the endogenous Hsp75 levels in the
control cells, as indicated by the results of the western blot
analysis (Fig. 3).
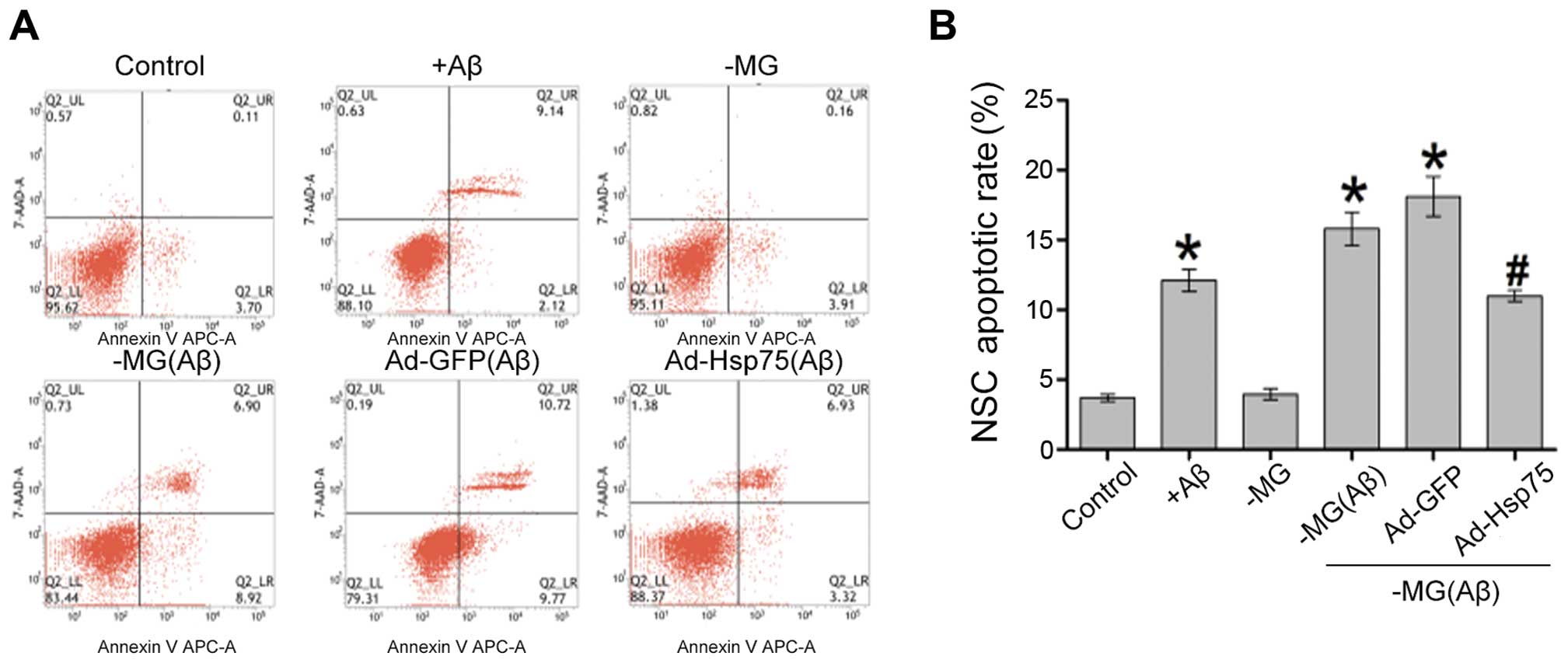
Hsp75 overexpression protects C17.2 cells
against soluble factor-induced neurotoxicity
As Hsp75 expression in the NSCs increased following
treatment with soluble factors (Aβ1–42), we thus
performed experiments to determine whether this increase in Hsp75
expression exerts a protective effect against the neurotoxicity
induced by diffusible soluble factors. To investigate the effects
of Hsp75 overexpression on neurotoxicity, a C17.2 cell line
overexpressing human Hsp75 was established. We then investigated
the effects of Hsp75 overexpression on soluble factor-induced
apoptosis in the C17.2 cells. Apoptotic-like cell death was assayed
by flow cytometry using Annexin V-APC and 7-AAD (Fig. 4). Following treatment of the C17.2
cells with Aβ1–42, the percentage of apoptotic cells
increased to approximately 12% compared with only 4% in the control
group (p<0.05), which demonstrated that Aβ1–42 at a
concentration of 10 µM induced cell apoptosis. In the
co-culture system, the proportion of apoptotic NSCs was
approximately 4% in the NSC-MG cell co-culture system without
Aβ1–42 treatment (-MG group), and there was no apparent
difference (p>0.05) between the cells in the NSC-MG co-culture
system without treatment with Aβ1–42 and the control
group. When the MG cells were treated with Aβ1–42 for 36
h in the co-culture system, the percentage of early and late
apoptotic cells markedly increased, and also increased in the
Ad-GFP group, compared with the control group (p<0.05) (Fig. 4B). By contrast, Hsp75
overexpression markedly prevented MG-derived soluble factor-induced
apoptosis. These results indicate that Hsp75 overexpression
provides protection against MG-derived soluble factor-induced
neurotoxicity in NSCs.
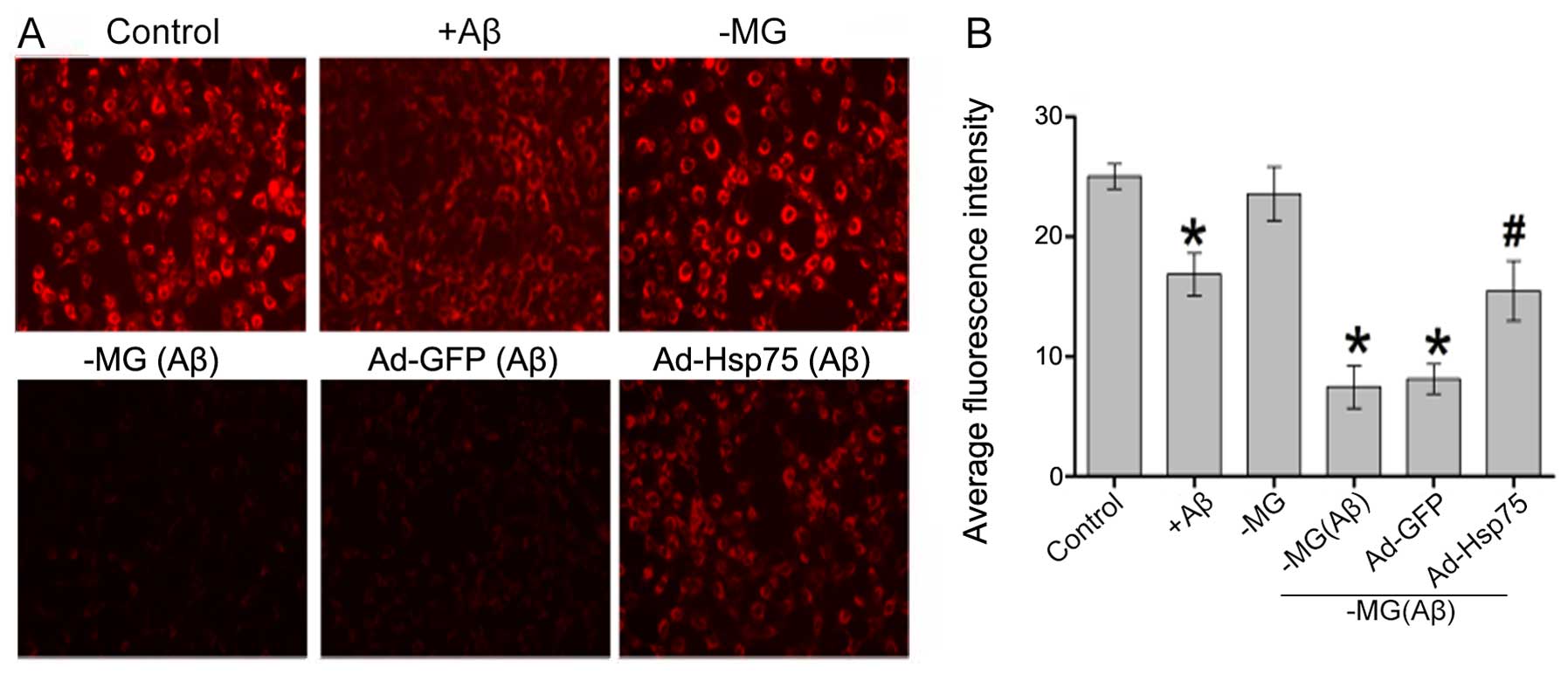
Hsp75 overexpression attenuates soluble
factor-induced mitochondrial dysfunction
As mitochondrial membrane potential is a key
indicator of cell apoptosis, we speculated that soluble
factor-induced neurotoxicity correlates with mitochondrial
dysfunction. To examine the effects of Hsp75 overexpression on
mitochondrial dysfunction, mitochondrial membrane potential was
monitored using the fluorescent dye, TMRE. As shown in Fig. 5, compared with the control group,
mitochondrial membrane potential significantly decreased in the
C17.2 cells that were exposed directly to Aβ1–42.
However, there was no apparent difference between the NSC-MG cell
group (in the co-culture system) without treatment with
Aβ1–42 and the control group. Additionally, in the
co-culture system, mitochondrial membrane potential in the presence
of Hsp75 overexpression was protected against diffusible soluble
factor-mediated neurotoxicity. By contrast, the TMRE signal was
reduced in the NSC-MG cells treated with Aβ1–42 and in
the adenoviral vector-infected C17.2 cells (Ad-GFP group). The
above-mentioned results indicate that Hsp75 overexpression can
effectively prevent soluble factor-induced mitochondrial
dysfunction.
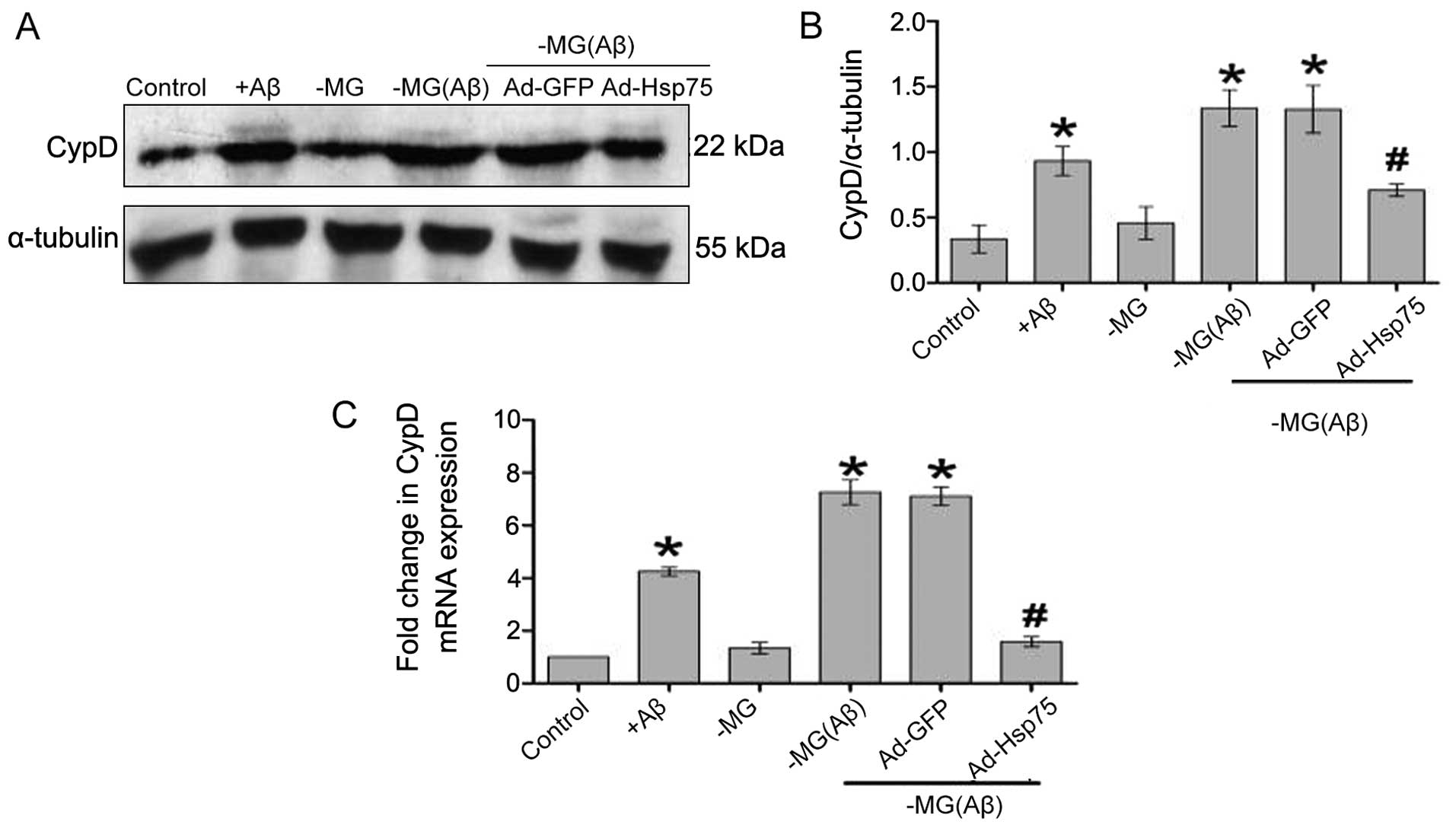
Hsp75 overexpression reduces soluble
factor-induced CypD-dependent mPTP opening
Previous studies have confirmed that mitochondrial
membrane potential dissipation is accompanied by an increase in
mPTP opening and indirectly reflects the state of mPTP opening
(24–26). Additionally, it has been
demonstrated that CypD is an important regulatory component of mPTP
and plays a vital role in regulating mPTP opening (27–29). Increasing evidence suggests that
an increased abundance of CypD directly reflects the state of mPTP
opening and that CypD-mediated mPTP opening is closely related to
cell viability (30–32). Thus, in order to determine whether
Hsp75 overexpression protects NSCs against MG-derived soluble
factor-induced neurotoxicity by regulating mPTP opening, we
estimated the relative abundance of CypD (Fig. 6). Briefly, compared with the
control group, the CypD expression levels were upregulated in the
C17.2 cells following direct exposure to Aβ1–42, which
is consistent with the results of a previous study (33). According to the results of western
blot analysis, the CypD levels in the soluble factor-stimulated
group in our co-culture system (NSC-MG cell group with
Aβ1–42 treatment and the Ad-GFP group) were
significantly increased as compared to the non-soluble factor group
(control group). By contrast, the CypD levels were downregulated in
response to Hsp75 overexpression. We observed similar results at
the mRNA level.
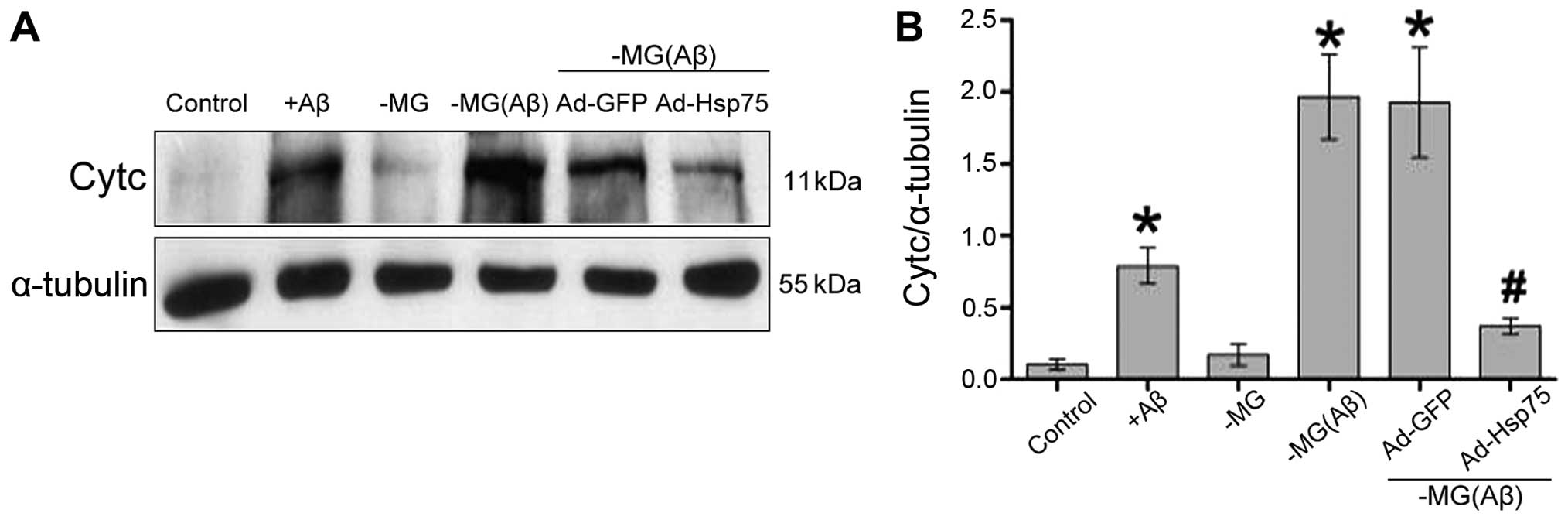
Hsp75 overexpression prevents the soluble
factor-induced release of Cytc
The opening of the mPTP ultimately results in
mitochondrial membrane potential dissipation, mitochondrial
swelling, rupture of the outer membrane and the release of the
pro-apoptotic factor, Cytc, from the mitochondrial matrix into the
cytoplasm (13,34). We hypothesized that Hsp75
overexpression may reduce the soluble factor-induced release of
Cytc.
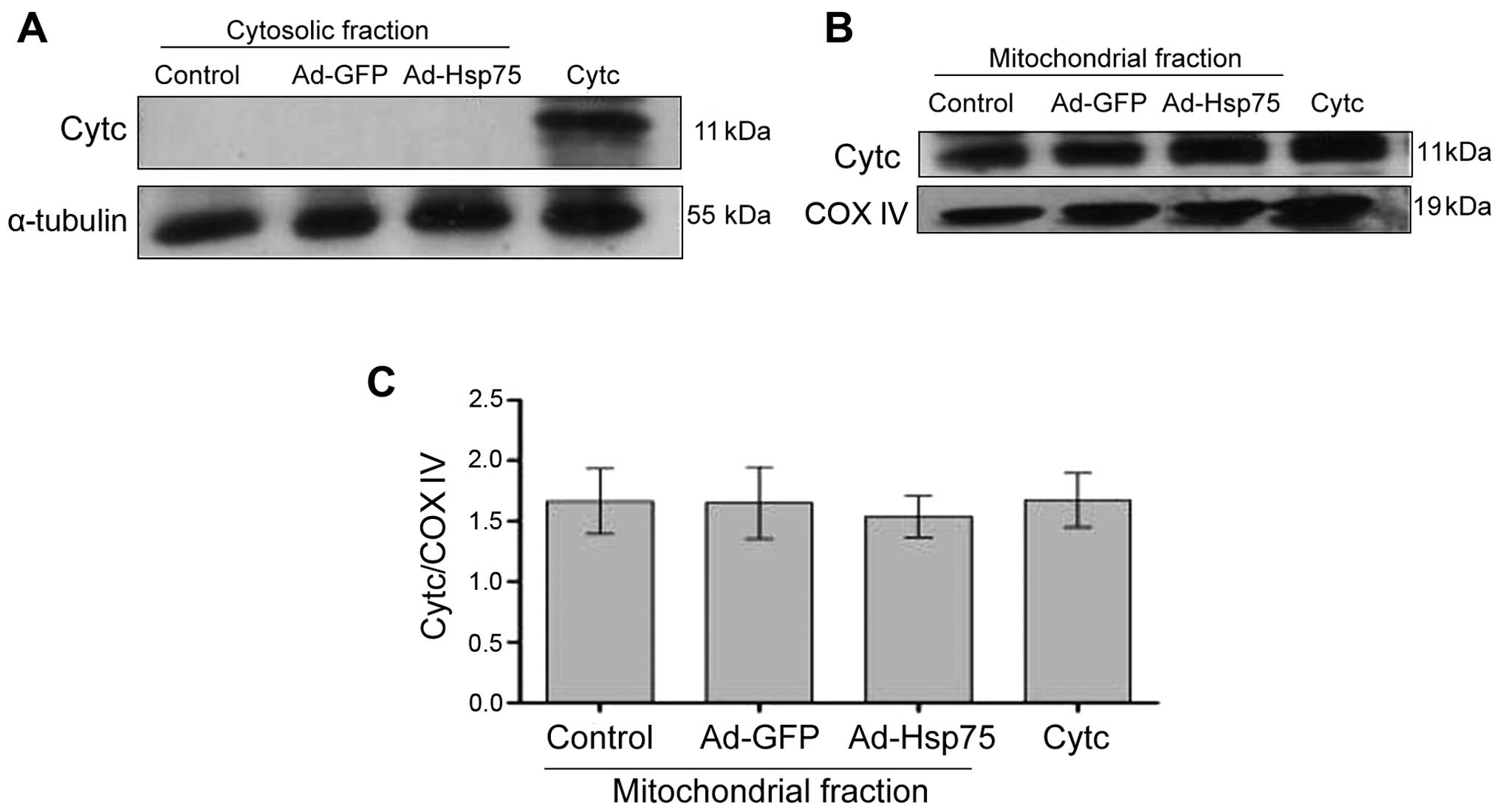
The total amount of Cytc expression was measured by
western blot analysis using NSCs (C17.2 cells) transfected with
adenovirus. In the cytosolic fraction, no signal was observed in
the NSCs (Fig. 7A), which is
proof of the successful separation of the cytosolic fraction. In
the mitochondrial fraction, there were no significant differences
among the groups, and a strong signal was detected (Fig. 7B and C). These results indicated
that the mitochondria were successfully separated, and that the
total amount of Cytc protein expression was not altered following
transfection with adenovirus.
To further determine whether Hsp75 overexpression
reduces the soluble factor-induced release of Cytc, the release of
mitochondrial Cytc into the cytosol was assessed by western blot
analysis. Indeed, compared with the control group, the levels of
Cytc increased significantly in the C17.2 cells that were directly
exposed to Aβ1–42. However, there was no clear
difference between the NSC-MG cells (in our co-culture system; MG
group) without Aβ1–42 treatment and the control group.
In addition, in the co-culture system, soluble factors
significantly increased the release of Cytc in the NSC-MG cells
treated with Aβ1–42 [MG(Aβ) group] and in the
vector-infected C17.2 cells (Ad-GFP group) compared to the control
group, whereas cells overexpressing Hsp75 exhibited a marked
decrease in the release of Cytc. These data demonstrate that Hsp75
overexpression prevents the release of Cytc induced by soluble
factors (Fig. 8).
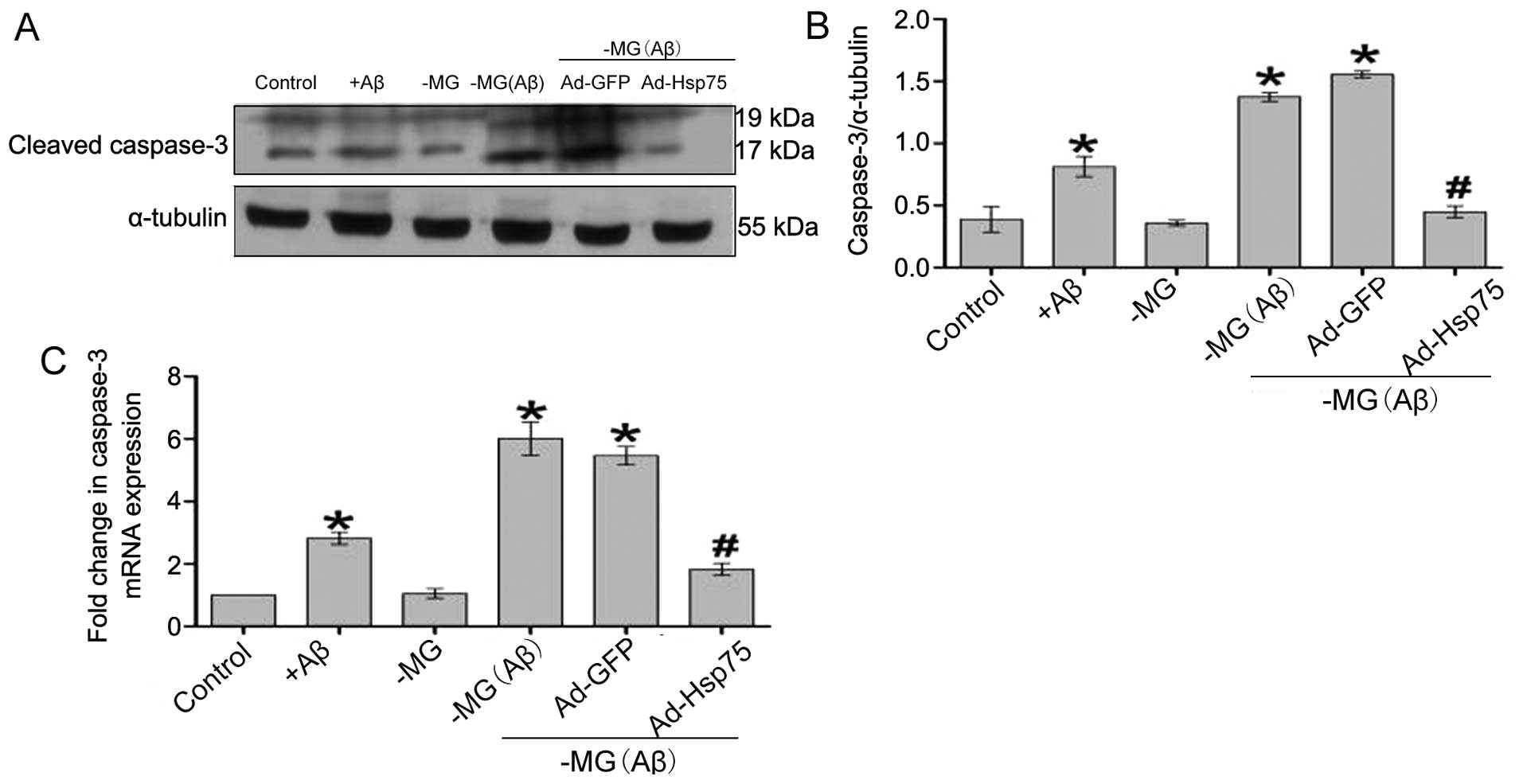
Hsp75 overexpression prevents the soluble
factor-induced activation of caspase-3
It has previously been demonstrated that
mitochondrial Cytc release promotes apoptotic signaling and the
activation of caspase-dependent apoptotic pathways (13,35). Caspase-3 activation results in DNA
breakage, nuclear chromatin condensation and ultimately leads to
apoptosis. In the present study, the results obtained for caspase-3
activation were similar to the above-mentioned results obtained for
Cytc release (Fig. 9). Thus,
Hsp75 overexpression also led to a significant attenuation of
soluble factor-induced caspase-3 activation, indicating that Hsp75
overexpression protects C17.2 cells against soluble factor-induced
neurotoxicity by inhibiting the mitochondrial caspase-dependent
apoptosis pathway.
Discussion
In the present study, we verified that C17.2 cells
were nestin-positive and had relatively high transfection rates
with the recombinant adenoviral vector. In addition, we found that
the morphology of the C17.2 cells was not altered following
transfection. Therefore, the use of a recombinant adenoviral vector
is a suitable approach for determining whether Hsp75 provides
protection against MG-derived soluble factor-induced neurotoxicity.
Moreover, we demonstrated that treatment with Aβ1–42 in
a co-culture system induced a time-dependent increase in the
protein levels of Hsp75. Thus, MG-derived soluble factors induce an
increase in the levels of Hsp75. Our data also indicate that the
overproduction of Hsp75 inhibits soluble factor-induced
depolarization of mitochondrial membrane potential and cell death,
reducing the abundance of mPTP, regulating CypD protein and
inhibiting mitochondrial Cytc release and caspase-3 activity. In
short, MG-derived soluble factors induce mPTP opening, and
concurrently, the pore opening results in the loss of mitochondrial
membrane potential, the release of Cytc into the cytosol and,
finally, the activation of a caspase cascade, leading to apoptosis.
These results suggest that Hsp75 overexpression protects against
soluble factor-induced apoptosis by blocking the consequent
apoptotic cascade which is mediated by mitochondria, as shown by
the inhibition of CypD-dependent mPTP opening, reduced
mitochondrial Cytc release and decreased caspase-3 activity.
Collectively, to the best of our knowledge, these results
constitute the first evidence (relating to AD) that the protective
effects of Hsp75 overexpression on soluble factor-induced
neurotoxicity are attributable to the role which Hsp75 plays in
maintaining mitochondrial function.
Hsp75 is a heat shock protein, and like other Hsp70
members, it has been shown to be upregulated by diverse damaging
stimuli: a previous study demonstrated that hypoxic injury
increases Hsp75 levels in a time-dependent manner in cardiomyocytes
(21). Moreover, deferoxamine, an
iron chelator, has been shown to markedly decrease Hsp75 levels in
a time- and dose-dependent manner in a normal human hepatocyte cell
line (36). However, the changes
in Hsp75 expression in NSCs following exposure to MG-derived
soluble factors remain unclear. Based on the results of the present
study, we concluded that soluble factors induce a time-dependent
increase in Hsp75 levels in NSCs.
Although we verified that soluble factors induce
increased levels of Hsp75, the effect of the increase in Hsp75
remains to be explored. Therefore, we constructed NSCs
overexpressing human Hsp75 protein in order to determine whether
the increase in Hsp75 is a protective reaction in NSCs. Previous
studies have demonstrated that Hsp75 is a mitochondrial molecular
chaperone and anti-apoptotic protein; Hsp75 overexpression can
suppress apoptosis induced by hypoxia in cardiomyocytes (21), ischemic injury in primary
astrocytes (37), glucose
deprivation in PC12 cells (16)
and CHL cells (38), mercury
exposure in renal cells (39),
arsenite exposure in lung epithelial cells (40), iron chelation in a human
hepatocyte cell line (36) and
Aβ1–42-induced neurotoxicity in the SH-SY5Y cell line
(22). We also demonstrated in
the present study that Hsp75 overexpression exerts a protective
effect against MG-derived soluble factor-induced neurotoxicity in
an NSC cell line (C17.2 cells).
Previous biochemical studies have indicated that
diverse damaging stimuli induce mPTP formation and that mPTP
formation is closely related to AD (41,42). The mPTP consists of the
voltage-dependent anion channel (VDAC), the adenine nucleotide
translocator (ANT) and CypD. Although its exact structure remains
controversial, CypD is generally accepted as a key regulatory
component of the mPTP. Increasing evidence indicates that CypD
contributes to the formation of mPTP and that mPTP formation
results in severe mitochondrial membrane potential dissipation and
the release of pro-apoptotic factors, which eventually leads to
cell damage. In addition, it should be noted that Hsp75 is a
mitochondrial molecular chaperone that maintains mitochondrial
functions and is associated with the mPTP. As previously shown by
Qu et al (23), Hsp75
over-expression improves cell viability and decreases apoptosis by
regulating mPTP opening. Similarly, Hsp75 overexpression also
protects cells against hypoxic injury by reducing mPTP opening
(21). Therefore, we hypothesized
that Hsp75 overexpression may also exert a protective effect during
exposure to MG-derived soluble factors by regulating CypD-dependent
mPTP opening in NSCs. In the present study, we verified that the
increase in CypD expression was prevented by Hsp75 overexpression,
and thus mPTP opening was decreased and the apoptotic pathways were
subsequently inhibited.
In conclusion, to the best of our knowledge, our
results provide the first evidence that Hsp75 is an effective
protective protein against soluble factor-induced neurotoxicity and
that the protective effects are related to the elimination of
mitochondrial dysfunction. However, our experiments were limited to
the assessment of Hsp75 overexpression and a cellular model of AD,
and therefore, further studies are required to determine whether
Hsp75 overexpression provides significant protection against AD in
animal models. In addition, the silencing of Hsp75 expression in
vivo and in vitro needs be assessed in future studies.
Finally, further investigations are warranted to examine whether
soluble factors promote CypD translocation to the inner membrane to
trigger mPTP opening and whether Hsp75 overexpression prevents CypD
translocation. It may thus be possible to develop a novel
therapeutic target for the treatment of AD based on the
manipulation of Hsp75 expression.
Acknowledgments
This study was supported in part by grants from the
National Natural Science Foundation of China (no. 30973162) and the
Natural Science Foundation of Guangdong Province (no.
S2013010015546). In addition, this study was supported in part by
the Guangdong Provincial Key Laboratory of Malignant Tumor
Epigenetics and Gene Regulation, the Sun Yat-Sen Memorial Hospital
and Sun Yat-Sen University.
References
|
1
|
Mattson MP: Pathways towards and away from
Alzheimer's disease. Nature. 430:631–639. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gómez-Isla T, Hollister R, West H, Mui S,
Growdon JH, Petersen RC, Parisi JE and Hyman BT: Neuronal loss
correlates with but exceeds neurofibrillary tangles in Alzheimer's
disease. Ann Neurol. 41:17–24. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sadigh-Eteghad S, Sabermarouf B, Majdi A,
Talebi M, Farhoudi M and Mahmoudi J: Amyloid-Beta: a Crucial Factor
in Alzheimer's Disease. Med Princ Pract. 2014.PubMed/NCBI
|
|
4
|
McGeer PL and McGeer EG: Targeting
microglia for the treatment of Alzheimer's disease. Expert Opin
Ther Targets. 19:497–506. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Floden AM and Combs CK: Microglia
demonstrate age-dependent interaction with amyloid-β fibrils. J
Alzheimers Dis. 25:279–293. 2011.
|
|
6
|
Westerlund U, Moe MC, Varghese M,
Berg-Johnsen J, Ohlsson M, Langmoen IA and Svensson M: Stem cells
from the adult human brain develop into functional neurons in
culture. Exp Cell Res. 289:378–383. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Temple S: The development of neural stem
cells. Nature. 414:112–117. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Henn A, Lund S, Hedtjärn M, Schrattenholz
A, Pörzgen P and Leist M: The suitability of BV2 cells as
alternative model system for primary microglia cultures or for
animal experiments examining brain inflammation. ALTEX. 26:83–94.
2009.PubMed/NCBI
|
|
9
|
Lundqvist J, El Andaloussi-Lilja J,
Svensson C, Gustafsson Dorfh H and Forsby A: Optimisation of
culture conditions for differentiation of C17.2 neural stem cells
to be used for in vitro toxicity tests. Toxicol In Vitro.
27:1565–1569. 2013. View Article : Google Scholar
|
|
10
|
Liu WG, Lu GQ, Li B and Chen SD:
Dopaminergic neuroprotection by neurturin-expressing c17.2 neural
stem cells in a rat model of Parkinson's disease. Parkinsonism
Relat Disord. 13:77–88. 2007. View Article : Google Scholar
|
|
11
|
Zamzami N, Larochette N and Kroemer G:
Mitochondrial permeability transition in apoptosis and necrosis.
Cell Death Differ. 12(Suppl 2): 1478–1480. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kim HS, Lee JH, Lee JP, Kim EM, Chang KA,
Park CH, Jeong SJ, Wittendorp MC, Seo JH, Choi SH and Suh YH:
Amyloid beta peptide induces cytochrome C release from isolated
mitochondria. Neuroreport. 13:1989–1993. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Brustovetsky N, Brustovetsky T, Jemmerson
R and Dubinsky JM: Calcium-induced cytochrome c release from CNS
mitochondria is associated with the permeability transition and
rupture of the outer membrane. J Neurochem. 80:207–218. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bhattacharyya T, Karnezis AN, Murphy SP,
Hoang T, Freeman BC, Phillips B and Morimoto RI: Cloning and
subcellular localization of human mitochondrial hsp70. J Biol Chem.
270:1705–1710. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lee AS: The glucose-regulated proteins:
stress induction and clinical applications. Trends Biochem Sci.
26:504–510. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liu Y, Liu W, Song XD and Zuo J: Effect of
GRP75/mthsp70/PBP74/mortalin overexpression on intracellular ATP
level, mitochondrial membrane potential and ROS accumulation
following glucose deprivation in PC12 cells. Mol Cell Biochem.
268:45–51. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Carette J, Lehnert S and Chow TY:
Implication of PBP74/mortalin/GRP75 in the radio-adaptive response.
Int J Radiat Biol. 78:183–190. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Massa SM, Longo FM, Zuo J, Wang S, Chen J
and Sharp FR: Cloning of rat grp75, an hsp70-family member, and its
expression in normal and ischemic brain. J Neurosci Res.
40:807–819. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tsuchiya D, Hong S, Matsumori Y, Shiina H,
Kayama T, Swanson RA, Dillman WH, Liu J, Panter SS and Weinstein
PR: Overexpression of rat heat shock protein 70 is associated with
reduction of early mitochondrial cytochrome c release and
subsequent DNA fragmentation after permanent focal ischemia. J
Cereb Blood Flow Metab. 23:718–727. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Rokutan K, Hirakawa T, Teshima S, Nakano
Y, Miyoshi M, Kawai T, Konda E, Morinaga H, Nikawa T and Kishi K:
Implications of heat shock/stress proteins for medicine and
disease. J Med Invest. 44:137–147. 1998.PubMed/NCBI
|
|
21
|
Xiang F, Huang YS, Shi XH and Zhang Q:
Mitochondrial chaperone tumour necrosis factor receptor-associated
protein 1 protects cardiomyocytes from hypoxic injury by regulating
mitochondrial permeability transition pore opening. FEBS J.
277:1929–1938. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Qu M, Zhou Z, Xu S, Chen C, Yu Z and Wang
D: Mortalin overexpression attenuates beta-amyloid-induced
neurotoxicity in SH-SY5Y cells. Brain Res. 1368:336–345. 2011.
View Article : Google Scholar
|
|
23
|
Qu M, Zhou Z, Chen C, Li M, Pei L, Yang J,
Wang Y, Li L, Liu C, Zhang G, et al: Inhibition of mitochondrial
permeability transition pore opening is involved in the protective
effects of mortalin overexpression against beta-amyloid-induced
apoptosis in SH-SY5Y cells. Neurosci Res. 72:94–102. 2012.
View Article : Google Scholar
|
|
24
|
Sugrue MM, Wang Y, Rideout HJ,
Chalmers-Redman RM and Tatton WG: Reduced mitochondrial membrane
potential and altered responsiveness of a mitochondrial membrane
mega-channel in p53-induced senescence. Biochem Biophys Res Commun.
261:123–130. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Saotome M, Katoh H, Satoh H, Nagasaka S,
Yoshihara S, Terada H and Hayashi H: Mitochondrial membrane
potential modulates regulation of mitochondrial Ca2+ in
rat ventricular myocytes. Am J Physiol Heart Circ Physiol.
288:H1820–H1828. 2005. View Article : Google Scholar
|
|
26
|
Lee CS, Park SY, Ko HH, Song JH, Shin YK
and Han ES: Inhibition of MPP+-induced mitochondrial
damage and cell death by trifluoperazine and W-7 in PC12 cells.
Neurochem Int. 46:169–178. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Leung AW and Halestrap AP: Recent progress
in elucidating the molecular mechanism of the mitochondrial
permeability transition pore. Biochim Biophys Acta. 1777:946–952.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Halestrap AP: What is the mitochondrial
permeability transition pore? J Mol Cell Cardiol. 46:821–831. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gutiérrez-Aguilar M and Baines CP:
Structural mechanisms of cyclophilin D-dependent control of the
mitochondrial permeability transition pore. Biochim Biophys Acta.
pii: S0304-4165(14)00386-9. 2014.
|
|
30
|
Baines CP, Kaiser RA, Purcell NH, Blair
NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA,
Dorn GW, et al: Loss of cyclophilin D reveals a critical role for
mitochondrial permeability transition in cell death. Nature.
434:658–662. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Du H, Guo L, Fang F, Chen D, Sosunov AA,
McKhann GM, Yan Y, Wang C, Zhang H, Molkentin JD, et al:
Cyclophilin D deficiency attenuates mitochondrial and neuronal
perturbation and ameliorates learning and memory in Alzheimer's
disease. Nat Med. 14:1097–1105. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
32
|
Matas J, Young NT, Bourcier-Lucas C, Ascah
A, Marcil M, Deschepper CF and Burelle Y: Increased expression and
intra-mitochondrial translocation of cyclophilin-D associates with
increased vulnerability of the permeability transition pore to
stress-induced opening during compensated ventricular hypertrophy.
J Mol Cell Cardiol. 46:420–430. 2009. View Article : Google Scholar
|
|
33
|
Du H, Guo L, Zhang W, Rydzewska M and Yan
S: Cyclophilin D deficiency improves mitochondrial function and
learning/memory in aging Alzheimer disease mouse model. Neurobiol
Aging. 32:398–406. 2011. View Article : Google Scholar
|
|
34
|
Kim JS, He L and Lemasters JJ:
Mitochondrial permeability transition: a common pathway to necrosis
and apoptosis. Biochem Biophys Res Commun. 304:463–470. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yuan J, Murrell GA, Trickett A and Wang
MX: Involvement of cytochrome c release and caspase-3 activation in
the oxidative stress-induced apoptosis in human tendon fibroblasts.
Biochim Biophys Acta. 1641:35–41. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Im CN, Lee JS, Zheng Y and Seo JS: Iron
chelation study in a normal human hepatocyte cell line suggests
that tumor necrosis factor receptor-associated protein 1 (TRAP1)
regulates production of reactive oxygen species. J Cell Biochem.
100:474–486. 2007. View Article : Google Scholar
|
|
37
|
Voloboueva LA, Duan M, Ouyang Y, Emery JF,
Stoy C and Giffard RG: Overexpression of mitochondrial Hsp70/Hsp75
protects astrocytes against ischemic injury in vitro. J Cereb Blood
Flow Metab. 28:1009–1016. 2008. View Article : Google Scholar :
|
|
38
|
Zheng DH, Zuo J, Yang ZJ, Xia BL and Zhang
XN: grp75 protects cells from injuries caused by glucose
deprivation. Yi Chuan Xue Bao. 27:666–671. 2000.In Chinese.
|
|
39
|
Stacchiotti A, Ricci F, Rezzani R, Li
Volti G, Borsani E, Lavazza A, Bianchi R and Rodella LF: Tubular
stress proteins and nitric oxide synthase expression in rat kidney
exposed to mercuric chloride and melatonin. J Histochem Cytochem.
54:1149–1157. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lau AT, He QY and Chiu JF: A proteome
analysis of the arsenite response in cultured lung cells: evidence
for in vitro oxidative stress-induced apoptosis. Biochem J.
382:641–650. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Du H and Yan SS: Mitochondrial
permeability transition pore in Alzheimer's disease: cyclophilin D
and amyloid beta. Biochim Biophys Acta. 1802:198–204. 2010.
View Article : Google Scholar
|
|
42
|
Rao VK, Carlson EA and Yan SS:
Mitochondrial permeability transition pore is a potential drug
target for neurodegeneration. Biochim Biophys Acta. 1842:1267–1272.
2014. View Article : Google Scholar :
|