Introduction
Inducible nitric oxide synthase (iNOS or NOS2)
expression can be induced by a variety of inflammatory cytokines
(1). NOS2 exerts its functions by
catalyzing L-arginine to nitric oxide (NO), resulting in large
amounts of free radicals (2). The
primary function of NOS2 is macrophage-mediated non-specific immune
defense against intracellular bacteria (3) and certain tumor cells (4). In pathophysiological cases,
uncontrolled NOS2 released at the wrong sites has been associated
with allograft rejection (5),
neurodegeneration (6) and septic
shock (7).
As a signature cytokine of M1 macrophages,
interferon-γ (IFN-γ) plays a key role in activation, inflammation
and host defense against the intracellular pathogens of macrophages
(8). Moreover, IFN-γ is also an
inducer of NOS2, and promotes NOS2 expression by activating several
related transcription factors, such as nuclear factor-κB (NF-κB)
and signal transducer and activator of transcription 1 (STAT1) and
causing them to bind to the NOS2 promoter (9,10).
Interleukin-17 (IL-17) is a signature cytokine of
Th17 cells (11). Aberrant
production of IL-17 is associated with autoimmune and inflammatory
diseases: for example, delayed onset, reduced maximum severity
scores, and early recovery have been observed in IL-17-deficient
mice in a model of experimental autoimmune encephalomyelitis (EAE)
(12). Previously it was noted
that blockade of IL-17 in ApoE-deficient mice induces impaired
monocyte/macrophage recruitment to the aortic wall, leading to
reduced atherosclerosis (13). It
has been reported that IL-17 facilitates the expression of
inflammatory chemokines and cytokines through the NF-κB, p38
mitogen-activated protein kinase (MAPK) and extracellular
signal-regulated kinase (ERK) pathways (14,15). These secreted factors are known to
be responsible for the recruitment of monocytes and lymphocytes,
which eventually aggravate inflammation (16). Previous research has also revealed
that the inflammatory effect of IL-17 is partially related to the
synergistic effects it exerts with other cytokines, including tumor
necrosis factor-α (TNF-α) (17).
Increased IFN-γ has been noted in a mouse model of
atherosclerosis, where IL-17 plays a proinflammatory role (18). Moreover, it has also been noted
that IL-17 synergistically acts with IFN-γ to induce an
inflammatory response in vascular smooth muscle cells by enhancing
the expression of inflammatory cytokines and chemokines (19). In the present study, we aimed to
investigate whether synergistic effects between IL-17 and IFN-γ in
the inflammatory response could be noted in macrophages, especially
in relation to NOS2 expression.
Materials and methods
Reagents
The recombinant murine IFN-γ and IL-17 were
purchased from PeproTech (Rock Hill, NJ, USA). The STAT1 inhibitor
fludarabine (Flu), JAK inhibitor AG-490, NF-κB inhibitor SN50,
phosphorylated (p-)p38 inhibitor SB203580, p-ERK1/2 inhibitor
PD98059 and also antibody against p-p38 MAPK(Thr180/Tyr182)
(sc-17852-R) were all supplied by Santa Cruz Biotechnology, Inc.
(Shanghai, China). The antibodies against NOS(pan) (#2977),
p-STAT1(Y701) (#7649), p-STAT1(S727) (#8826),
p-ERK1/2(Thr202/Tyr204) (#4370) and p65 (#8242) were all purchased
from Cell Signaling Technology (Danvers, MA, USA). Antibody against
STAT1 (#21044-1) was supplied by Signalway Antibody (Baltimore, MD,
USA). Antibodies against β-actin (20536-1-AP) and histone H3
(17168-1-AP) were obtained from Proteintech Group (Wuhan, China).
The antibody against IκBα (6A920) was purchased from Novus
Biologicals (Littleton, CO, USA). Horseradish peroxidase-conjugated
goat anti-rabbit (A21020) and goat anti-mouse (A21010) secondary
antibodies were both obtained from Abbkine (Redlands, CA, USA).
Cell culture
A murine macrophage cell line (RAW 264.7) was
obtained from the American Type Culture Collection (Manassas, VA,
USA) and maintained in Dulbecco's modified Eagle's medium (DMEM)
supplemented with 10% fetal bovine serum (both from Life
Technologies, Carlsbad, CA, USA). Cells were incubated in
serum-free medium overnight before treatment. In order to explore
the roles of the signaling factors involved, cells were pretreated
with the previously mentioned specific inhibitors for specified
periods of time prior to cytokine exposure.
Animal and treatment protocol
All animal procedures were approved by the
Institutional Ethics Committee for Animal Experiments of Xi'an
Jiaotong University. All surgical and experimental procedures were
carried out in accordance with the National Institutes of Health
Guide for the Care and Use of Laboratory Animals (NIH publications
number 23-80) revised in 2011. Animal maintenance was in accordance
with The National Institutes of Health guidelines on care and use
of animal subjects in research (National Academy of Science, 1996).
All mice had readily access to normal chow and were housed
collectively on a 12 h light-dark cycle (lights on from 8:00 a.m.
to 8:00 p.m.) in a facility accredited by the Association for the
Assessment and Accreditation of Laboratory Animal Care. All
experiments were carried out using 6 to 8-week-old C57BL/6J mice
purchased from the Center of Laboratory Animal Science of Xi'an
Jiaotong University (Shaanxi, China) weighing 20–30 g. Four mice
were intraperitoneally injected with Brewer thioglycollate medium
48 h before being sacrificed by cervical dislocation. The
peritoneal macrophages were acquired by intraperitoneal lavage of
the sacrificed mice with DMEM on a clean bench.
NO detection in medium
RAW 264.7 cells were stimulated with cytokines for
24 or 48 h. Nitrite concentration, a measure of NO production, was
determined by Griess reaction using a QuantiChrom™ Nitric Oxide
Assay kit (BioAssay Systems, Hayward, CA, USA). Briefly, 100
µl supernatant was mixed with an equal volume of Griess
reagent (1% sulfanilamide, 5% phosphoric acid and 0.1%
N-(1-naphthyl)-ethylenediamine), and the mixture was incubated at
room temperature for 10 min. Absorbance was measured at a
wavelength of 540 nm using a microplate reader (Molecular Devices
Corporation, Sunnyvale, CA, USA). Nitrite concentration was then
calculated from a NaNO2 standard curve.
Quantitative PCR (qPCR)
Total RNA was extracted from cells using TRIzol
reagent (Invitrogen, Carlsbad, CA, USA) according to the
manufacturer's instructions. First-strand cDNA was synthesized
using the RevertAid™ First Strand cDNA Synthesis kit (Fermentas,
Vilnius, Lithuania). qPCR was performed on the IQ5™ Multicolor
real-time PCR Detection System (Bio-Rad, Hercules, CA, USA) using
SYBR® Select Master Mix (Life Technologies). In
addition, β-actin was selected as the housekeeping gene. The primer
sequences are summarized in Table
I.
 | Table IPrimer sequences used for qPCR. |
Table I
Primer sequences used for qPCR.
| Gene/site | Forward
primer
(5′→3′) | Reverse
primer
(5′→3′) | Product
length (bp) |
|---|
| For qPCR
(mouse) |
| NOS2 |
TGTGGCTACCACATTGAAGA |
GCCCCTCACCATTATCTTTAC | 234 |
| β-actin |
CTAAGGCCAACCGTGAAAAG |
ACCAGAGGCATACAGGGACA | 104 |
| For ChIP
(mouse) |
| Binding sites on
NOS2 promoter |
| GAS (STAT1) |
ACACGAGGCTGAGCTGACTT |
CACACATGGCATGGAATTTT | 151 |
| NF-κB element |
CACACAGACTAGGAGTGTCCATCAT |
CATAACTGTTCCCAAAGGGAGAGT | 79 |
| Binding site on
GAPDH promoter |
| RNA polymerase
II |
TACTCGCGGCTTTACGGG |
TGGAACAGGGAGGAGCAGAGAGCA | 169 |
Western blot analysis
Whole cell extracts from treated RAW 264.7 cells or
peritoneal macrophages were prepared using RIPA buffer (Cybrdi
Inc., Gaithersburg, MD, USA) supplemented with 1% Halt Protease and
Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) according to the manufacturer's instructions.
Cytoplasmic and nuclear extracts from treated RAW 264.7 cells were
prepared using NE-PER Nuclear and Cytoplasmic Extraction Reagents
(Thermo Fisher Scientific, Inc.) supplemented with 1% Halt Protease
and Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific, Inc.)
according to the manufacturer's instructions. Protein concentration
was determined using a BCA Protein Assay Reagent kit (Thermo Fisher
Scientific, Inc.). Proteins were separated by electrophoresis on an
SDS-PAGE gel (4–10%) and then electrotransferred onto a PVDF
membrane (Roche Diagnostics, Inc., Indianapolis, IN, USA).
Subsequently, the PVDF membrane was incubated with various primary
antibodies and then their corresponding secondary antibodies.
Immunoreactive bands were visualized using ECL substrate in a
ChemiDoc XRS Imaging system (Bio-Rad). In addition, β-actin and
histone H3 were used as loading controls.
Chromatin immunoprecipitation (ChIP)
assay
In the present study, the ChIP assay was performed
using a Pierce™ Agarose ChIP kit (purchased from Thermo Fisher
Scientific, Inc.) according to the manufacturer's instructions.
Briefly, cells were cross-linked with 1% formaldehyde and then
pre-cleared with Protein A/G PLUS Agarose (Thermo Fisher
Scientific, Inc.). For the immunoprecipitation assay, cell lysates
were incubated with antibodies against p-STAT1(Y701) or p65 (Cell
Signaling Technology) at 4°C overnight. Rabbit IgG was used as a
negative control, whereas anti-RNA polymerase II antibody
(#1862243; Thermo Fisher Scientific, Inc.) was employed as a
positive control. As loading controls, 10% total input samples were
used. The primer sequences used for ChIP are shown in Table I.
Statistical analysis
Data were analyzed by one-way analysis of variance
(one-way ANOVA) with SPSS 18.0 software. All values are expressed
as the means ± SD unless otherwise stated, and a p-value <0.05
was considered to indicate a statistically significant
difference.
Results
IL-17 intensifies NOS2 upregulation and
NO production induced by IFN-γ in macrophages
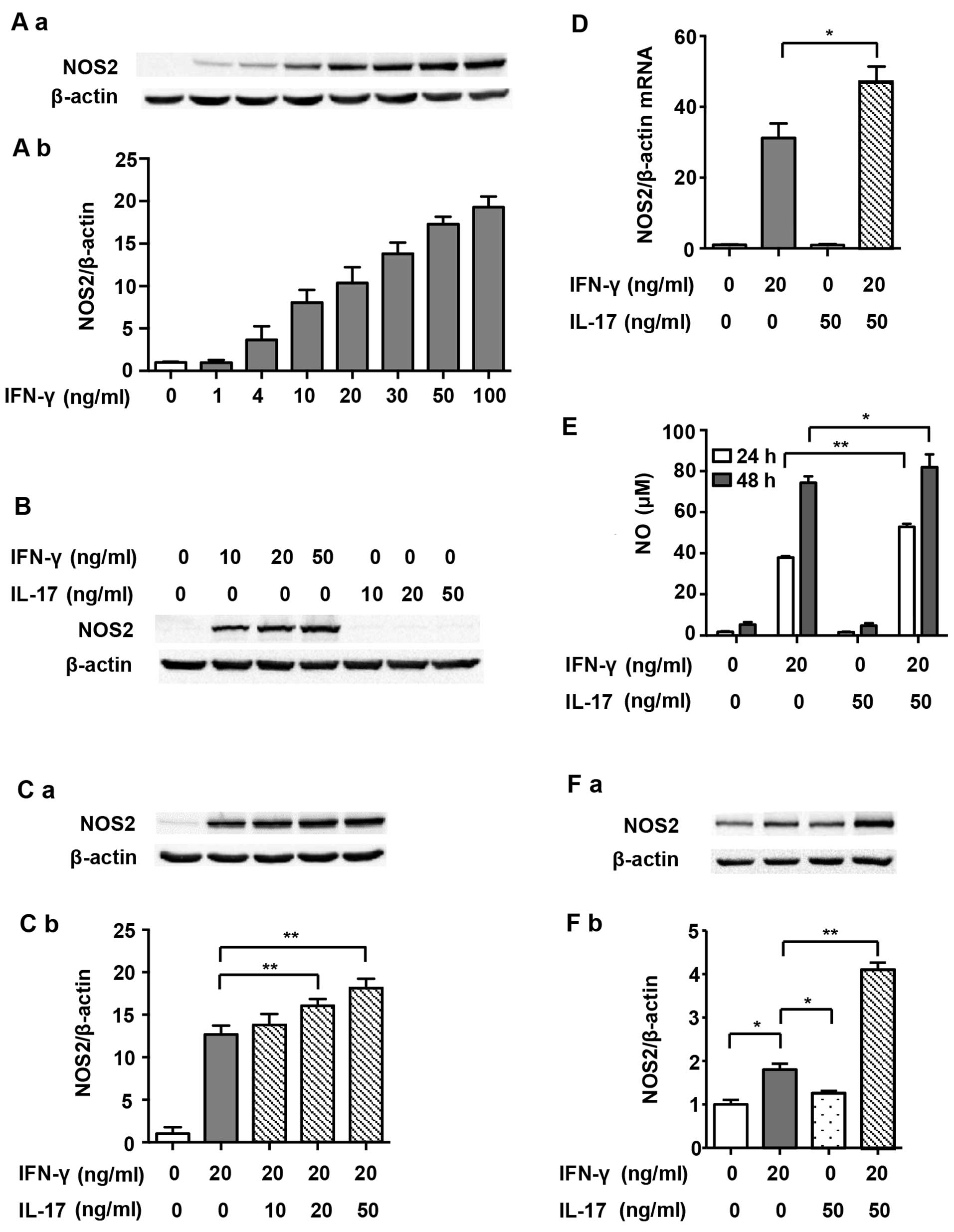
RAW 264.7 cells expressed undetectable levels of
NOS2 without stimulus. In order to determine the optimal dosage,
IFN-γ at various concentrations (1, 4, 10, 20, 30, 50 and 100
ng/ml) was used initially in the present study. Fig. 1A and B show that NOS2 expression
increased in a dose-dependent manner after IFN-γ treatment, and
levels reached half maximal expression when exposed to 20 ng/ml
IFN-γ for 24 h. IL-17 alone did not induce NOS2 expression at any
dosage (Fig. 1B). However, after
RAW 264.7 cells were stimulated with a combination of IL-17 (20 and
50 ng/ml) and IFN-γ, NOS2 expression was further augmented, as
compared with IFN-γ alone (Fig.
1C; 1.27- and 1.43-fold). At the mRNA level, NOS2 upregulation
was also significantly increased by treatment with IL-17 (50 ng/ml)
in combination with IFN-γ (20 ng/ml) for 12 h compared with IFN-γ
alone (1.50-fold), suggesting that IL-17 intensified IFN-γ-induced
NOS2 transcription (Fig. 1D).
Based on these findings, we decided to treat cells with 20 ng/ml
IFN-γ and 50 ng/ml IL-17 in the subsequent experiments. We also
detected the release of NO, the prime effective product of NOS2, in
the supernatant. Fig. 1E shows
that IL-17 significantly increased IFN-γ-induced NO synthesis after
treatment for 24 h (1.39-fold) and 48 h (1.10-fold). Moreover,
IL-17 significantly intensified IFN-γ-induced NOS2 upregulation in
peritoneal macrophages (2.41-fold), proving that the effect was not
a phenomenon peculiar to RAW 264.7 cells (Fig. 1F). The following experiments
carried out to determine the mechanisms involved used RAW 264.7
cells only.
STAT1 is involved in the effect of IL-17
on intensifying NOS2 upregulation induced by IFN-γ in RAW 264.7
cells
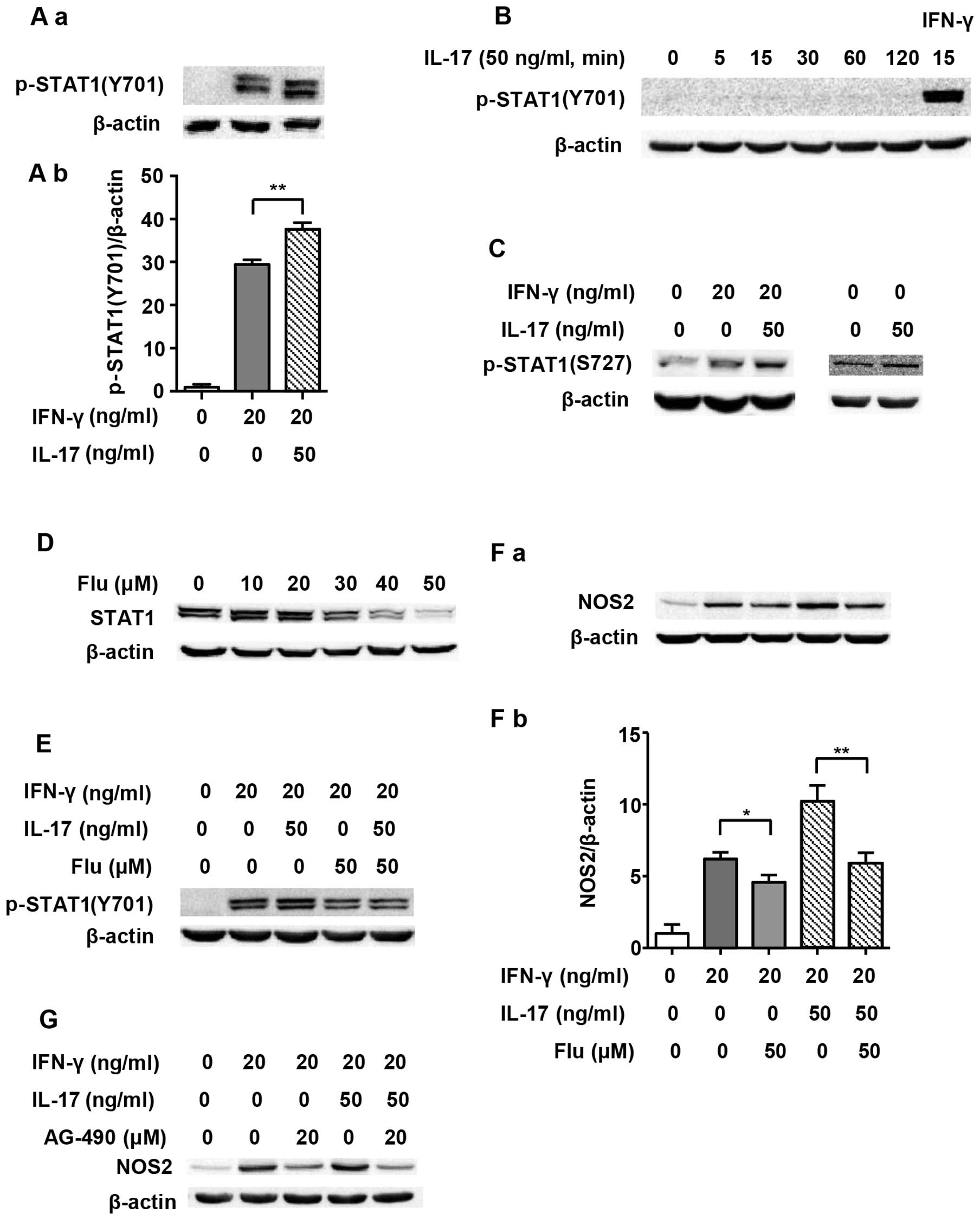
As one of the main transcription factors, STAT1 is
believed to play a vital role in the expression of the NOS2 gene
(20). Therefore, we evaluated
STAT1 activation in order to explore the underlying mechanism of
IL-17-mediated upregulation of NOS2 induced by IFN-γ. Previous
research has shown that Tyr701-p-STAT1 is pivotal in
STAT1-dependent regulation of NOS2 expression (21). Fig.
2A and B illustrate that although 50 ng/ml IL-17 per se
did not induce phosphorylation of STAT1(Y701), it intensified
IFN-γ-induced p-STAT1(Y701) expression as early as 5 min after
treatment (1.29-fold). We employed Flu, which is an effective
inhibitor of STAT1, to explore whether the increased NOS2
upregulation caused by IL-17 was through p-STAT1(Y701). It was
confirmed that Flu (50 µM) significantly inhibited the
expression of STAT1 in RAW 264.7 cells (Fig. 2D). Fig. 2E and F show that when STAT1
expression was inhibited by Flu, p-STAT1(Y701) was obviously
decreased, and so was NOS2 expression (0.58-fold in IFN-γ/IL-17
group). As Janus kinase (JAK) is a direct activator of STAT1, we
also used JAK inhibitor AG-490 to evaluate the role of the
JAK/STAT1 pathway in NOS2 expression. Fig. 2G shows that AG-490 markedly
inhibited NOS2 expression.
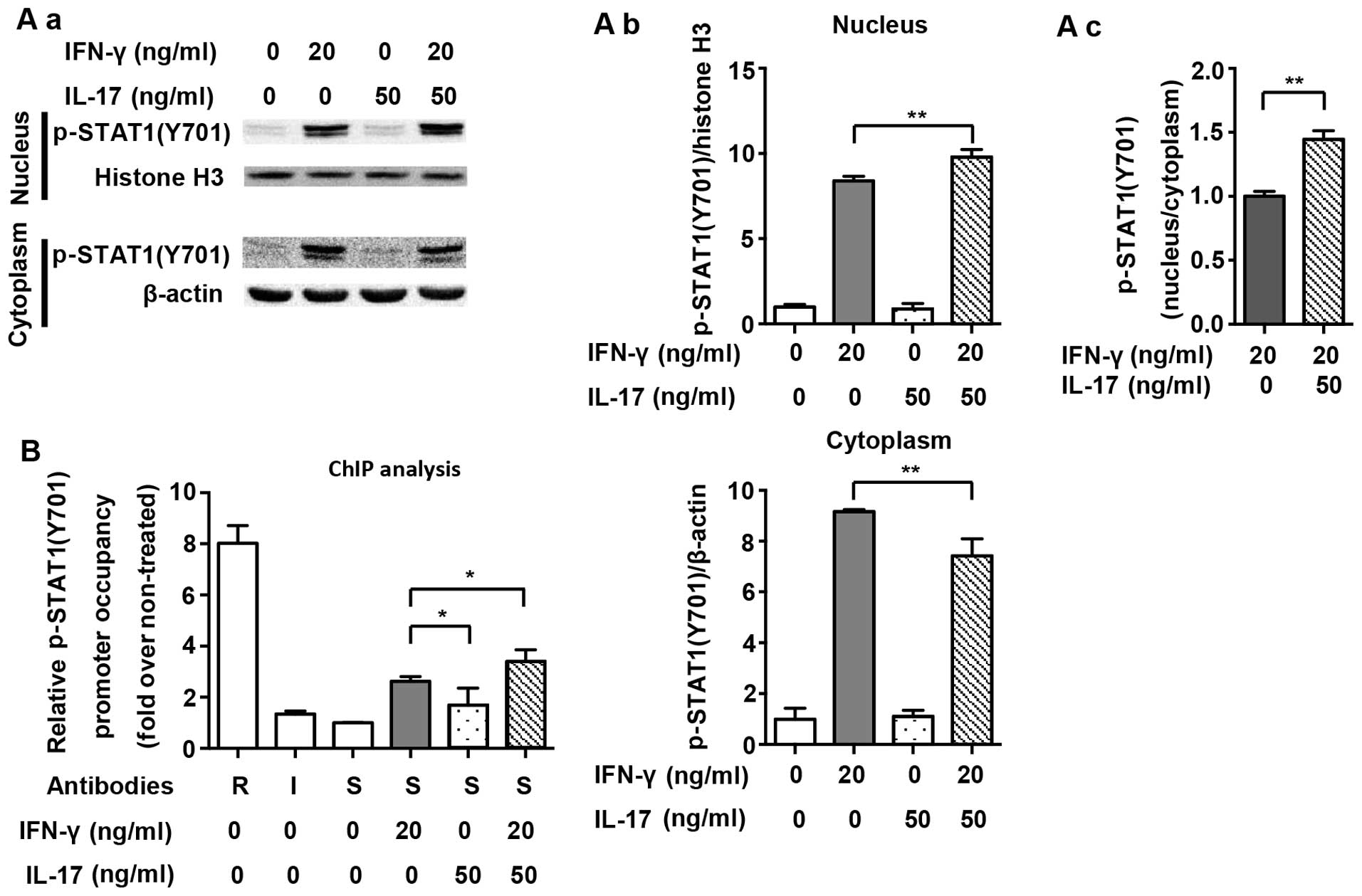
The nuclear translocation of p-STAT1(Y701) and its
binding to IFN-gamma-activated sites (GASs) in the NOS2 promoter
are necessary for the expression of NOS2 (20). Fig.
3A demonstrates that translocation of p-STAT1(Y701) to the
nucleus was significantly increased after IFN-γ treatment for 1 h
(2.6-fold). A combination of IFN-γ and IL-17 further increased the
amount of p-STAT1(Y701) in the nucleus (1.21-fold vs. IFN-γ group).
Proximal GAS in the NOS2 promoter was targeted for amplification in
a ChIP assay, as previously described (9). Fig.
3B shows that the binding of p-STAT1(Y701) to GAS was markedly
increased after IFN-γ treatment for 1 h (2.63-fold). Although IL-17
alone did not induce the binding of p-STAT1(Y701) to GAS, it
considerably enhanced IFN-γ-induced binding (1.29-fold vs. IFN-γ
group). The results revealed that supernumerary increases in
phosphorylation, nuclear translocation and binding to GAS of
p-STAT1(Y701) were closely related to the effect of IL-17-mediated
upregulation of NOS2 which was induced by IFN-γ.
NF-κB is also involved in the effect of
IL-17 on intensifying the NOS2 upregulation induced by IFN-γ in RAW
264.7 cells
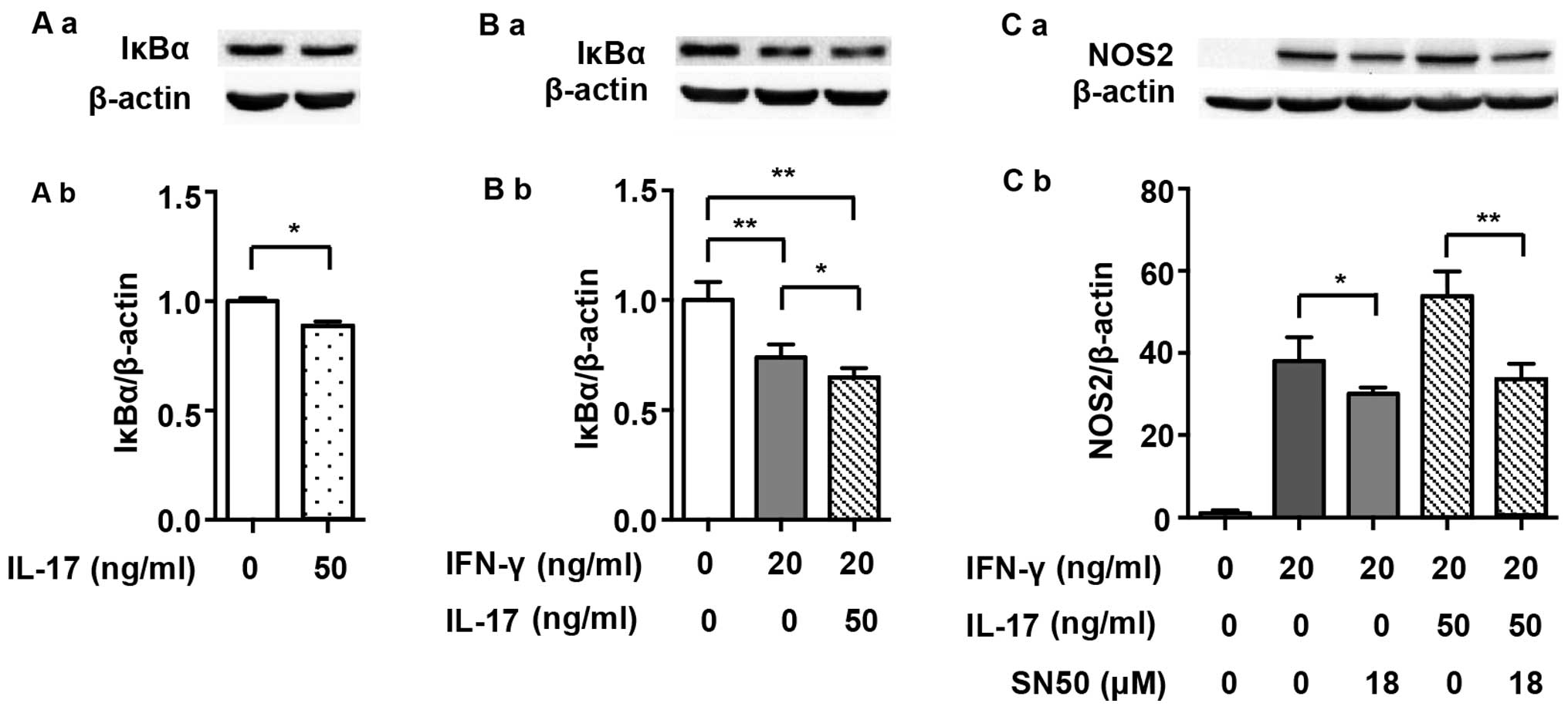
It has previously been demonstrated that NF-κB,
which is another prime transcription factor, also modulates NOS2
expression in mouse macrophages (20). Firstly, we detected the expression
of IκBα, which is an intrinsic inhibitor that binds to p65 and p50
and then degrades after the NF-κB pathway is activated. Fig. 4A demonstrates that IL-17 mildly
intensified IκBα degradation (0.89-fold) and, as has also
previously been shown, that IL-17 activated the NF-κB pathway
(22). IκBα degradation was
induced by 20 ng/ml IFN-γ alone (Fig.
4B; 0.74-fold), and it became more significant when treated
with a combination of IFN-γ and IL-17 (0.65-fold vs. non-treated).
SN50, an extrinsic NF-κB inhibitor, partially abolished the NOS2
upregulation induced by IFN-γ (0.79-fold) or IFN-γ/IL-17 (Fig. 4C; 0.62-fold).
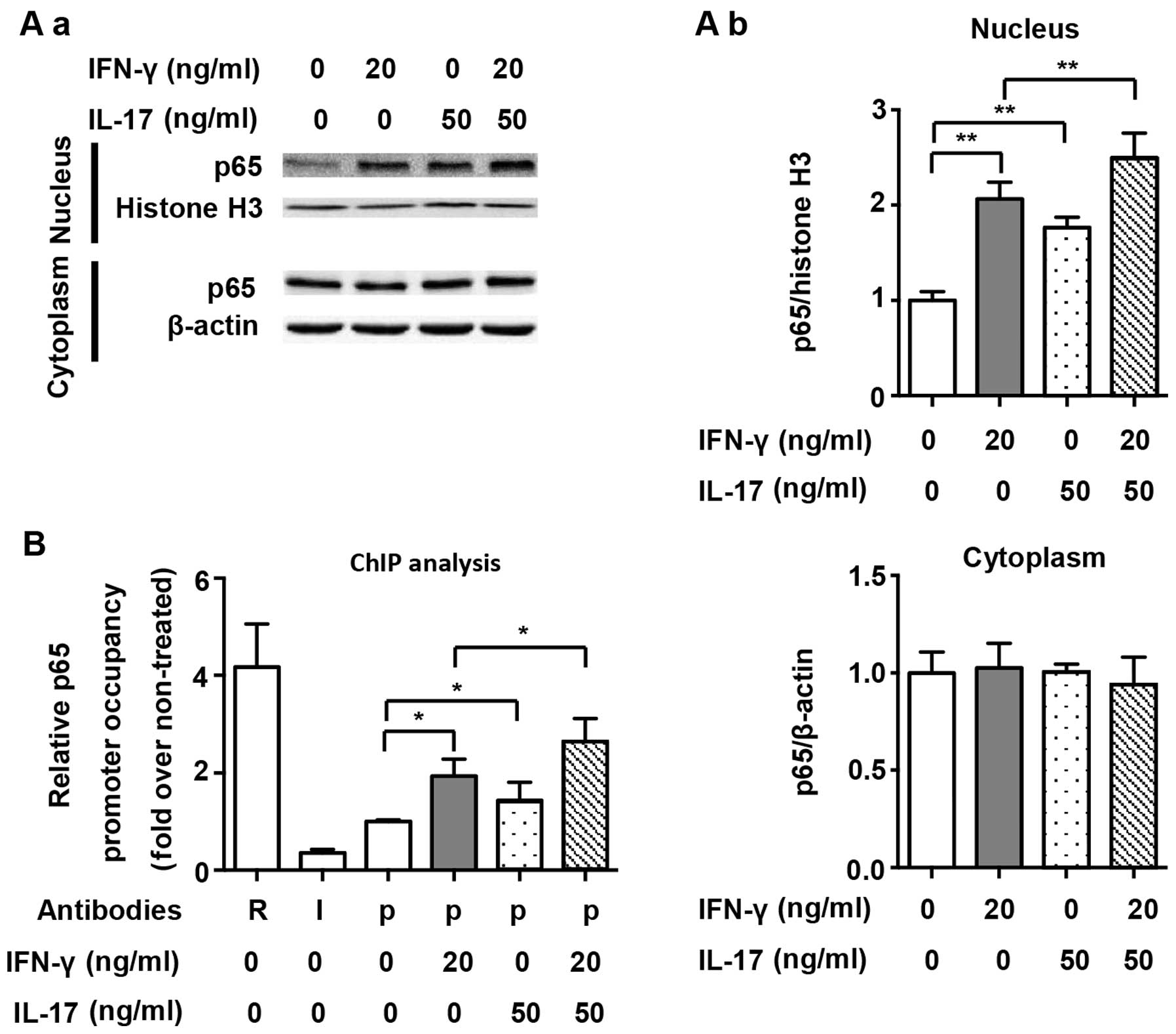
We also assessed the nuclear translocation of NF-κB
and its binding to the NOS2 promoter, and p65 was selected as the
representative transcription factor of the NF-κB pathway. As shown
in Fig. 5A, we demonstrated that
both IFN-γ and IL-17 individually and significantly induced p65
translocation into the nucleus (2.06- and 1.76-fold). However, the
combination of IFN-γ and IL-17 enhanced p65 translocation into the
nucleus (Fig. 5A; 2.50-fold vs.
non-treated). The proximal NF-κB element in the NOS2 promoter was
targeted for amplification in a ChIP assay, as has also been
previously described (9). ChIP
analysis demonstrated that IFN-γ and IL-17 alone significantly
increased the binding of p65 to the NF-κB element (1.93- and
1.43-fold); however, we noted that there was no significant
difference between these two groups. Compared with treatment with
IFN-γ or IL-17 individually, the combination of IFN-γ and IL-17
further augmented the binding of p65 to the NF-κB element in RAW
264.7 cells (Fig. 5B; 1.37- and
1.84-fold). These results indicate that IL-17 upregulated NOS2
expression by increasing IκBα degradation, enhancing p65
translocation into the nucleus and accelerating the binding of p65
to the NF-κB element. Interestingly, we noted that although IL-17
alone activated the NF-κB pathway, it did not induce NOS2
expression by itself. This finding suggests that the collaboration
between several important transcription factors, including NF-κB
and STAT1, is necessary for NOS2 expression in RAW 264.7 cells.
p-p38 MAPK promotes NOS2 upregulation
which is induced by IFN-γ alone or in combination with IL-17 in RAW
264.7 cells
We noted that NF-κB is modulated by a range of
upstream signaling factors, and it has been noted that p38 MAPK is
involved in such modulation (23). Thus, we questioned whether p38
MAPK played a role in the IL-17-mediated upregulation of NOS2
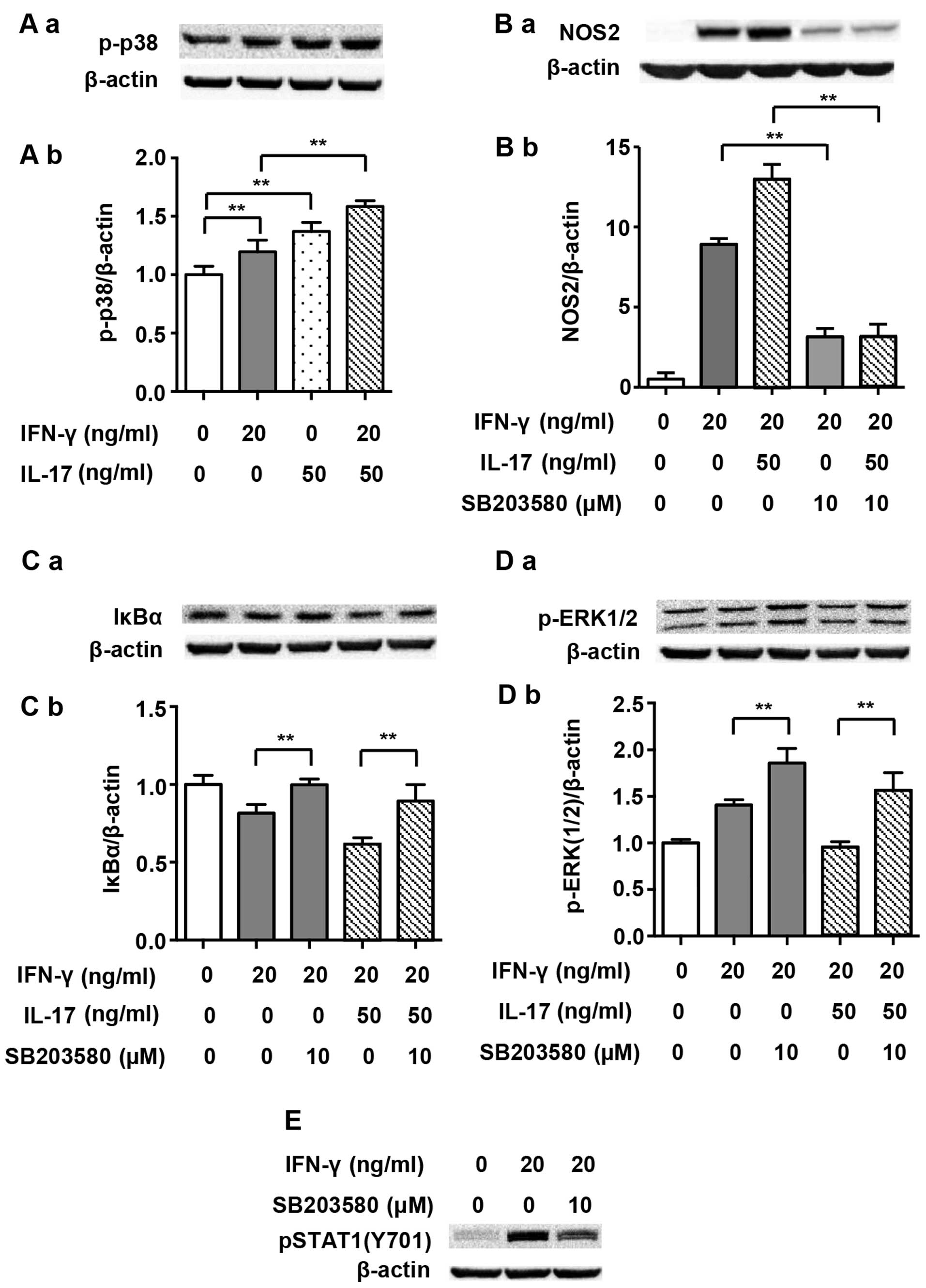
induced by IFN-γ. Fig. 6A shows
that both IFN-γ (20 ng/ml) and IL-17 (50 ng/ml) individually
enhanced the phosphorylation of p38 MAPK markedly after 5 min of
treatment (1.20- and 1.37-fold). The combination of IFN-γ and IL-17
further enhanced the phosphorylation of p38 MAPK as expected
(1.58-fold vs. non-treated). Moreover, p-p38 MAPK inhibitor
SB203580 considerably inhibited the NOS2 upregulation induced by
IFN-γ alone (0.35-fold) or that induced by the combination of IFN-γ
and IL-17 (Fig. 6B; 0.24-fold).
In addition, SB203580 almost completely reversed the IκBα
degradation induced by IFN-γ alone (1.23-fold) or in combination
with IL-17 (Fig. 6C; 1.45-fold).
These data suggest that p-p38 MAPK is an upstream activator of
NF-κB and is involved in the regulation of NOS2 expression.
p-ERK1/2 inhibits NOS2 upregulation
induced by IFN-γ alone or in combination with IL-17 in RAW 264.7
cells
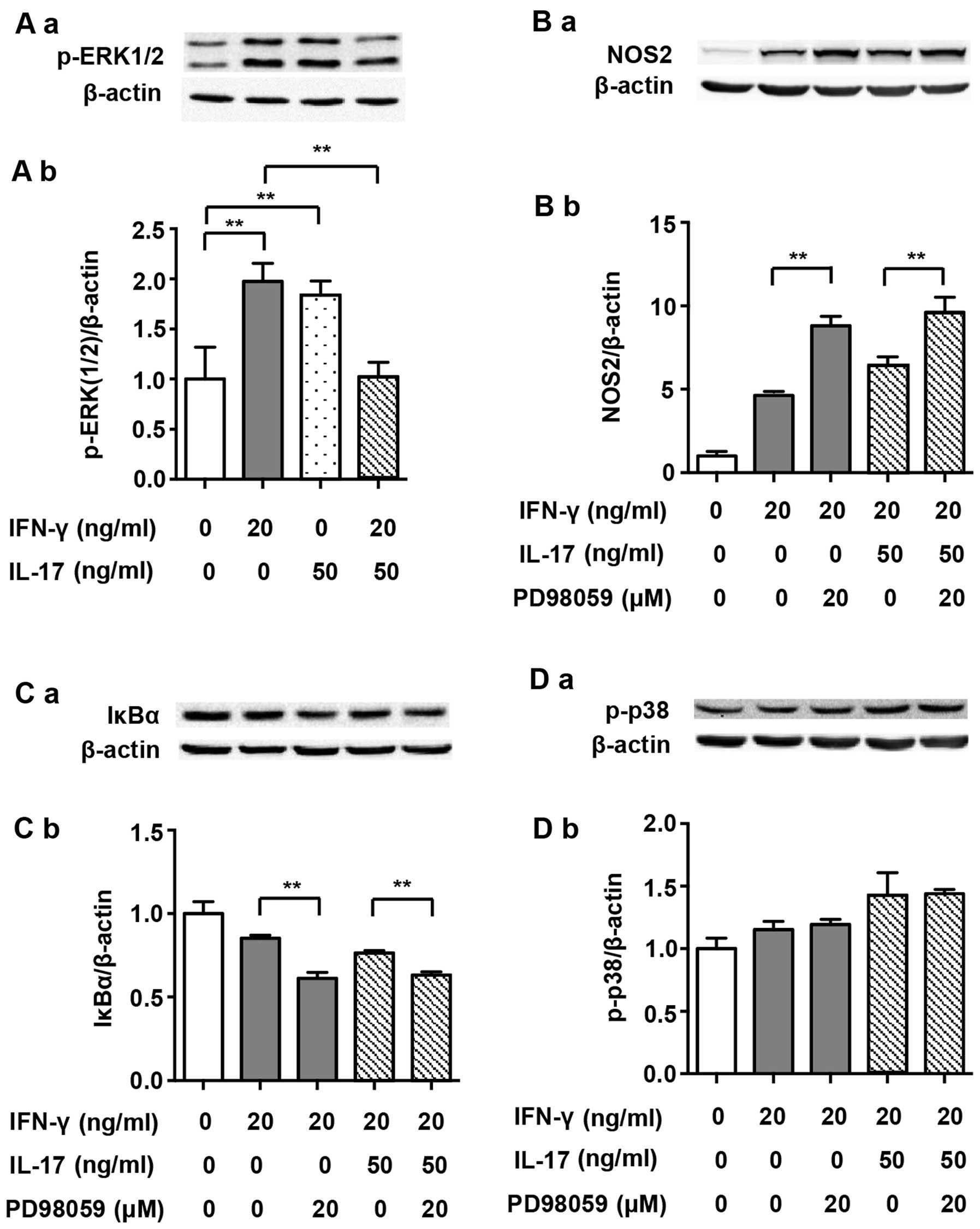
It has previously been demonstrated that ERK1/2
participates in the regulation of the NF-κB pathway (23). It has also been noted that
IFN-γ-induced phosphorylation of ERK1/2 increases NOS2 expression
in J774A.1 mouse macrophages (24). Therefore, we investigated the
roles of ERK1/2 in NOS2 upregulation induced by IFN-γ alone or by
its combination with IL-17 in RAW 264.7 cells. Although IFN-γ and
IL-17 alone enhanced the phosphorylation of ERK1/2 after treatment
for 30 min (1.98- and 1.84-fold), the combination of these two
cytokines significantly dephosphorylated ERK1/2 almost to its
baseline level (Fig. 7A; 0.52-
and 0.56-fold). The p-ERK1/2 inhibitor PD98059 markedly enhanced
NOS2 expression when cells were exposed to IFN-γ alone (Fig. 7B; 1.90-fold). More importantly,
the NOS2 upregulation induced by a combination of IFN-γ and IL-17
was also enhanced by PD98059 (1.54-fold). Furthermore, PD98059
intensified the IκBα degradation induced by IFN-γ alone (0.72-fold)
or in combination with IL-17 (Fig.
7C; 0.83-fold). These data indicate that unlike J774A.1 mouse
macrophages, IFN-γ-induced phosphorylation of ERK1/2 plays a
negative role in the regulation of NOS2 expression in RAW 264.7
cells. Thus, we suggest that IL-17 alleviates the inhibition of
ERK1/2 in cases of IFN-γ-induced NOS2 upregulation by restricting
p-ERK1/2 phosphorylation.
Discussion
In the present study, we found that although IL-17
alone was unable to induce NOS2 expression, it synergized with
IFN-γ to amplify NOS2 expression and NO production through the
STAT1 and NF-κB pathways in RAW 264.7 cells. The phenomenon of IL-7
on intensifying IFN-γ-induced NOS2 upregulation does not exist
solely in RAW 264.7 cells. It is also found in other macrophages,
such as peritoneal macrophages, which shows greater significance in
exploring the underlying mechanisms.
Macrophages play a key role in inflammation, and
NOS2 is a potent inflammatory factor which is regulated by a
variety of cytokines. In the present study NOS2 expression and NO
production induced by IFN-γ were enhanced by IL-17, which
intensified host defense reactions, such as the clearance of
pathogens. On the other hand, however, the excessive release of NO
eventually leads to uncontrolled inflammation and injury of
adjacent tissues (25). These
data indicate that modulating interaction between different
cytokines is a feasible way to acquire the optimal host defense
reaction.
As a key molecule of the canonical JAK/STAT1
pathway, p-STAT1(Y701) is phosphorylated by JAK1/2 and is critical
to the upregulation of NOS2 induced by IFN-γ (20,26). IL-17 alone did not activate
p-STAT1(Y701), but it enhanced the activation, nuclear
translocation and binding to the NOS2 promoter of p-STAT1(Y701)
induced by IFN-γ. It has previously been reported that STAT1 is
also phosphorylated at Ser727 (27). Dual phosphorylation of STAT1 at
Tyr701 and Ser727 is required to induce the full expression of
IFN-γ-activated genes, including NOS2 (27). NOS2 expression is partially
inhibited by STAT1(S727A) site mutation (28). We also examined the expression of
p-STAT1(S727) induced by IFN-γ and IL-17. Fig. 2C shows that IFN-γ and IL-17 alone
increased the expression of p-STAT1(S727). The combination of IFN-γ
and IL-17 further enhanced the phosphorylation of STAT1 at S727
compared with IFN-γ alone. However, the exact role of p-STAT1(S727)
in the expression of NOS2 induced by IFN-γ and IL-17 remains
unclear.
NF-κB is another important transcription factor in
NOS2 expression (20). In the
present study, NF-κB was activated by IFN-γ or IL-17 individually.
The combination of these two cytokines further activated NF-κB,
which was demonstrated not only by IκBα degradation and p65
translocation into the nucleus, but also the binding of p65 to the
NOS2 promoter. The NF-κB inhibitor SN50 partially abolished the
NOS2 upregulation induced by IFN-γ alone or in combination with
IL-17. These results demonstrate that IL-17 enhanced IFN-γ-induced
NOS2 upregulation by further activating the NF-κB pathway.
In the present study, IFN-γ and IL-17 individually
activated p38 MAPK, and in combination they further intensified
this activation. Furthermore, the p-p38 MAPK inhibitor SB203580
partially inhibited NOS2 expression in cells treated with IFN-γ
alone or in combination with IL-17. Inhibition of p-p38 MAPK by
SB203580 reversed the IκBα degradation induced by IFN-γ alone or in
combination with IL-17, indicating that IκBα degradation and the
activation of NF-κB pathway are regulated by p-p38 MAPK. We also
noted that SB203580 restrained IFN-γ-induced phosphorylation of
STAT1(Y701) (Fig. 6E). This
result suggested that p38 MAPK was a positive regulator of
p-STAT1(Y701), which has been previously reported in dendritic
cells (29). As a result, we
suggest that the increased phosphorylation of STAT1(Y701) by IL-17
is closely linked with increased p-p38 MAPK activity.
We noted that ERK1/2 played unpredictable and
interesting roles in the NOS2 upregulation induced by IFN-γ alone
or in combination with IL-17 in RAW 264.7 cells. IFN-γ and IL-17
individually increased ERK1/2 phosphorylation; however, the
combination of these two cytokines markedly decreased ERK1/2
phosphorylation compared with IFN-γ alone. Moreover, the p-ERK1/2
inhibitor PD98059 enhanced IκBα degradation and increased the NOS2
upregulation induced by IFN-γ alone or in combination with IL-17,
indicating that p-ERK1/2 plays an inhibitive role in IFN-γ-induced
NOS2 upregulation in RAW 264.7 cells. It has previously been
reported that p-ERK1/2 promoted NOS2 expression in murine
macrophage cell lines, including J774A.1 (30). The RAW 264.7 cell line was
established through intraperitoneal injection of Abelson murine
leukemia virus (A-MuLV, an RNA virus) into male BAB/14 mice and
then extracting cells from ascites (31). By contrast, the J774A.1 cell line
was established from cells in ascites, which were acquired by
percutaneous inoculation of plasmacytoma cells (induced by
intraperitoneal injection of saturated hydrocarbons such as Bayol
F) into female BALB/c mice (32–34). Therefore, the discrepant functions
of p-ERK1/2 may be explained by the different methods used to
acquire these two cell lines. Thus, it is necessary to be cautious
when interpreting the role of p-ERK1/2 in the inflammatory response
of macrophages.
NOS2 is an inflammatory factor, and its excessive
expression induces cell apoptosis (35). We believe that the simultaneous
activation of p38 MAPK and ERK1/2 induced by IFN-γ alone restricts
the overexpression of NOS2 and over-production of NO in RAW 264.7
cells, which results in the avoidance of self-damage and possible
apoptosis. IL-17 enhanced the NOS2 upregulation induced by IFN-γ by
restricting ERK1/2 phosphorylation, in other words, through
limiting the inhibitive effect of p-ERK1/2.
It has previously been reported that inhibition of
the p38 MAPK pathway upregulates ERK1/2 activity in M1 macrophages
(36). Our study also
demonstrated that inhibition of p38 MAPK facilitated the
phosphorylation of ERK1/2 induced by IFN-γ (1.21-fold) alone or in
combination with IL-17 (Fig. 6D)
(1.64-fold). However, inhibition of p-ERK1/2 did not markedly alter
p38 MAPK activity (Fig. 7D).
These data indicated that p38 MAPK phosphorylation induced by IFN-γ
or IFN-γ/IL-17 relieved the inhibitive effect of p-ERK1/2 on NOS2
expression in RAW 264.7 cells by restricting its
phosphorylation.
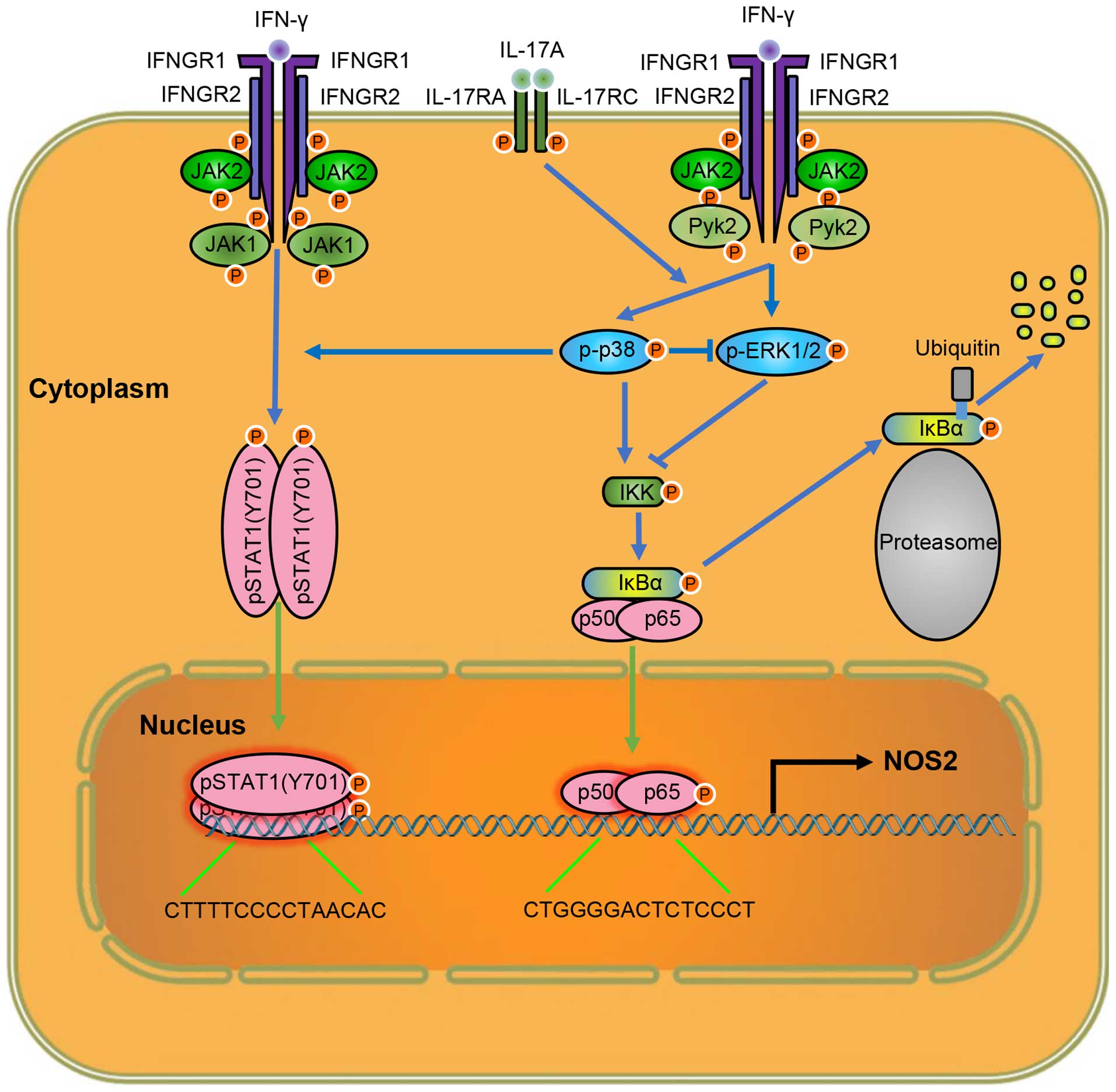
Taken together, our results suggest that IL-17
intensifies IFN-γ-induced NOS2 upregulation and NO production by
increasing the transcriptional activity of p-STAT1(Y701) and NF-κB
(Fig. 8). Thus, lowering the
level of IL-17 is a target for attenuating inflammation, and
anti-IL-17 monoclonal antibodies have yielded positive results in
inflammatory diseases (37). In
addition, considering the roles of p38 MAPK in NOS2 expression,
inhibiting the activity of p38 MAPK may also help control excessive
inflammation due to tissue injury. As the functions of ERK1/2 on
NOS2 expression vary in different macrophages, the cell-type
specific regulation of ERK1/2 activity may provide new ideas for
the tissue-specific control of inflammation, which may mitigate
inflammation and avoid impaired host defense, simultaneously. These
important indications provide potential targets for clinical
therapies of related diseases. Further exploration of the mechanism
of IL-17 on intensifying IFN-γ-induced NOS2 upregulation may reveal
more information on how IL-17 exerts its inflammatory effects, and
provide us with more options to regulate inflammation.
Acknowledgments
This study was supported by the National Natural
Science Fund (nos. 91339116, 81170138 and 81200541) and the
National Basic Research Program of China ('973 Project' no.
2012CB517804).
References
|
1
|
Rao KM: Molecular mechanisms regulating
iNOS expression in various cell types. J Toxicol Environ Health B
Crit Rev. 3:27–58. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Aktan F: iNOS-mediated nitric oxide
production and its regulation. Life Sci. 75:639–653. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Green SJ, Mellouk S, Hoffman SL, Meltzer
MS and Nacy CA: Cellular mechanisms of nonspecific immunity to
intracellular infection: cytokine-induced synthesis of toxic
nitrogen oxides from L-arginine by macrophages and hepatocytes.
Immunol Lett. 25:15–19. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Li LM, Kilbourn RG, Adams J and Fidler IJ:
Role of nitric oxide in lysis of tumor cells by cytokine-activated
endothelial cells. Cancer Res. 51:2531–2535. 1991.PubMed/NCBI
|
|
5
|
Langrehr JM, Hoffman RA, Billiar TR, Lee
KK, Schraut WH and Simmons RL: Nitric oxide synthesis in the in
vivo allograft response: a possible regulatory mechanism. Surgery.
110:335–342. 1991.PubMed/NCBI
|
|
6
|
Brown GC and Neher JJ: Inflammatory
neurodegeneration and mechanisms of microglial killing of neurons.
Mol Neurobiol. 41:242–247. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Annane D, Sanquer S, Sébille V, Faye A,
Djuranovic D, Raphaël JC, Gajdos P and Bellissant E:
Compartmentalised inducible nitric-oxide synthase activity in
septic shock. Lancet. 355:1143–1148. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhu J, Yamane H and Paul WE:
Differentiation of effector CD4 T cell populations (*). Annu Rev
Immunol. 28:445–489. 2010. View Article : Google Scholar
|
|
9
|
Goldring CE, Reveneau S, Algarté M and
Jeannin JF: In vivo footprinting of the mouse inducible nitric
oxide synthase gene: inducible protein occupation of numerous sites
including Oct and NF-IL6. Nucleic Acids Res. 24:1682–1687. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Burke SJ, Updegraff BL, Bellich RM, Goff
MR, Lu D, Minkin SC Jr, Karlstad MD and Collier JJ: Regulation of
iNOS gene transcription by IL-1β and IFN-γ requires a coactivator
exchange mechanism. Mol Endocrinol. 27:1724–1742. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gu C, Wu L and Li X: IL-17 family:
cytokines, receptors and signaling. Cytokine. 64:477–485. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Komiyama Y, Nakae S, Matsuki T, Nambu A,
Ishigame H, Kakuta S, Sudo K and Iwakura Y: IL-17 plays an
important role in the development of experimental autoimmune
encephalomyelitis. J Immunol. 177:566–573. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Smith E, Prasad KM, Butcher M, Dobrian A,
Kolls JK, Ley K and Galkina E: Blockade of interleukin-17A results
in reduced atherosclerosis in apolipoprotein E-deficient mice.
Circulation. 121:1746–1755. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Laan M, Lötvall J, Chung KF and Lindén A:
IL-17-induced cytokine release in human bronchial epithelial cells
in vitro: role of mitogen-activated protein (MAP) kinases. Br J
Pharmacol. 133:200–206. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Do H, Pyo S and Sohn EH: Suppression of
iNOS expression by fucoidan is mediated by regulation of p38 MAPK,
JAK/STAT, AP-1 and IRF-1, and depends on upregulation of scavenger
receptor B1 expression in TNF-alpha- and IFN-gamma-stimulated C6
glioma cells. J Nutr Biochem. 21:671–679. 2010. View Article : Google Scholar
|
|
16
|
Ouyang W, Kolls JK and Zheng Y: The
biological functions of T helper 17 cell effector cytokines in
inflammation. Immunity. 28:454–467. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Griffin GK, Newton G, Tarrio ML, Bu DX,
Maganto-Garcia E, Azcutia V, Alcaide P, Grabie N, Luscinskas FW,
Croce KJ and Lichtman AH: IL-17 and TNF-α sustain neutrophil
recruitment during inflammation through synergistic effects on
endothelial activation. J Immunol. 188:6287–6299. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Danzaki K, Matsui Y, Ikesue M, Ohta D, Ito
K, Kanayama M, Kurotaki D, Morimoto J, Iwakura Y, Yagita H, et al:
Interleukin-17A deficiency accelerates unstable atherosclerotic
plaque formation in apolipoprotein E-deficient mice. Arterioscler
Thromb Vasc Biol. 32:273–280. 2012. View Article : Google Scholar
|
|
19
|
Eid RE, Rao DA, Zhou J, Lo SF, Ranjbaran
H, Gallo A, Sokol SI, Pfau S, Pober JS and Tellides G:
Interleukin-17 and interferon-gamma are produced concomitantly by
human coronary artery-infiltrating T cells and act synergistically
on vascular smooth muscle cells. Circulation. 119:1424–1432. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pautz A, Art J, Hahn S, Nowag S, Voss C
and Kleinert H: Regulation of the expression of inducible nitric
oxide synthase. Nitric Oxide. 23:75–93. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Trinh B, Ko SY, Haria D, Barengo N and
Naora H: The homeo-protein DLX4 controls inducible nitric oxide
synthase-mediated angiogenesis in ovarian cancer. Mol Cancer.
14:972015. View Article : Google Scholar
|
|
22
|
Ruddy MJ, Wong GC, Liu XK, Yamamoto H,
Kasayama S, Kirkwood KL and Gaffen SL: Functional cooperation
between interleukin-17 and tumor necrosis factor-alpha is mediated
by CCAAT/enhancer-binding protein family members. J Biol Chem.
279:2559–2567. 2004. View Article : Google Scholar
|
|
23
|
Cargnello M and Roux PP: Activation and
function of the MAPKs and their substrates, the MAPK-activated
protein kinases. Microbiol Mol Biol Rev. 75:50–83. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jaramillo M, Naccache PH and Olivier M:
Monosodium urate crystals synergize with IFN-gamma to generate
macrophage nitric oxide: involvement of extracellular
signal-regulated kinase 1/2 and NF-kappa B. J Immunol.
172:5734–5742. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Abramson SB, Amin AR, Clancy RM and Attur
M: The role of nitric oxide in tissue destruction. Best Pract Res
Clin Rheumatol. 15:831–845. 2001. View Article : Google Scholar
|
|
26
|
Hu X and Ivashkiv LB: Cross-regulation of
signaling pathways by interferon-gamma: implications for immune
responses and autoimmune diseases. Immunity. 31:539–550. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
van Boxel-Dezaire AH and Stark GR: Cell
type-specific signaling in response to interferon-gamma. Curr Top
Microbiol Immunol. 316:119–154. 2007.PubMed/NCBI
|
|
28
|
Varinou L, Ramsauer K, Karaghiosoff M,
Kolbe T, Pfeffer K, Müller M and Decker T: Phosphorylation of the
Stat1 transactivation domain is required for full-fledged
IFN-gamma-dependent innate immunity. Immunity. 19:793–802. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Takauji R, Iho S, Takatsuka H, Yamamoto S,
Takahashi T, Kitagawa H, Iwasaki H, Iida R, Yokochi T and Matsuki
T: CpG-DNA-induced IFN-alpha production involves p38 MAPK-dependent
STAT1 phosphorylation in human plasma-cytoid dendritic cell
precursors. J Leukoc Biol. 72:1011–1019. 2002.PubMed/NCBI
|
|
30
|
Beurel E and Jope RS: Glycogen synthase
kinase-3 promotes the synergistic action of interferon-gamma on
lipopolysaccharide-induced IL-6 production in RAW264.7 cells. Cell
Signal. 21:978–985. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Raschke WC, Baird S, Ralph P and Nakoinz
I: Functional macrophage cell lines transformed by Abelson leukemia
virus. Cell. 15:261–267. 1978. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ralph P and Nakoinz I: Phagocytosis and
cytolysis by a macrophage tumour and its cloned cell line. Nature.
257:393–394. 1975. View
Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ralph P, Prichard J and Cohn M: Reticulum
cell sarcoma: an effector cell in antibody-dependent cell-mediated
immunity. J Immunol. 114:898–905. 1975.PubMed/NCBI
|
|
34
|
Potter M and Lieberman R: Common
individual antigenic determinants in five of eight BALB-c IgA
myeloma proteins that bind phosphoryl choline. J Exp Med.
132:737–751. 1970. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sharshar T, Gray F, Lorin de la
Grandmaison G, Hopkinson NS, Ross E, Dorandeu A, Orlikowski D,
Raphael JC, Gajdos P and Annane D: Apoptosis of neurons in
cardiovascular autonomic centres triggered by inducible nitric
oxide synthase after death from septic shock. Lancet.
362:1799–1805. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hall JP and Davis RJ: Inhibition of the
p38 pathway upregulates macrophage JNK and ERK activities, and the
ERK, JNK, and p38 MAP kinase pathways are reprogrammed during
differentiation of the murine myeloid M1 cell line. J Cell Biochem.
86:1–11. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Leonardi C, Matheson R, Zachariae C,
Cameron G, Li L, Edson-Heredia E, Braun D and Banerjee S:
Anti-interleukin-17 monoclonal antibody ixekizumab in chronic
plaque psoriasis. N Engl J Med. 366:1190–1199. 2012. View Article : Google Scholar : PubMed/NCBI
|