Introduction
Alzheimer's disease (AD), which has a high
prevalence in the elderly, is a progressive and fatal
neurodegenerative disease characterized by loss of synapses and
neurons, amyloid plaque deposition, neurofibrillary tangle
aggregation and neuroinflammation; previous research has confirmed
that the accumulation of intracellular β-amyloid (Aβ) is an early
event in the development of AD (1,2).
Evidence from in vivo and in vitro experiments have
demonstrated that Aβ induces the inflammatory response, oxidative
stress and neuronal apoptosis, resulting in loss of neurons,
particularly in the cerebral cortex and hippocampal cortex
(3,4). Despite considerable progress that
has been made in exploring the complicated underlying mechanisms of
AD, there is still no effective treatment which is able to reverse,
prevent or even halt the development of AD (5).
Ginseng, the root of Panax ginseng C.A.
Meyer, has been used extensively as a drug in traditional Chinese
medicine for over 2,000 years. Currently, more than 40 kinds of
ginsenoside constituents have been extracted from ginseng. Among
them, ginsenoside Rg1 (Rg1) is considered an important constituent
which exerts important pharmacological effects and has also been
proven to potentiate prominent neuroprotective properties both
in vivo and in vitro (6–9).
Our previous study has reported that the steroid receptor-dependent
anti-protein tyrosine nitration pathway plays a vital role in the
protection of Rg1 against Aβ-induced toxicity (7). However, the molecular mechanism of
how Rg1 suppresses nitric oxide (NO) production and nitrotyrosine
formation remains ambiguous, and needs to be studied further.
Nuclear factor κ-light-chain-enhancer of activated B
cells (NF-κB) is a protein complex which is in charge of DNA
transcription, cytokine secretion and cell survival (10). NF-κB is present in almost all cell
types in the nervous system. Incorrect modulation of NF-κB has been
connected to inflammation, cancer, neurodegenerative diseases and
improper immune development (11–13). Autopsy results have demonstrated
that the NF-κB activity in neurons and astrocytes near amyloid
plaques was abnormally increased in the brains of patients with AD
(14–16), suggesting that Aβ plays a critical
role in the activation of NF-κB in AD. Furthermore, it has been
shown that Rg1 downmodulated LPS-induced proinflammatory cytokines
release, and prevented NF-κB nuclear translocation and DNA binding
activity in RAW264.7 and A549 cells (17). In PC12 cells, Rg1 has been shown
to prevent the cell lesions caused by hydrogen peroxide
(H2O2) solution attack via modulation of the
NF-κB and MAPK signaling pathways (18). Therefore, we hypothesized that
NF-κB serves as an important pharmacological target in the
neuroprotective mechanisms of Rg1.
In the present study, we used an in vitro
model of aggregated Aβ25–35-induced AD to investigate
the potential cellular and molecular mechanisms by which Rg1
counteracts mitochondrial dysfunction and
Aβ25–35-mediated apoptosis in primary cultured rat
cortical neurons.
Materials and methods
Reagents and antibodies
Rg1 was purchased from the National Institute for
the Control of Pharmaceutical and Biological Products, with more
than 99% purity (China). β-amyloid peptide 25–35
(Aβ25–35), s-methylisothiourea hemisulfate (SMT) salt,
ammonium pyrrolidine dithiocarbamate (PDTC),
H2O2, dimethyl sulfoxide (DMSO),
poly-D-lysine,
3-(4,5-dimeth-ylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
(MTT), 4-hydroxy-2,2,6,6-tetramethylpiperidine 1-oxyl (tempol), and
5-(and-6)-carboxy-2′,7′-dichlorofluo rescein diacetate
(carboxy-DCFDA) were purchased from Sigma-Aldrich (St. Louis, MO,
USA). Dulbecco's modified Eagle's medium (DMEM), fetal bovine
serum, B27, and neurobasal medium were all purchased from Gibco-BRL
(Burlington, ON, Canada). Primary antibodies against NF-κB (p65)
(sc-372), IκB-α (sc-371), phosphorylated (p-)IκB-α (sc-8404), lamin
B (sc-6217), glyceraldehyde 3-phosphate dehydrogenase (GAPDH)
(sc-47724), inducible nitric oxide synthase (iNOS) (sc-651), Bcl-2
(sc-7382), Bax (sc-7480) were all purchased from Santa Cruz
Biotechnology, Inc. (Santa Cruz, CA, USA). Primary antibodies
against cytochrome c (#4272), cytochrome c oxidase
(COX IV) (#11967), pro-caspase-9 (#9508), cleaved caspase-3
(#9661), and pro-caspase-3 (#9662) were all purchased from Cell
Signaling Technology, Inc. (Beverly, MA, USA). Secondary antibodies
against mouse IgG (HRP-conjugated) (BA1051), rabbit IgG
(HRP-conjugated; BA1054), DyLight 488-conjugated (BA1127), goat IgG
(HRP-conjugated) (BA1060) were all from Boster, Inc. (Wuhan,
China).
Cell culture
Primary cultured cortical neurons were prepared from
embryonic day (D17–18) Sprague Dawley (SD) rat fetuses as
previously described (7).
Briefly, pregnant rats were anesthetized with halothane and
sacrificed by cervical dislocation. All of the fetal rats were
seperated from the maternal body. The brain cortex of the fetuses
was then dissected in D-Hank's buffer (137 mmol/l NaCl, 5.4 mmol/l
KCl, 0.4 mmol/l KH2PO4, 0.34 mmol/l
Na2HPO4•7H2O, 10 mmol/l glucose
and 10 mmol/l Hepes) containing 0.125% trypsin. After incubation at
37℃ for 8-10 min, cortical tissues were dissociated by passing
through a series of fire-polished constricted Pasteur pipettes.
Approximately 5×105 cells/ml were seeded onto
poly-D-lysine (10 µg/ml) coated 96-well or 6-well plates.
Cells were routinely cultured in Neurobasal medium supplemented
with 2% B27, 10 U/ml penicillin, 10 U/ml streptomycin and 0.5
mmol/l glutamine at 37°C in an atmosphere with 5% CO2
and observed by inverted phase-contrast microscopy. Neuronal
cultures were maintained for 7 days in vitro before the
various chemical treatments. After identification with a phase
contrast microscope and immunocytochemical analysis for neuronal
markers, the primary neuronal cultures were found to be more than
90% pure. All animals were purchased from the Laboratory Animal
Center of Zhejiang University. The experiments were conducted under
a protocol approved by the Institutional Animal Care and Use
Committee of Zhejiang University (Hangzhou, China).
Immunocytochemical analysis
After treatments, cells were rinsed with ice-cold
PBS buffer solution and immediately fixed with 100% methanol (−2°C)
for 15 min. Fixed cells were washed with PBS, blocked with 10%
normal fetal serum for 1 h, and then incubated at 4°overnight with
primary antibodies. After this, the cells were washed three times
in PBS solution for 5 min each. Cells were then incubated with
fluorescent secondary antibodies (1:400). The cultures were then
incubated in 2 mg/ml DAPI solution in PBS for 1 min to label the
nuclei, and immunoreactivity was visualized by microscopic
examination carried out using a Leica inverted microscope equipped
for fluorescence analysis (Leica Microsystems, Wetzlar,
Germany).
Experimental treatment of cultures
Aβ25–35 was dissolved in sterile
distilled water at a concentration of 0.5 mg/ml as a stock
solution. The stock solution was incubated at 37°C for 72 h for the
aggregation of Aβ25–35 and then stored at 4°C. After the
medium was refreshed, the cortical neuronal cultures at day 7 were
preincubated with the ROS scavenger tempol (10 µM),
iNOS-selective inhibitor SMT (100 µM), NF-κB-selective
inhibitor PDTC (1 µM) or Rg1 (20 µM), for 36 h,
followed by exposure to 10 µM aggregated Aβ25–35
for 72 h. All the chemicals were dissolved in DMSO. Control rats
were treated with the same volume of vehicle (DMSO) as the
experimental groups; the final concentration was 0.1%, which had no
toxic effect on cell viability.
Exposure to
H2O2
A total of 30% H2O2 solution
(Sigma-Aldrich) was diluted with sterile distilled water to 0.15
mol/l as a working solution. As a positive control to release ROS,
H2O2 solution was added to the culture medium
at a final concentration of 150 µM for 24 h.
Cell viability assay
The viability of cell cultures was detected by MTT
assay, as previously described, (19) after Aβ25–35 treatment
for 72 h. Primary neurons were incubated with MTT stock solution
(0.5 mg/ml) for 3 h at 37°C, washed in PBS three times and then
shaken for at least 20 min at room temperature to dissolve the
formazane crystals in DMSO. The absorbance was measured using a
microplate reader at 570 nm (DTX880; Beckman Coulter, Inc., La
Brea, CA, USA). The cell viability of the control was defined as
100%. Assays were repeated in three independent culture
preparations, each performed in triplicate.
Measurement of intracellular reactive
oxygen species (ROS) generation
The production of intracellular ROS was evaluated in
primary neurons with an oxidation sensitive fluorescent dye,
carboxy-DCFDA. The intracellular ROS oxidize non-fluorescent
intracellular DCFDA into the highly fluorescent
dichlorofluorescein. An increase in the green fluorescence
intensity was used to quantify the generation of intracellular ROS.
After carboxy-DCFDA was added at a final concentration of 15
µM to culture medium for 30 min at 37°C, neurons were
photographed with a fluorescence microscope. The fluorescence
intensity was measured by a microplate reader (DTX880; Beckman
Coulter, Inc.) with an argon laser with 488 and 525 nm bandpass
filters.
NO production assay
After primary neuronal cells were exposed to
Aβ25–35 for 24 h, the supernatant was collected from
plates and NO production was determined spectrophotometrically
using a Griess assay reagent kit (Jiancheng Bioengineering
Institute, Nanjing, China) (20).
Briefly, 100 µl supernatant was mixed with 100 µl
Griess reagent (0.1% N naphthyl ethylethylenediamine
dihydrochloride in 5% phosphoric acid and 1% sulfanilamide were
mixed according to the proportion of 1:1). The absorbance was then
detected spectrophotometrically at 550 nm using the microplate
reader. Nitrite production in the control group was defined as a
value of 1.0.
Western blot analysis
After the various treatments, medium was removed and
cells were harvested at 4°C. Total proteins were obtained by cell
lysis (Tris-HCl 50 mM, pH 7.5, NaCl 150 mM, EGTA 20 mM, 1% Triton
X-100, 0.5% sodium deoxycholate, DTT 1 mM, NaF 20 mM, sodium
vandate 1 mM, PMSF 1 mM, leupeptin 10 µg/ml and aprotinin 30
µg/ml) as previously described (19). To detect the cytosolic release of
cytochrome c, the untreated and drug-treated cells were
harvested by centrifugation at 1,000 × g for 5 min at 4°C. Cell
pellets were washed once with ice-cold PBS and resuspended with 5
volumes of EGTA (1 mM), DTT (1 mM), HEPES-KOH (pH 7.5, 20 mM), KCl
(10 mM), MgCl2 (1.5 mM), PMSF (0.1 mM), sucrose (250
mM), and EDTA (1 mM). Cells were homogenized and centrifuged at 750
× g for 10 min at 4°C. The sediments were the fraction of
cytoplasmic protein and were lysed in lysis buffer. Supernatants
were further centrifuged at 100,000 × g for 15 min at 4°C. The
obtained supernatants were the fraction of mitochondrial protein
and were lysed in lysis buffer as well. The concentration of
protein was determined by a modified Lowry assay (DC Protein assay;
Bio-Rad Laboratories, Inc., Hercules, CA, USA). SDS-PAGE and
western blot analysis were operated under standard protocols. GAPDH
was used as a housekeeping protein for cytosolic fraction and total
protein. COX IV was used as a housekeeping protein for
mitochondrial fraction.
For nucleoprotein extraction, the nuclear protein
was extracted with a ReadyPrep™ Protein Extraction kit
(Cytoplasmic/Nuclear) (#163-2089, Bio-Rad Laboratories, Inc.).
Briefly, for each 0.05 ml packed cells, 0.5 ml ice-cold cytoplasmic
protein extraction buffer was added. This was followed by vortexing
to suspend the cell pellet, and the cells were then incubated on
ice for 30 min to lyse the cells without damaging the nuclei. The
cell lysate was centrifuged at 1000 × g for 10 min at 4°C. The
supernatant contained the cytoplasmic proteins and the pellet in
the tube contained nuclei. The nuclei was suspended in 0.5 ml
protein solubilization buffer. The tube was vortexed to solubilize
the nuclear proteins and centrifuged at maximum speed (12–16,000 ×
g) for 15–20 min at room temperature. The clarified supernatant was
the nuclear protein fraction.
Statistical analysis
All data are expressed as the means ± standard
deviation of at least three independent experiments. Student's
t-test was examined for statistical analysis and a P-value <0.05
was considered to indicate a statistically significant
difference.
Results
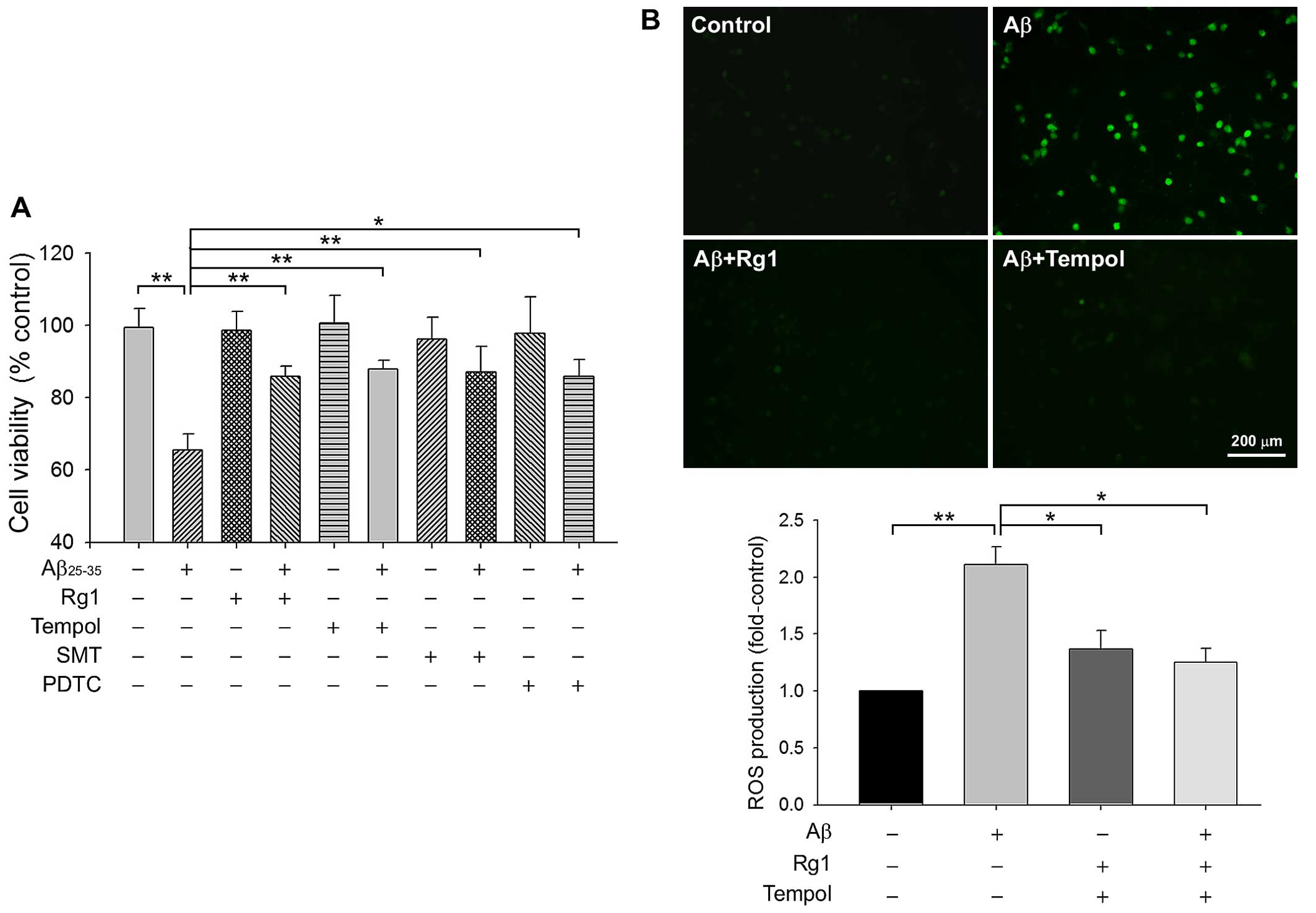
Rg1 inhibits Aβ25–35-induced
neuronal cell death by decreasing ROS accumulation
Primary rat cortical neuronal cultures were
preincubated with Rg1 or different inhibitors for 24 h, followed by
exposure to Aβ25–35 10 µM for 72 h. We reported
previously that Rg1 attenuated neural injury induced by 10
µM Aβ25–35 in a dose-dependent manner, and the
20-µM dose which was shown to be the optimal concentration
for Rg1 was thus used in subsequent experiments (7). The viability of neuronal cultures,
established by MTT metabolism, decreased by nearly 35% in
Aβ25–35-treated neurons compared to non-treated
controls. Tempol, a superoxide scavenger, was used as the
antioxidative positive control. Upon pretreatment with Rg1 and
tempol, MTT metabolism rose from 65.6±4.3% of the model group to
86.0±2.8% and 87.9±2.5% in the presence of 20 µM Rg1 and 10
µM tempol, respectively (Fig.
1A). We subsequently evaluated changes in ROS production
following Aβ25–35 exposure using the fluorescent dye
carboxy-DCFDA. After exposure to Aβ25–35 for 24 h, the
fluorescence intensity of ROS was significantly augmented
(2.11±0.16-fold) (Fig. 1B)
compared with the control. Both Rg1 and tempol reduced
Aβ25-35-triggered ROS release almost to the baseline
level. These data suggest that Rg1 inhibited
Aβ25–35-induced neuronal death, at least in part through
the ROS-scavenging pathway.
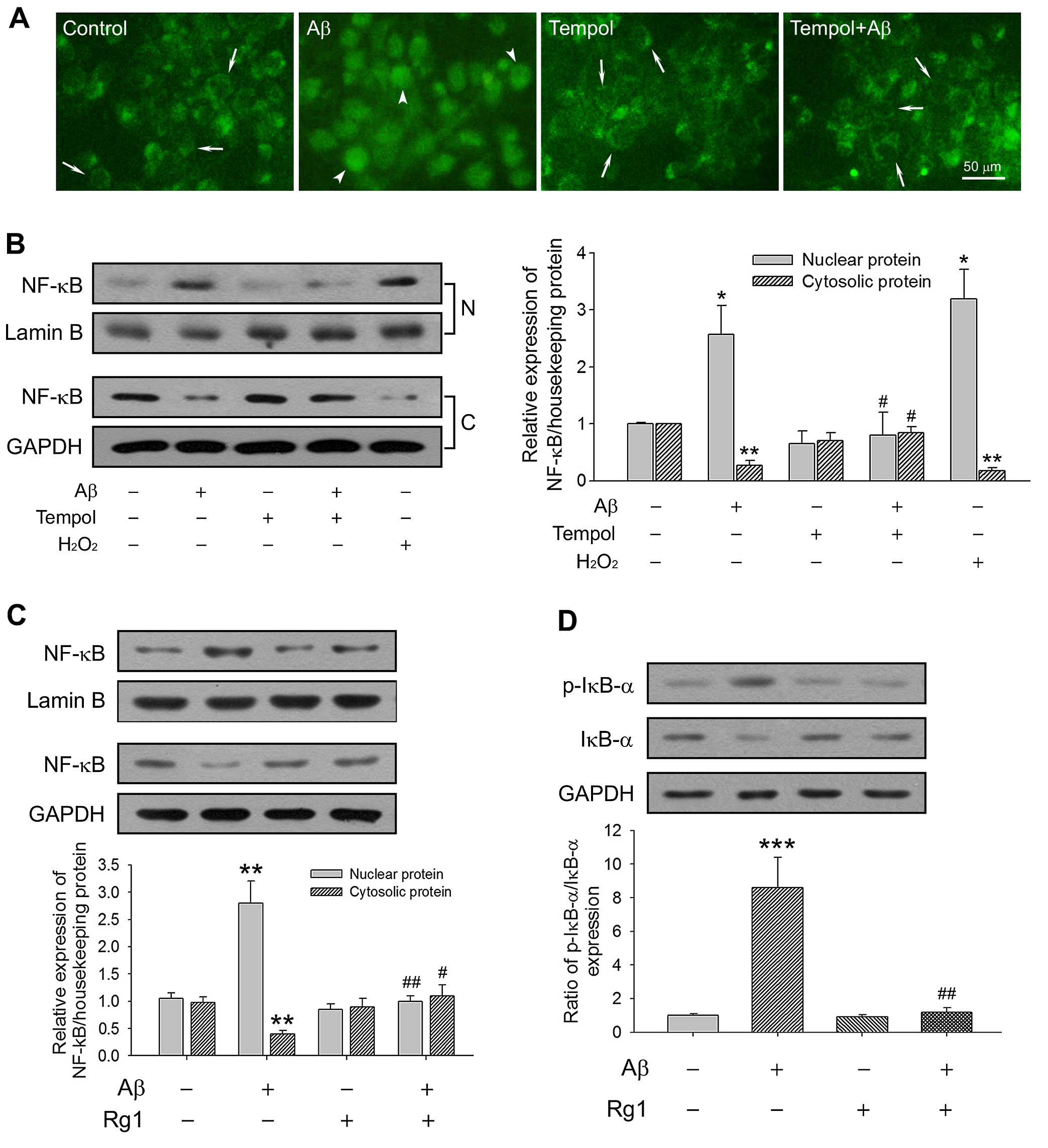
Rg1 compromises Aβ-triggered NF-κB
activation through its antioxidative effects
NF-κB is regarded as a reduction/oxidation
(redox)-sensitive factor. Under healthy physiological conditions,
as a passive form, NF-κB combines with its inhibitor IκB-α to form
a cytoplasmic complex. When suffering from stress such as that
caused by ROS and Aβ, however, IκB-α is phosphorylated and
degraded, subsequently freeing NF-κB to translocate to the nucleus
and stimulate the expression of target genes (21,22). To confirm whether Aβ-induced NF-κB
activation is oxidative stress-dependent, we investigated NF-κB
nuclear translocation in primary cultured neurons. As shown in
Fig. 2A, immunofluorescence
imaging demonstrated that the level of NF-κB (p65) nuclear
translocation significantly increased after Aβ25–35
treatment for 24 h, and tempol pretreatment counteracted this
effect. The modulating effects of Rg1 and tempol on the expression
of NF-κB and its depressor IκB-α were further tested by western
blot analysis of the cytoplasmic and nuclear fractions. Consistent
with the immunofluorescence imaging results, Aβ25-35
markedly enhanced the NF-κB level in the nuclear fractions of
primary neurons, while the level was decreased in cytoplasmic
fractions (P<0.05 and P<0.01, respectively). Tempol
pretreatment decreased Aβ25–35-induced NF-κB activation,
while H2O2 exposure aggravated it (Fig. 2B). As an antioxidant, Rg1 also
exerted a positive effect in terms of the suppression NF-κB nuclear
translocation, and stabilized accumulated IκB-α stimulated by
Aβ25–35 through restraint of IκB-α phosphorylation
(Fig. 2C and D). Our results
indicated that Rg1 decreased Aβ25–35-stimulated NF-κB
activation by 'mopping up' redundant cellular ROS in primary
neurons.
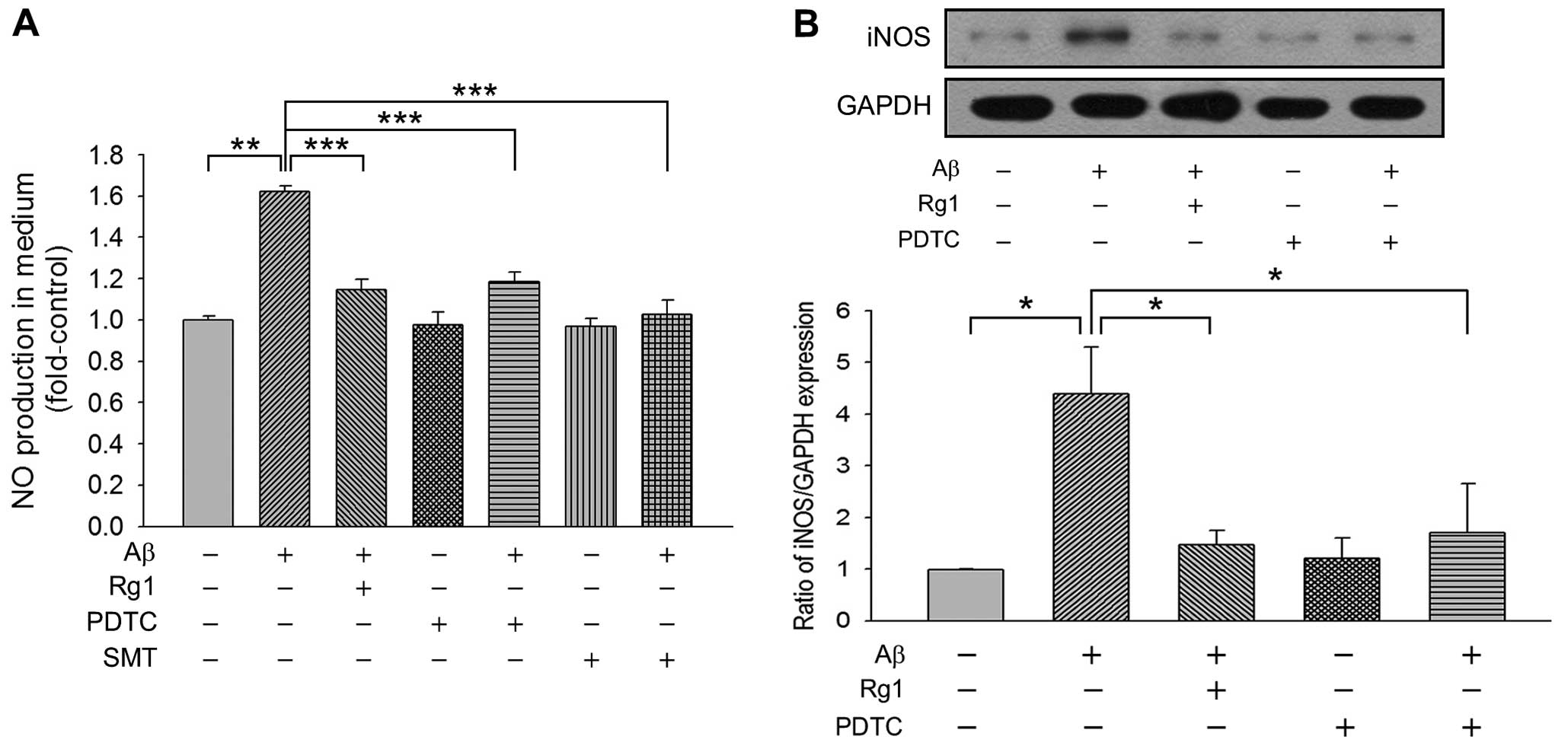
Rg1 represses Aβ-induced NO production in
an NF-κB-dependent manner
Since NO, ROS and endoplasmic reticulum (ER) stress
have been shown to be involved in Aβ25–35 insult, we
detected NO production using Griess reagent in primary cortical
neuronal cells. As shown in Fig.
3A, Aβ25–35 incubation for 24 h caused marked
upregulation in NO synthesis (1.62±0.08-fold vs. control,
P<0.01), whereas Rg1 significantly decreased
Aβ25–35-mediated NO generation to 1.16±0.10-fold.
Notably, the NF-κB inhibitor PDTC also decreased NO production, as
well as iNOS-specific inhibitor SMT, suggesting that iNOS is
responsible for Aβ25–35-induced NO release. We further
explored the expression of iNOS in primary cortical neurons and
found that the level of iNOS was markedly elevated after
Aβ25–35 treatment. Both Rg1 and PDTC exerted distinctly
negative effects on the expression of iNOS, suggesting that NF-κB
is a pivotal regulatory factor which is targeted by Rg1 in
precluding Aβ25–35-mediated NO production (Fig. 3B). In addition, as shown in
Fig. 1A, both PDTC and SMT
treatment reduced the cell death caused by Aβ25–35
injury, suggesting that the NF-κB-mediated decrease in NO
production is cardinal to the protective effect exerted by Rg1 in
cases of Aβ25–35-induced neurotoxicity.
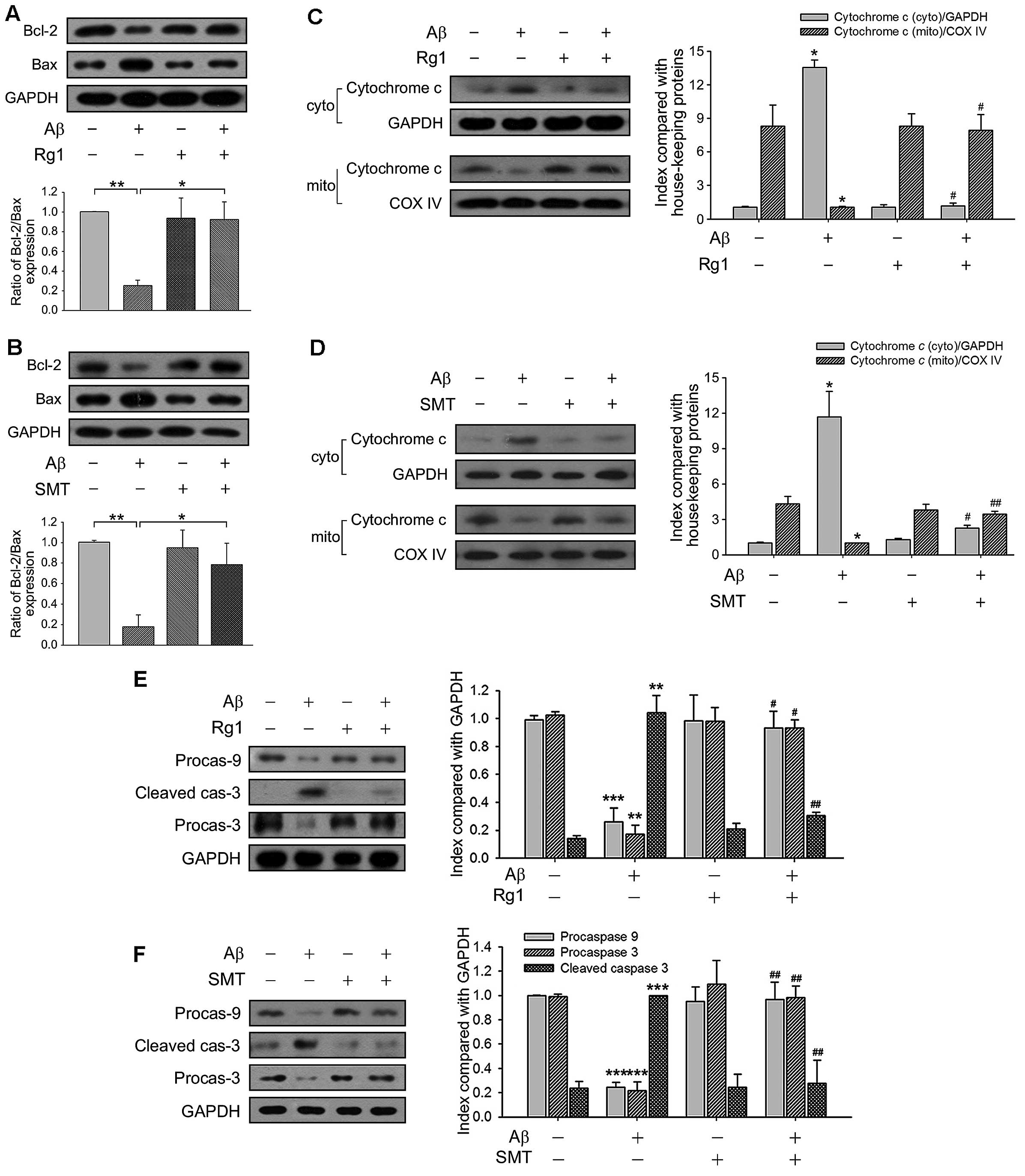
Rg1 hampers Aβ25–35-induced
mitochondrial apoptotic cascades by decreasing NO production
The ratio of Bcl-2/Bax expression is generally
recognized to be the controller of mitochondria permeability, which
modulates the release of cytochrome c from the mitochondria
to cytoplasm during mitochondrion-mediated apoptotic cascades
(23). Compared with the control,
Aβ25–35 exposure diminished the level of Bcl-2, while
raised the level of Bax, resulting in a low proportion of
Bcl-2/Bax. Rg1 exerted neuroprotective effects and elevated the
ratio of Bcl-2/Bax, as did SMT (Fig.
4A and B). We subsequently prepared both the cytosolic and
mitochondria-rich fractions of primary neurons, in order to verify
the subcellular distribution of cytochrome c. In the
Aβ25–35-exposed group, the expression of cytochrome
c in the cytosol increased, whereas mitochondrial cytochrome
c content decreased. By contrast, endogenous cytochrome
c expression in the control and Rg1-treated group was
detected to be mostly in mitochondria (Fig. 4C). The decrease in NO production
by SMT also potentiated the inhibition of cytochrome c
release from the mitochondria (Fig.
4D). The full-length pro-form of caspase-9 and caspase-3 were
used to assess caspase-9 and caspase-3 activation through their
cleavage, indicated here by the loss of signal for the pro-form.
Both Rg1 and SMT markedly affected Aβ25–35-induced
caspase activation (Fig. 4E and
F), indicating that Rg1 blocked Aβ25–35-induced
mitochondrion-mediated apoptosis through suppression of NO
generation.
Discussion
Previous studies have suggested that Rg1 exerts
neuroprotective properties in vitro and in vivo
(7,24–26), but the underlying mechanisms are
yet been fully understood. In the present study, we indicated that
Rg1 protects primary cultured rat cortical neuronal cells from
Aβ25–35-ignited apoptosis via the NF-κB/NO signaling
pathway.
NF-κB, a typical redox-sensitive factor, plays a
central role in the regulation of transcription, inflammation,
oxidative stress, and apoptosis (27). Much evidence has shown that
inflammation is relevant to AD. The deposition of Aβ has been
revealed to activate neuroinflammatory responses by triggering the
expression of inflammatory cytokines, chemokines and mediators
through the NF-κB and mitogen-activated protein kinase (MAPK)
signaling pathways (28).
Moreover, it has previously been reported that the level of NF-κB
(p65) is markedly elevated in the brains of patients with AD, and
NF-κB (p65) activation further leads to upregulated β-secretase
cleavage and Aβ deposition (29),
suggesting that NF-κB plays a pivotal role in AD pathological
processes. In the present study, we found that both Aβ and
H2O2 triggered the activation of NF-κB
(Fig. 2). Furthermore, Rg1 was
also proven to exert neuroprotective effects (Fig. 1). Thus, it is reasonable to
suggest that Rg1 reduced Aβ-induced NF-κB activation by
upregulating intracellular antioxidation.
NO, an important cell signaling molecule, is
generated from the amino acid L-arginine by NOSs, and it has been
suggested that iNOS is the main source of pathological NO
generation (30,31). However, it has also been
demonstrated that Aβ exposure increases the level of iNOS and the
production of NO (32), but the
signaling cascades involved in Aβ stimulation and NO generation
remain ambiguous. Certain studies have documented close interaction
between NO and NF-κB, but the modes of action between the two in
different cell types were quite different. It has been reported
that NO is the pivotal controller of the signaling pathways,
facilitating IL-1 to NF-κB activation in chondrocytes (33). On the other hand, the level of
iNOS is dependent on the activation of NF-κB in microglial cells
(34). Futhermore, it has also
been reported that iNOS gene expression directly blocked the
phosphorylation and the subsequent degradation of IκB-α, and NO
inhibits cytokine-induced NF-κB activation in rat vascular smooth
muscle cells (35). In the
present study, we demonstrated that Rg1 and the NF-κB inhibitor
PDTC considerably reduced iNOS expression and NO generation, which
were stimulated by Aβ25–35 (Fig. 3), suggesting that the decline of
NO synthesis through inactivation of NF-κB is one of the most
important mechanisms of Rg1 in the defense of primary neurons
against Aβ25–35.
Mitochondria play a major role in apoptosis
triggered by many stimuli, which is relevant to various
neurodegenerative disorders, such as AD, Parkinson's disease (PD)
and Huntington's disease (HD) (36). Neuronal exposure to Aβ impairs
mitochondrial dynamics and function; loss of mitochondrial membrane
potential (ΔΨm) in primary neurons was observed after incubation
with Aβ25–35 (7). This
was a hallmark of mitochondrial dysfunction and may subsequently
stimulate mitochondrial apoptotic signaling (37). The Bcl-2 family is made up of pro-
and anti-apoptotic members. As an antiapoptotic protein, Bcl-2 has
the ability to suppress the release of cytochrome c from
mitochondria, to hinder apop-tosis, whereas Bax is a proapoptotic
protein and acts as a promoter of apoptosis. As a result, the
proportion of Bcl-2/Bax serves as a kind of an on-off switch,
regulating cytochrome c release from mitochondria and the
downstream mitochondrial apoptotic pathway (38). The findings of our present study
demonstrated that Rg1 increased the ratio of Bcl-2/Bax and
decreased the following release of cytochrome c from the
mitochondrion in primary cultured neurons after Aβ25–35
exposure, while the effects were imitated by iNOS inhibitor SMT
(Fig. 4). Thus, the reduction of
NO generation likely contributed to the antiapoptotic activities of
Rg1 in primary cultured neuronal cells after Aβ25–35
exposure.
In conclusion, our study demonstrated that Rg1
exerts neuro-protective effects on primary cultured rat cortical
neuronal cells against Aβ25–35 injury by interfering
with mitochondrial apoptotic pathways via downregulation of the
NF-κB/NO signaling pathway.
Abbreviations:
|
AD
|
Alzheimer's disease
|
|
Aβ25–35
|
amyloid β-peptide 25–35
|
|
carboxy-DCFDA
|
5-(and-6)-carboxy-2′,7′dichloro-fluorescein diacetate
|
|
DMSO
|
dimethyl sulfoxide
|
|
GAPDH
|
glyceraldehyde 3-phosphate
dehydrogenase
|
|
H2O2
|
hydrogen peroxide
|
|
iNOS
|
inducible nitric oxide synthase
|
|
MTT
|
3-(4,5-dimeth-ylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
|
|
NF-κB
|
nuclear factor κ-light-chain-enhancer
of activated B cells
|
|
NO
|
nitric oxide
|
|
PDTC
|
ammonium
pyrrolidinedithiocarbamate
|
|
Rg1
|
ginsenoside Rg1
|
|
ROS
|
reactive oxygen species
|
|
SMT
|
s-methylisothiourea hemi-sulfate
salt
|
|
tempol
|
4-hydroxy-2,2,6,6-tetramethyl-piperidine 1-oxyl
|
Acknowledgments
The present study was supported by the National
Natural Sciences Foundation of China (no. 81402907), the Zhejiang
Provincial Natural Science Foundation of China (no. LQ14H310002)
and the Zhejiang Medical Technology Program (no. 201474924).
References
|
1
|
Cummings JL: Alzheimer's disease. N Engl J
Med. 351:56–67. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hampel H: Amyloid-β and cognition in aging
and Alzheimer's disease: molecular and neurophysiological
mechanisms. J Alzheimers Dis. 33(Suppl 1): S79–S86. 2013.
|
|
3
|
Ingelsson M, Fukumoto H, Newell KL,
Growdon JH, Hedley-Whyte ET, Frosch MP, Albert MS, Hyman BT and
Irizarry MC: Early Abeta accumulation and progressive synaptic
loss, gliosis, and tangle formation in AD brain. Neurology.
62:925–931. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mormino EC, Brandel MG, Madison CM, Marks
S, Baker SL and Jagust WJ: Aβ deposition in aging is associated
with increases in brain activation during successful memory
encoding. Cereb Cortex. 22:1813–1823. 2012. View Article : Google Scholar :
|
|
5
|
Huang Y and Mucke L: Alzheimer mechanisms
and therapeutic strategies. Cell. 148:1204–1222. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ardah MT, Paleologou KE, Lv G, Menon SA,
Abul Khair SB, Lu JH, Safieh-Garabedian B, Al-Hayani AA, Eliezer D,
Li M and El-Agnaf OM: Ginsenoside Rb1 inhibits fibrillation and
toxicity of alpha-synuclein and disaggregates preformed fibrils.
Neurobiol Dis. 74:89–101. 2015. View Article : Google Scholar
|
|
7
|
Wu J, Pan Z, Wang Z, Zhu W, Shen Y, Cui R,
Lin J, Yu H, Wang Q, Qian J, et al: Ginsenoside Rg1 protection
against β-amyloid peptide-induced neuronal apoptosis via estrogen
receptor α and glucocorticoid receptor-dependent anti-protein
nitration pathway. Neuropharmacology. 63:349–361. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Xie CL, Li JH, Wang WW, Zheng GQ and Wang
LX: Neuroprotective effect of ginsenoside-Rg1 on cerebral
ischemia/reperfusion injury in rats by downregulating
protease-activated receptor-1 expression. Life Sci. 121:145–151.
2015. View Article : Google Scholar
|
|
9
|
Zhu J, Mu X, Zeng J, Xu C, Liu J, Zhang M,
Li C, Chen J, Li T and Wang Y: Ginsenoside Rg1 prevents cognitive
impairment and hippocampus senescence in a rat model of
D-galactose-induced aging. PLoS One. 9:e1012912014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gilmore TD: Introduction to NF-kappaB:
players, pathways, perspectives. Oncogene. 25:6680–6684. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mattson MP and Camandola S: NF-kappaB in
neuronal plasticity and neurodegenerative disorders. J Clin Invest.
107:247–254. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Merlo E, Freudenthal R and Romano A: The
IkappaB kinase inhibitor sulfasalazine impairs long-term memory in
the crab Chasmagnathus. Neuroscience. 112:161–172. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Karin M: NF-kappaB as a critical link
between inflammation and cancer. Cold Spring Harb Perspect Biol.
1:a0001412009. View Article : Google Scholar
|
|
14
|
Boissière F, Hunot S, Faucheux B,
Duyckaerts C, Hauw JJ, Agid Y and Hirsch EC: Nuclear translocation
of NF-kappaB in cholinergic neurons of patients with Alzheimer's
disease. Neuroreport. 8:2849–2852. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kaltschmidt B, Uherek M, Volk B, Baeuerle
PA and Kaltschmidt C: Transcription factor NF-kappaB is activated
in primary neurons by amyloid beta peptides and in neurons
surrounding early plaques from patients with Alzheimer disease.
Proc Natl Acad Sci USA. 94:2642–2647. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kitamura Y, Shimohama S, Ota T, Matsuoka
Y, Nomura Y and Taniguchi T: Alteration of transcription factors
NF-kappaB and STAT1 in Alzheimer's disease brains. Neurosci Lett.
237:17–20. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Du J, Cheng B, Zhu X and Ling C:
Ginsenoside Rg1, a novel glucocorticoid receptor agonist of plant
origin, maintains glucocorticoid efficacy with reduced side
effects. J Immunol. 187:942–950. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu Q, Kou JP and Yu BY: Ginsenoside Rg1
protects against hydrogen peroxide-induced cell death in PC12 cells
via inhibiting NF-κB activation. Neurochem Int. 58:119–125. 2011.
View Article : Google Scholar
|
|
19
|
Wang Z, Zhang X, Wang H, Qi L and Lou Y:
Neuroprotective effects of icaritin against beta amyloid-induced
neurotoxicity in primary cultured rat neuronal cells via
estrogen-dependent pathway. Neuroscience. 145:911–922. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cristina de Assis M, Cristina Plotkowski
M, Fierro IM, Barja-Fidalgo C and de Freitas MS: Expression of
inducible nitric oxide synthase in human umbilical vein endothelial
cells during primary culture. Nitric Oxide. 7:254–261. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Brasier AR: The NF-kappaB regulatory
network. Cardiovasc Toxicol. 6:111–130. 2006. View Article : Google Scholar
|
|
22
|
Perkins ND: Integrating cell-signalling
pathways with NF-kappaB and IKK function. Nat Rev Mol Cell Biol.
8:49–62. 2007. View Article : Google Scholar
|
|
23
|
Ola MS, Nawaz M and Ahsan H: Role of Bcl-2
family proteins and caspases in the regulation of apoptosis. Mol
Cell Biochem. 351:41–58. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li YB, Wang Y, Tang JP, Chen D and Wang
SL: Neuroprotective effects of ginsenoside Rg1-induced neural stem
cell transplantation on hypoxic-ischemic encephalopathy. Neural
Regen Res. 10:753–759. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wu J, Pan Z, Cheng M, Shen Y, Yu H, Wang Q
and Lou Y: Ginsenoside Rg1 facilitates neural differentiation of
mouse embryonic stem cells via GR-dependent signaling pathway.
Neurochem Int. 62:92–102. 2013. View Article : Google Scholar
|
|
26
|
Li W, Chu Y, Zhang L, Yin L and Li L:
Ginsenoside Rg1 attenuates tau phosphorylation in SK-N-SH induced
by Aβ-stimulated THP-1 supernatant and the involvement of p38
pathway activation. Life Sci. 91:809–815. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kabe Y, Ando K, Hirao S, Yoshida M and
Handa H: Redox regulation of NF-kappaB activation: distinct redox
regulation between the cytoplasm and the nucleus. Antioxid Redox
Signal. 7:395–403. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tuppo EE and Arias HR: The role of
inflammation in Alzheimer's disease. Int J Biochem Cell Biol.
37:289–305. 2005. View Article : Google Scholar
|
|
29
|
Chen CH, Zhou W, Liu S, Deng Y, Cai F,
Tone M, Tone Y, Tong Y and Song W: Increased NF-κB signalling
up-regulates BACE1 expression and its therapeutic potential in
Alzheimer's disease. Int J Neuropsychopharmacol. 15:77–90. 2012.
View Article : Google Scholar
|
|
30
|
Aktan F: iNOS-mediated nitric oxide
production and its regulation. Life Sci. 75:639–653. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Surh YJ1, Chun KS, Cha HH, Han SS, Keum
YS, Park KK and Lee SS: Molecular mechanisms underlying
chemopreventive activities of anti-inflammatory phytochemicals:
down-regulation of COX-2 and iNOS through suppression of NF-kappa B
activation. Mutat Res. 480–481:243–268. 2001. View Article : Google Scholar
|
|
32
|
Zara S, Di Stefano A, Nasuti C, Rapino M,
Patruno A, Pesce M, Sozio P, Cerasa LS and Cataldi A: NOS-mediated
morphological and molecular modifications in rats infused with Aβ
(1–40), as a model of Alzheimer's disease, in response to a new
lipophilic molecular combination codrug-1. Exp Gerontol.
46:273–281. 2011. View Article : Google Scholar
|
|
33
|
Mendes AF, Carvalho AP, Caramona MM and
Lopes MC: Role of nitric oxide in the activation of NF-kappaB, AP-1
and NOS II expression in articular chondrocytes. Inflamm Res.
51:369–375. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Pahan K, Sheikh FG, Liu X, Hilger S,
McKinney M and Petro TM: Induction of nitric-oxide synthase and
activation of NF-kappaB by interleukin-12 p40 in microglial cells.
J Biol Chem. 276:7899–7905. 2001. View Article : Google Scholar
|
|
35
|
Katsuyama K, Shichiri M, Marumo F and
Hirata Y: NO inhibits cytokine-induced iNOS expression and
NF-kappaB activation by interfering with phosphorylation and
degradation of IkappaB-alpha. Arterioscler Thromb Vasc Biol.
18:1796–1802. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Camins A, Pallas M and Silvestre JS:
Apoptotic mechanisms involved in neurodegenerative diseases:
experimental and therapeutic approaches. Methods Find Exp Clin
Pharmacol. 30:43–65. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Tillement L, Lecanu L and Papadopoulos V:
Alzheimer's disease: effects of β-amyloid on mitochondria.
Mitochondrion. 11:13–21. 2011. View Article : Google Scholar
|
|
38
|
Burlacu A: Regulation of apoptosis by
Bcl-2 family proteins. J Cell Mol Med. 7:249–257. 2003. View Article : Google Scholar : PubMed/NCBI
|