Introduction
Retinitis pigmentosa (RP; MIM: #268000) refers to a
heterogeneous group of inherited retinal diseases (IRDs) caused by
the loss of photoreceptors, which is characterized by night
blindness, progressive loss of peripheral vision in the early
stages, and complete loss of vision in the end stages. The
worldwide prevalence of RP is reported as approximately one case in
3,500–5,000 individuals (1). RP
is classified as non-syndromic and syndromic RP. Non-syndromic RP
is inherited in different patterns. Approximately 30–40% of cases
are autosomal dominant, 5–15% are inherited through X-linkage and
50–60% of cases are most likely autosomal recessive (2). Rarer forms also exist, such as
mitochondrial and digenic RP. Digenic RP occurs in individuals who
are heterozygous for both a retinal outer segment membrane protein
1 (ROM1) mutation and a peripherin 2 (retinal degeneration,
slow) (PRPH2) mutation (3).
RP is extremely heterogeneous. To date, mutations in
77 genes have been found to cause non-syndromic RP [Retinal
Information Network (RetNet) database, https://sph.uth.edu/retnet/sum-dis.htm]. There may be
many different disease-causing mutations in each gene. Different
mutations in the same gene may cause different diseases and the
same mutation in different individuals may produce different
clinical consequences, even among cases within the same family.
Moreover, the spectrum of mutations within a given gene may vary
between populations (1,3,4).
The extent of heterogeneity among patients with RP is a cause of
confusion to both patients and clinicians, and is a confounding
factor in the diagnosis of RP. However, the known causal genes
explain no more than half of the clinical cases of RP. Novel
causative genes remain to be discovered (2). The identification of the genetic
mutations causing RP improves our understanding of the disease
process and promotes the development of novel treatments. Several
methods are used to detect genetic variations and of these, Sanger
sequencing remains the gold-standard. Due to the heterogeneity of
RP, it is time consuming and expensive to screen all known genes.
Novel and efficient methods of screening mutations are therefore
necessary to aid in the detection of variations in known causal
genes and in the discovery of the remaining fraction of RP genes.
The development of high-throughput sequencing techniques over the
past 10 years has increased the power to detect variations
(5). Next-generation sequencing,
in particular whole-exome sequencing (WES), enables investigators
to perform the analysis of the coding regions of the human genome
in individuals or in small families, including patients in whom a
clear genotype-phenotype correlation is absent as well as to
analyze clinically and genetically heterogeneous conditions
(5). Previous research has
demonstrated that WES provides a promising alternative method for
the molecular diagnosis and identification of disease genes in
Mendelian disorders (6–9).
In the present study, we successfully used the WES
approach to determine the genetic basis of sporadic RP in two
Chinese patients. We have identified a causative mutation in the
prominin 1 (PROM1) gene that has not been previously
detected, to the best of our knowledge.
Subjects and methods
Subjects and clinical assessment
Two patients with sporadic RP (case#1 and case#2) as
well as family members (namely parents and siblings) from China
were recruited into this study. The present study was performed in
accordance with the Code of Ethics of the World Medical Association
(Declaration of Helsinki) for medical research involving human
subjects. The Institutional Review Boards of the Hospital of the
University of Electronic Science and Technology of China and
Sichuan Provincial People's Hospital (Chengdu, China) approved this
study. Written informed consent was obtained from all participants
or their guardians. All subjects were evaluated by a retina
specialist. A complete ophthalmological assessment of each family
member was performed, including best-corrected visual acuity
(BCVA), slit-lamp biomicroscopy, fundus photography, visual field
tests (Octopus; Interzeag, Schlieren, Switzerland), and
electroretinography (ERG). A clinical diagnosis of RP was based on
the presence of night blindness, severe defects of the peripheral
visual field, lesions in the fundus (bone-spicule-shaped pigment
deposits, vessel attenuation and various degrees of retinal
atrophy) and abnormal ERG measurements (marked diminution in the
amplitude of a- and b-waves or the complete absence of a response)
as well as family history.
The control subjects were recruited from the
Hospital of the University of Electronic Science and Technology of
China and Sichuan Provincial People's Hospital. All subjects
provided informed consent prior to participating in the study. The
200 normal matched controls underwent an eye examination and no
signs of eye diseases were observed.
DNA extraction
Blood (5 ml) from the probands, their family members
and the controls was collected in ethylenediaminetetraacetic acid
(EDTA) Vacutainer tubes. All genomic DNA was isolated from the
peripheral leukocytes using a QIAamp DNA Blood Midi kit (Qiagen,
Hilden, Germany) according to the manufacturer's instructions. The
DNA samples were stored at −20°C until use. DNA integrity was
evaluated by performing 1% agarose gel electrophoresis.
WES and data analysis
WES was performed on the genomic DNA samples
obtained from the two patients with sporadic RP by Axeq
Technologies, Inc. (Seoul, Korea). The samples for sequencing were
prepared according to the Illumina protocols. Illumina Exome
Enrichment and quality control analysis for the enriched library
were performed by Axeq Technologies Inc. A TruSeq Exome Enrichment
kit was used to enrich the coding regions of the human genome. It
covered 20,794 genes and 201,121 exons in the Consensus Coding
Sequence (CCDS) Region database, and approximately 97.2% of CCDS
exons or 96.4% of Reference Sequence (RefSeq) exons were captured
(http://www.illumina.com/products/truseq-exome.html).
WES data analysis was performed as described
previously (10,11). Briefly, raw data were processed by
Illumina base-calling software 1.7 using default parameters. The
sequencing reads were aligned to the human reference genome [hg19;
University of California, Santa Cruz (UCSC) Genome Browser;
http://genome.ucsc.edu/] using Burrows-Wheeler
Aligner (BWA) software (http://bio-bwa.sourceforge.net/). Sequence variants
[single nucleotide polymorphisms (SNPs) and short insertions or
deletions (indels)] were listed. The variants were annotated and
filtered against the following four public databases and one
in-house database: dbSNP138 (http://www.ncbi.nlm.nih.gov/projects/SNP/), 1000
Genomes Project (ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp), HapMap
Project (ftp://ftp.ncbi.nlm.nih.gov/hapmap), Exome Variant
Server for the NHLBI Exome Sequencing Project (http://evs.gs.washington.edu/EVS/), Exome
Aggregation Consortium (ExAC) database (http://exac.broadinstitute.org/) and our in-house
database generated from 1,977 WES samples. Two major steps were
performed to prioritize all the variants: i) synonymous variants,
intronic variants (>5 bp from exon boundaries) and common
variants (minor allele frequency, >1%) were excluded from
downstream analysis; ii) possible deleterious effects of each
variant on protein structure/function were predicted using the SIFT
algorithm (http://sift.bii.a-star.edu.sg/) and Polymorphism
Phenotyping v2 (Polyphen2; http://genetics.bwh.harvard.edu/pph2/). The variants
were classified as potentially pathogenic variants, variants of
unknown clinical significance or benign variants, in accordance
with the interpretation guidelines of the American College of
Medical Genetics and Genomics (ACMG) (12). Deleterious mutations and variants
of unknown clinical significance were further classified as being
related or unrelated to the proband phenotype. In the present
study, we focused on only 238 disease-causing genes for IRDs, and
prioritized 77 disease-causing genes for non-syndromic RP using the
RetNet database (https://sph.uth.edu/retnet/sum-dis.htm).
Validation of variants
After filtering the variants against multiple
databases, Sanger sequencing was used to determine whether any of
the candidate variants co-segregated with the disease phenotype in
these families. The primers were designed according to the genomic
sequences of the Human Genome database and synthesized by Sangon
Biotech (Shanghai, China). Sanger sequencing was performed
according to the manufacturer's protocols and the processed samples
were analyzed on an ABI 3730XL Genetic Analyzer (Applied Biosystems
Life Technologies, Foster City, CA, USA).
Results
Clinical data of the patients
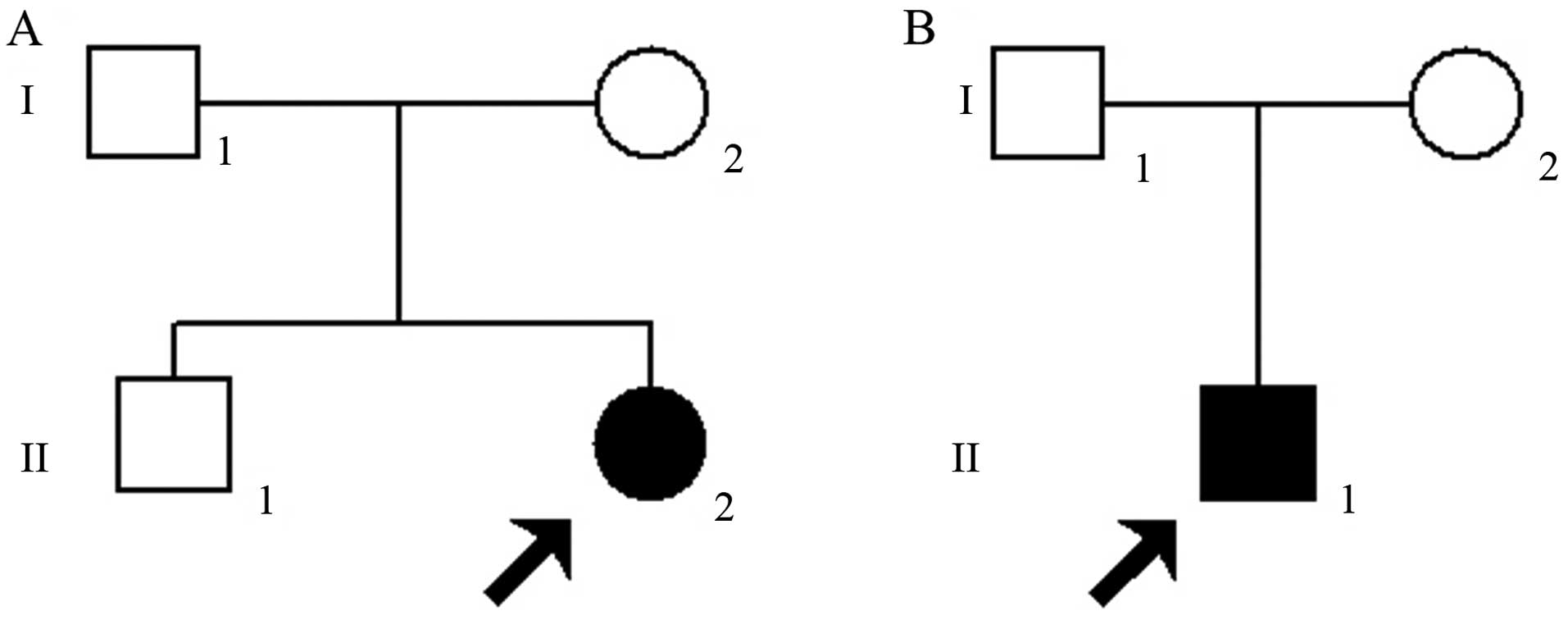
Case#1 (Fig. 1A)
developed night blindness at the age of 8. She presented with
severely impaired visual acuity [oculus dexter (OD), right eye,
0.05; oculus sinister (OS), left eye: 0.05] and severe defects of
the peripheral visual field at 26 years of age. Case#2 (Fig. 1B) experienced defective dark
adaptation at 14 years of age. At the age of 23, the patient
presented with markedly decreased visual acuity (OD, 0.04; OS,
0.04) in both eyes, and severe defects of the peripheral visual
field.
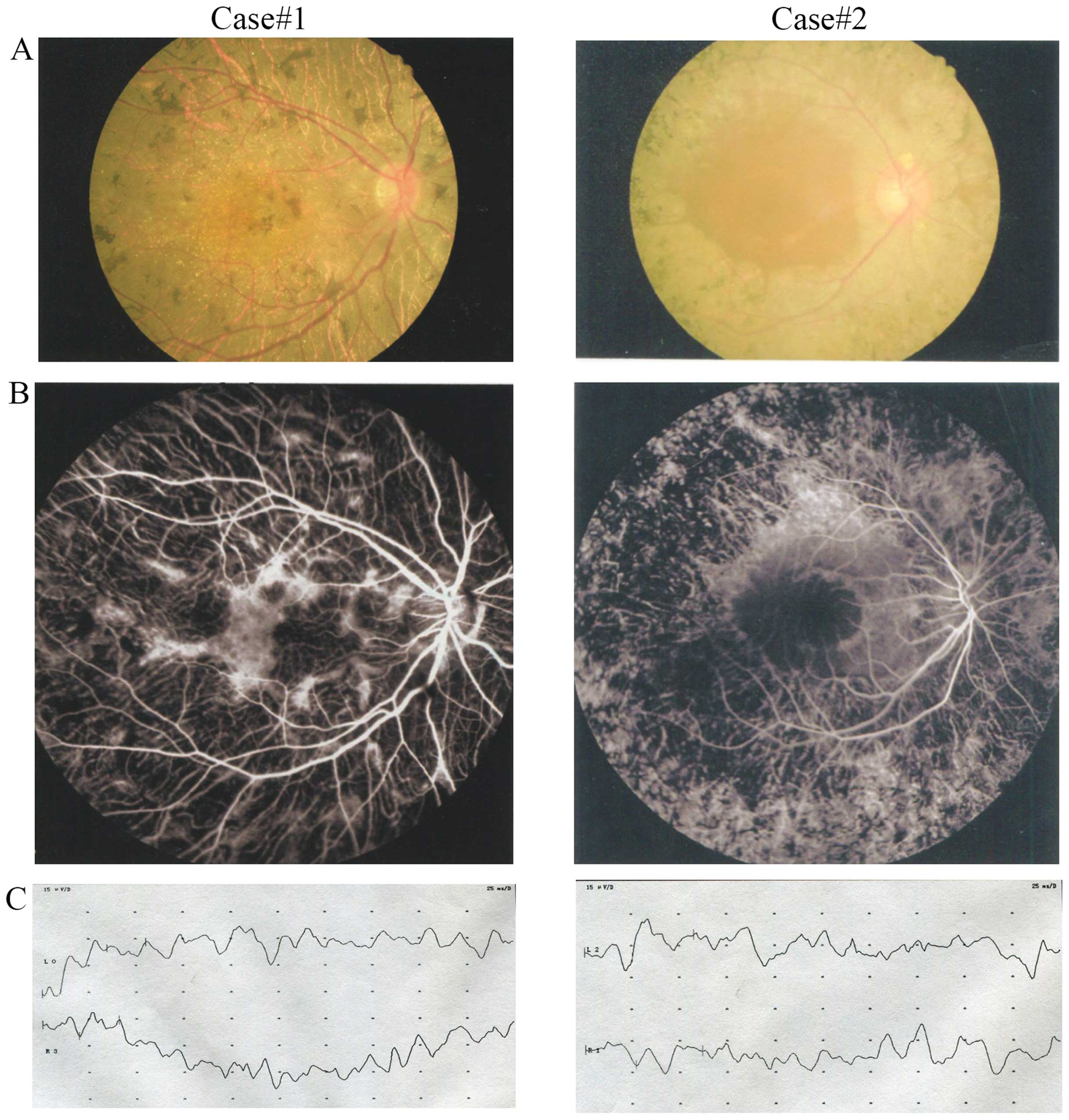
Fundus examination of the probands showed
bone-spicule-shaped pigment deposits, retinal vessel attenuation as
well as retinal atrophy (Fig. 2A and
B). Electroretinograms revealed no recordable responses under
either scotopic or photopic conditions, indicating a significant
loss of function of both the rods and the cones (Fig. 2C). An ophthalmological examination
of the other family members confirmed that they did not exhibit
symptoms of RP (data not shown). Since the parents of the probands
had no apparent symptoms of RP, the disease shows a pattern of
recessive inheritance in these families. In addition, regarding
de novo mutation, it is still possible that the disease
shows a pattern of autosomal dominant inheritance in these
families.
Identification of disease-causing
candidate variants
To identify the causative mutation in the patients
affected by RP, WES was performed on the genomic DNA with mean read
depths across the targeted regions (42.3× and 57.6×) for each
proband. In the genomic sample from case#1, 69,584 variants were
initially identified, including 18,753 variants in the exonic and
splicing regions. WES data analysis of the sample from case #2
identified 72,048 variants, including 20,690 variants in the exonic
and splicing regions. We prioritized the functional SNPs/indels in
homozygous or heterozygous mutations, including non-synonymous (NS)
variants, splice acceptor and donor site mutations (SS), and
frameshift indels in coding regions, which were more likely to be
pathogenic mutations. These variants were then compared with the
databases (dbSNP138, 1000 Genomes Project, HapMap Project, ExAC
database and our in-house database generated from 1,977 WES
samples). We focused on only 238 disease-causing genes for IRDs and
prioritized 77 disease-causing genes for non-syndromic RP.
Homozygous or compound heterozygous variants were filtered for
potential autosomal recessive RP-causing mutations. Regarding de
novo mutations, it is still possible that the disease shows a
pattern of autosomal dominant inheritance in these families.
Heterozygous variants in the disease-causing genes for autosomal
dominant retinal diseases were also filtered for potential
causative mutations. The filtered data were narrowed down to 4
variants in case#1 and 10 variants in case#2 (Table I).
 | Table ICandidate exome sequence variants in
retinal disease-causing genes identified after filtering case#1 and
case#2 data against various databases. |
Table I
Candidate exome sequence variants in
retinal disease-causing genes identified after filtering case#1 and
case#2 data against various databases.
| Case no. | Gene | Chromosome
position | Transcript ID | Gene region | Transcript
variant | Protein variant | Function
prediction | Hom/Het | 1000 G frequency | Inheritance | Phenotype |
|---|
| 1 | IDH3B | chr20:2640199 | NM_174855 | Exon11 | c.1043C>T | p.A348V | Damaging | Het | – | AR | RP46 |
| PROM1 |
chr4:16002224-16002225 | NM_001145851 | Exon12 | c.1445dupT | p.F482fs | – | Hom | – | AD/AR |
CORD10/STGD4/RP41 |
| ABCA4 | chr1:94495135 | NM_000350 | Exon30 | c.4405A>G | p.N1469D | Tolerated | Het | – | AR | CORD3/STGD1/RP19 |
| TULP1 | chr6:35477095 | NM_003322 | Intron7 | c.719-6G>A | – | – | Het | – | AR | LCA15/RP14 |
| 2 | PROM1 |
chr4:16002224-16002225 | NM_001145851 | Exon12 | c.1445dupT | p.F482fs | – | Hom | – | AD/AR |
CORD10/STGD4/RP41 |
| CEP290 | chr12:88453691 | NM_025114 | Exon48 | c.6629G>A | p.R2210H | Probably
damaging | Het | – | AR | BBS14/LCA10 |
| CNGB1 | chr16:58001141 | NM_001135639 | Exon2 | c.50G>A | p.R17Q | Tolerated | Het | – | AR | RP45 |
| EYS | chr6:65596602 | NM_001142800 | Exon19 | c.2980C>G | p.P994A | Probably
damaging | Het | 0.0010 | AR | RP25 |
| HARS | chr5:140056309 | NM_001289093 | Exon7 | c.782G>A | p.R261H | Tolerated | Het | 0.0010 | AD/AR | UHS3B |
| NR2E3 | chr15:72106440 | NM_014249 | Exon8 | c.1082A>G | p.H361R | Tolerated | Het | 0.0023 | AD/AR | RP37 |
| OPA1 | chr3:193336676 | NM_130831 | Exon4 | c.467C>T | p.A156V | Probably
damaging | Het | 0.0103 | AD | OPA1 |
| SEMA4A | chr1:156126297 | NM_001193300 | Exon3 | c.232G>A | p.V78M | Probably
damaging | Het | – | AD/AR | CORD10/RP35 |
| USH2A | chr1:215822102 | NM_206933 | Exon66 | c.14350G>A | p.E4784K | Probably
damaging | Het | – | AR | RP39 |
| WFS1 | chr4:6302507 | NM_001145853 | Exon8 | c.985T>A | p.F329I | Probably
damaging | Het | – | AD/AR | Wolfram
syndrome |
Mutation detection and validation by
Sanger sequencing
Three variants in case#1 (DH3B, ABCA4
and TULP1) were excluded as autosomal recessive-IRD-causing
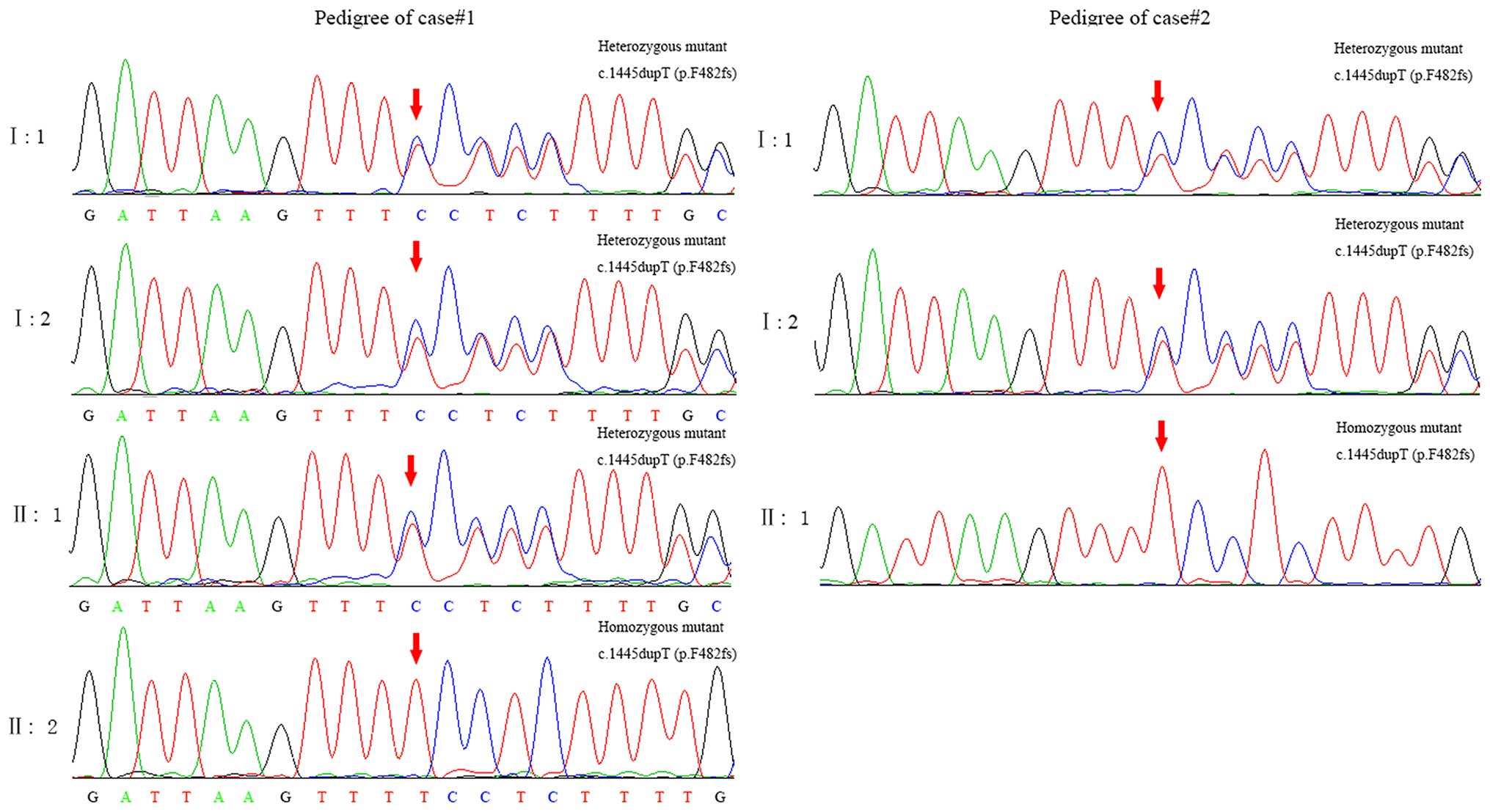
mutations. A homozygous frameshift mutation, c.1445dupT (p.F482fs)
in exon 12 of PROM1 (MIM: 604365) satisfied a recessive
inheritance model. Sanger sequencing confirmed that the affected
sibling (case#1 proband, II: 2) was homozygous for c.1445dupT
(p.F482fs) whereas her parents (I: 1 and I:2) and older brother
(II: 1) were unaffected heterozygous carriers of c.1445dupT
(p.F482fs), showing complete co-segregation of the mutation with
the disease phenotype in this family (Fig. 3). Five candidate variants for
autosomal dominant-IRD-causing mutations (HARS,
NR2E3, OPA1, SEMA4A and WFS1) were
found in the samples from the father or the mother of case#2 by
Sanger sequencing. The c.1445dupT mutation was also observed in
case#2 (Fig. 3). Furthermore, the
parents of case#2 were found to carry the same heterozygous
mutation.
This mutation was not detected by Sanger sequencing
in 200 ethnically-matched control samples (data not shown). Taken
together with the clinical presentation of the patients with RP,
these data demonstrate that the homozygous mutation, c.1445dupT
(p.F482fs) in the PROM1 gene is a causative mutation for RP.
The frame-shift mutation disrupts the amino acid sequence starting
at position 482; this region is highly evolutionarily conserved for
PROM1, which was confirmed by the analysis of orthologs from
8 different species (Fig. 4).
Discussion
In the present study, we have identified a novel
homozygous frameshift mutation, c.1445dupT (p.F482fs), in the
PROM1 gene that appears to have caused autosomal recessive
RP in two Chinese patients with sporadic RP. The mutation
co-segregated with the disease as the two affected individuals were
homozygous, whereas the parents of case#1 and case#2 carried only
one mutation. The mutation was absent in the 200 normal controls.
The affected individuals presented with classic RP disease and
rapidly progressed to show severe visual impairment.
The PROM1 gene (MIM: 604365) is located at
4p15.32 and encodes a five-transmembrane (TM) domain glycoprotein
with two short N (extracellular)- and C (cytoplasmic)-terminal
tails, and two large N-glycosylated extracellular loops (between
TM2 and -3, and TM4 and -5). PROM1 has been identified as a
hematopoietic, neuroepithelial and cancer stem cell marker
(13–15). PROM1 expression is
widespread throughout human tissues, including rod and cone
photoreceptors. In the retina, PROM1 is found at the base of
the photoreceptor outer segments (OSs). The function of
PROM1 in the retina remains unknown; however, it has been
demonstrated that mutations in PROM1 result in autosomal
recessive RP, autosomal dominant macular degeneration and cone-rod
dystrophy (16–23). To date, 35 different PROM1
mutations have been reported (Human Gene Mutation Database,
http://www.hgmd.cf.ac.uk/). Notably, there is an
association between the mutation type in PROM1 and the
phenotype. Missense mutations are associated with autosomal
dominant bull's eye maculopathy (20). Nonsense and frameshift mutations
have been associated with autosomal recessive RP and severe
cone-rod dystrophy with macular degeneration and night blindness
(17,19,23).
In the present study, we have identified a novel
homozygous frameshift mutation in the PROM1 gene that is
responsible for severe RP in two patients with sporadic RP from
China. Sanger sequencing of case#1 confirmed that the unaffected
sibling (II: 1) of the proband, her father (I: 1) and mother (I: 2)
were found to carry the mutation c.1445dupT (p.F482fs) in a
heterozygous state. The mutation shows complete co-segregation with
the disease phenotype in this family. The same mutation was then
found in an unrelated patient with RP (case#2). It remains
difficult to define a disease-causing mutation, particularly as
there are no readily available functional assays to determine the
phenotypic effects of specific variants. The potential
pathogenicity of the filtered variants was then interpreted
according to the existing and proposed ACMG guidelines (24). This frame-shift mutation resulted
in a completely different amino acid sequence from 483-amino acid
residues of PROM1 gene. The frame-shift mutation occurred in
half of the gene in a region that is highly conserved for
PROM1, which was demonstrated by analyzing orthologs from 9
different species, namely Homo sapiens, Pan troglodytes, Macaca
mulatta, Canis lupus, Mus musculus, Rattus norvegicus, Gallus
gallus and Danio rerio. Such a high degree of conservation
indicates the functional importance of the relevant amino acid
sequence. This mutation was absent in the 200 normal controls and
public databases including 1000 Genomes Project, Exome Variant
Server for the NHLBI Exome Sequencing Project (http://evs.gs.washington.edu/EVS/), ExAC database
(http://exac.broadinstitute.org/) and our
in-house database generated from 1,977 WES samples, excluding them
as common polymorphisms.
In conclusion, a homozygous mutation c.1445dupT
(p.F482fs) in the PROM1 gene was identified in two Han
Chinese subjects with RP by WES. The novel homozygous mutation in
PROM1 appears to be the cause of RP. The mutation further
expands the existing spectrum of PROM1 mutations in patients
with RP, thereby assisting in the molecular diagnosis of RP and
enhancing our understanding of genotype-phenotype correlations in
order to provide effective genetic counseling.
Acknowledgments
The present study was supported by the National
Natural Science Foundation of China (grant no. 81271048) and the
Foundation for Transfer of Scientific and Technological
Achievements of Sichuan Province (grant no. 15010118).
References
|
1
|
Chizzolini M, Galan A, Milan E, Sebastiani
A, Costagliola C and Parmeggiani F: Good epidemiologic practice in
retinitis pigmentosa: from phenotyping to biobanking. Curr
Genomics. 12:260–266. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hartong DT, Berson EL and Dryja TP:
Retinitis pigmentosa. Lancet. 368:1795–1809. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ferrari S, Di Iorio E, Barbaro V, Ponzin
D, Sorrentino FS and Parmeggiani F: Retinitis pigmentosa: genes and
disease mechanisms. Curr Genomics. 12:238–249. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Schorderet DF and Escher P: NR2E3
mutations in enhanced S-cone sensitivity syndrome (ESCS),
Goldmann-Favre syndrome (GFS), clumped pigmentary retinal
degeneration (CPRD), and retinitis pigmentosa (RP). Hum Mutat.
30:1475–1485. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ku CS, Cooper DN, Polychronakos C, Naidoo
N, Wu M and Soong R: Exome sequencing: dual role as a discovery and
diagnostic tool. Ann Neurol. 71:5–14. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ng SB, Buckingham KJ, Lee C, Bigham AW,
Tabor HK, Dent KM, Huff CD, Shannon PT, Jabs EW, Nickerson DA, et
al: Exome sequencing identifies the cause of a Mendelian disorder.
Nat Genet. 42:30–35. 2010. View
Article : Google Scholar :
|
|
7
|
Worthey EA, Mayer AN, Syverson GD,
Helbling D, Bonacci BB, Decker B, Serpe JM, Dasu T, Tschannen MR,
Veith RL, et al: Making a definitive diagnosis: successful clinical
application of whole exome sequencing in a child with intractable
inflammatory bowel disease. Genet Med. 13:255–262. 2011. View Article : Google Scholar
|
|
8
|
Yang Y, Muzny DM, Reid JG, Bainbridge MN,
Willis A, Ward PA, Braxton A, Beuten J, Xia F, Niu Z, et al:
Clinical whole-exome sequencing for the diagnosis of Mendelian
disorders. N Engl J Med. 369:1502–1511. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yang Y, Muzny DM, Xia F, Niu Z, Person R,
Ding Y, Ward P, Braxton A, Wang M, Buhay C, et al: Molecular
findings among patients referred for clinical whole-exome
sequencing. JAMA. 312:1870–1879. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhou Y, Tao S, Chen H, Huang L, Zhu X, Li
Y, Wang Z, Lin H, Hao F, Yang Z, et al: Exome sequencing analysis
identifies compound heterozygous mutation in ABCA4 in a Chinese
family with Stargardt disease. PLoS One. 9:e919622014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gong B, Wei B, Huang L, Hao J, Li X, Yang
Y, Zhou Y, Hao F, Cui Z, Zhang D, et al: Exome sequencing
identified a recessive RDH12 mutation in a family with severe
early-onset retinitis pigmentosa. J Ophthalmol.
2015(942740)2015.PubMed/NCBI
|
|
12
|
Richards CS, Bale S, Bellissimo DB, Das S,
Grody WW, Hegde MR and Lyon E: ACMG recommendations for standards
for interpretation and reporting of sequence variations: Revisions
2007. Genet Med. 10:294–300. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bauer N, Fonseca AV, Florek M, Freund D,
Jászai J, Bornhäuser M, Fargeas CA and Corbeil D: New insights into
the cell biology of hematopoietic progenitors by studying
prominin-1 (CD133). Cells Tissues Organs. 188:127–138. 2008.
View Article : Google Scholar
|
|
14
|
Walker TL, Wierick A, Sykes AM, Waldau B,
Corbeil D, Carmeliet P and Kempermann G: Prominin-1 allows
prospective isolation of neural stem cells from the adult murine
hippocampus. J Neurosci. 33:3010–3024. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Akita M, Tanaka K, Matsumoto S, Komatsu K
and Fujita K: Detection of the hematopoietic stem and progenitor
cell marker CD133 during angiogenesis in three-dimensional collagen
gel culture. Stem Cells Int. 2013(927403)2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang Q, Zulfiqar F, Xiao X, Riazuddin SA,
Ahmad Z, Caruso R, MacDonald I, Sieving P, Riazuddin S and
Hejtmancik JF: Severe retinitis pigmentosa mapped to 4p15 and
associated with a novel mutation in the PROM1 gene. Hum Genet.
122:293–299. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Maw MA, Corbeil D, Koch J, Hellwig A,
Wilson-Wheeler JC, Bridges RJ, Kumaramanickavel G, John S,
Nancarrow D, Röper K, et al: A frameshift mutation in prominin
(mouse)-like 1 causes human retinal degeneration. Hum Mol Genet.
9:27–34. 2000. View Article : Google Scholar
|
|
18
|
Yang Z, Chen Y, Lillo C, Chien J, Yu Z,
Michaelides M, Klein M, Howes KA, Li Y, Kaminoh Y, et al: Mutant
prominin 1 found in patients with macular degeneration disrupts
photoreceptor disk morphogenesis in mice. J Clin Invest.
118:2908–2916. 2008.PubMed/NCBI
|
|
19
|
Permanyer J, Navarro R, Friedman J,
Pomares E, Castro-Navarro J, Marfany G, Swaroop A and
Gonzàlez-Duarte R: Autosomal recessive retinitis pigmentosa with
early macular affectation caused by premature truncation in PROM1.
Invest Ophthalmol Vis Sci. 51:2656–2663. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Michaelides M, Gaillard MC, Escher P, Tiab
L, Bedell M, Borruat FX, Barthelmes D, Carmona R, Zhang K, White E,
et al: The PROM1 mutation p.R373C causes an autosomal dominant
bull's eye maculopathy associated with rod, rod-cone, and macular
dystrophy. Invest Ophthalmol Vis Sci. 51:4771–4780. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mayer AK, Rohrschneider K, Strom TM,
Glöckle N, Kohl S, Wissinger B and Weisschuh N: Homozygosity
mapping and whole-genome sequencing reveals a deep intronic PROM1
mutation causing cone-rod dystrophy by pseudoexon activation. Eur J
Hum Genet. 24:459–462. 2016. View Article : Google Scholar
|
|
22
|
Khan AO and Bolz HJ: Pediatric cone-rod
dystrophy with high myopia and nystagmus suggests recessive PROM1
mutations. Ophthalmic Genet. 36:349–352. 2015. View Article : Google Scholar
|
|
23
|
Pras E, Abu A, Rotenstreich Y, Avni I,
Reish O, Morad Y, Reznik-Wolf H and Pras E: Cone-rod dystrophy and
a frameshift mutation in the PROM1 gene. Mol Vis. 15:1709–1716.
2009.PubMed/NCBI
|
|
24
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al ACMG
Laboratory Quality Assurance Committee: Standards and guidelines
for the interpretation of sequence variants: A joint consensus
recommendation of the American College of Medical Genetics and
Genomics and the Association for Molecular Pathology. Genet Med.
17:405–424. 2015. View Article : Google Scholar : PubMed/NCBI
|