Introduction
Glioblastoma is the most common primary malignant
brain tumor in adults with overall survival rates of <3.3% at 5
years (1). Few effective
treatments are available. Following maximal surgical tumor
resection, the current standard of care is based on a phase 3,
randomized clinical trial conducted by the European Organization
for Research and Treatment of Cancer and the National Cancer
Institute of Canada, which demonstrated that concurrent treatment
with daily doses of temozolomide (TMZ) and radiotherapy followed by
maintenance with TMZ was superior to radiotherapy alone (2). Even though this combined
chemoradiotherapy approach led to improved outcomes, few patients
survive beyond 5 years (3). Thus,
determining the molecular mechanisms responsible for the
development and progression of the disease, as well as the
development of radiation resistance, may broaden our understanding
of the pathogenesis and progression of the disease and may provide
novel therapeutic strategies.
The sucrose non-fermenting 1 (SNF1)/AMP-activated
protein kinase (AMPK) family functions to control the balance of
cellular metabolism, and is activated by the cellular AMP:ATP ratio
that is regulated by metabolic stresses, such as hypoxia and
glucose deprivation (4,5). Twelve protein kinases
[brain-specific serine/threonine-protein kinase (BRSK)1, BRSK2,
NUAK family, SNF1-like kinase, 1 (NUAK1), NUAK family, SNF1-like
kinase, 2 (NUAK2) which is also known as SNF1-like kinase (SNARK),
salt-inducible kinase 2 (QIK), serine/threonine-protein kinase SIK3
(QSK), salt-inducible kinase (SIK), MAP/microtubule
affinity-regulating kinase (MARK)1, MARK2, MARK3, MARK4 and
maternal embryonic leucine zipper kinase (MELK)] have been
identified as AMPK-α1- and AMPK-α2-related kinases in the human
kinome (6,7). AMPK-related kinases function as
critical sensors coupling cellular energy status to cell growth and
proliferation by modulating the cell-cycle machinery and, when
deregulated, they result in cancer development and tumor
progression in several types of cancer of different cell lineages
(4,8,9).
NUAK2, which resides at 1q32, is a member of the SNF1/AMPK family
(serine/threonine kinases) that is regulated by the putative tumor
suppressor, liver kinase B1 (LKB1; also known as serine/threonine
kinase 11), as well as by death receptor signaling through nuclear
factor (NF)-κB (6,10–12). The crucial role of NUAK2 has been
highlighted in cancer development and in tumor progression
(6,11,12). Although AMPK has anti-oncogenic
properties, its role in glioma has not yet been reported to date,
to the best of our knowledge.
MicroRNAs (miRs or miRNAs) are small, non-coding
RNAs that modulate protein expression by binding to complementary
or partially complementary sequences in the 3′ untranslated region
(3′UTR) of target mRNAs and thereby target mRNAs for degradation or
translational inhibition (13,14). miR-143 is downregulated in glioma
tissues and directly targets the neuroblastoma RAS viral oncogene
homolog (N-RAS) and functions as a tumor suppressor in the disease
(15). The overexpression of
miR-143 has been shwon to decrease the expression of N-RAS, inhibit
phosphoinositide 3-kinase (PI3K)/AKT and mitogen-activated protein
kinase (MAPK)/extracellular signal-regulated kinase (ERK)
signaling, and to attenuate the accumulation of p65 in the nuclei
of glioma cells, as well as to decrease migration, invasion, tube
formation and attenuate tumor growth and angiogenesis (15). miR-143 has also been shown to
sensitize glioma cells to TMZ, the first-line drug for glioma
treatment (15). However, the
mechanisms through which miR-143 functions as a tumor suppressor
gene have not yet been fully elucidated.
In this study, we demonstrate that NUAK2 expression
is upregulated in glioma tissues and that its expression is
associated with the advanced stages of the disease. In
vitro, NUAK2 overexpression promoted the proliferation,
migration and invasion of A172 glioblastoma cells, whereas the
silencing of NUAK2 with a plasmid carrying shRNA targeting NUAK2
inhibited glioma cell proliferation. Moreover, NUAK2 regulated
cancer stem cell (CSC)-related gene expression in glioma cells. We
also performed an analysis of potential miRNA target sites using 3
commonly used prediction algorithms, miRanda, TargetScan and
PicTar. All 3 algorithms predicted that miR-143 targets the 3′UTR
of NUAK2. Subsequent experiments confirmed this prediction.
Finally, we found that miR-143 inhibited the proliferation,
migration and invasion of glioblastoma cells. Thus, the findings of
our study demonstrate that miR-143 inhibits oncogenic traits by
degrading NUAK2 in glioblastoma.
Materials and methods
Glioma tissues, cells and NUAK2
expression plasmids
Sixteen tissue samples of human glioma tissue (4
samples of WHO grade I and 12 samples of WHO grade IV) and matched
adjacent normal tissue samples were obtained from the Department of
Neurosurgery, Yishui Central Hospital (Linyi, China). The mean
patient age was 56 years (range, 31–78 years). The use of human
tissue samples followed internationally recognised guidelines, as
well as local and national regulations. The medical ethics
committee approved the experiments undertaken. Informed consent was
obtained from each individual. The human glioblastoma cell line,
A172, was kindly donated by Dr Yong Yu (Hubei Cancer Center, Wuhan,
China). The A172 glioblastoma cells were maintained in DMEM
supplemented with 10% fetal bovine serum (FBS). The NUAK2
expression plasmid, the plasmid carrying shRNA targeting NUAK2
(shNUAK2), the empty vector and the scrambled shRNA were purchased
from Tiangen Biotech (Beijing, China).
miRNA precursors
The miR-143 miRNA precursor (pre-miR-143) and a
control precursor (control miR) were purchased from Ambion, Inc.
(Austin, TX, USA).
Cell transfection
For the transfection experiments, the cells were
cultured in serum-free medium without antibiotics at 60% confluence
for 24 h, and then transfected with the NUAK2 expression plasmid,
shNUAK2, pre-miR-143 or control miR using transfection reagent
(Lipofectamine 2000; Invitrogen, Carlsbad, CA, USA) according to
the manufacturer's instructions. Following incubation for 6 h, the
medium was removed and replaced with normal culture medium for 48
h, unless otherwise specified.
Western blot analysis
Western blot analysis was performed as described in
a previous study (16). Briefly,
following incubation with anti-NUAK2 (1:500; ab107287),
anti-enhancer of zeste 2 polycomb repressive complex 2 subunit
(EZH2; 1:500; ab186006), anti-L1 cell adhesion molecule (L1CAM;
1:500; ab208155), anti-CD133 (1:500; ab16518), anti-Bmi (1:500;
ab38295), anti-multidrug resistance protein 1 (MDR1; 1:500;
ab170904), anti-stage-specific embryonic antigen 1 (SSEA1; 1:500;
sc-21702), anti-signal transducer and activator of transcription 3
(STAT3; 1:500; ab68153) and anti-β-actin (1:500; ab179467/ab3280)
primary antibodies (all purchased from Abcam, Cambridge, MA, USA)
overnight at 4°C, IRDye™-800 conjugated anti-rabbit/anti-mouse
secondary antibodies (LI 926-32211/LI 926-32210; Li-COR
Biosciences, Lincoln, NE, USA) were used for 30 min at room
temperature. The specific proteins were visualized using the
Odyssey™ Infrared Imaging System (Li-COR Biosciences).
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium (MTT) cell
proliferation assay
Cell proliferation was assessed by MTT assay (Sigma,
St. Louis, MO, USA). The cells were seeded into a 96 well-plate at
4,000 cells per well and measured using an MTT kit according to the
manufacturer's instructions (Sigma). The absorbance was directly
proportional to the number of viable cells.
BrdU cell proliferation assay
Cell proliferation was also assessed using a
colorimetric BrdU proliferation kit according to the manufacturer's
instructions (Roche, Indianapolis, IN, USA). The transfected cells
were labeled with BrdU for 4 h. Genomic DNA was fixed and
denatured, followed by incubation with peroxidase-conjugated
anti-BrdU antibody for 90 min. A substrate for the conjugated
peroxidase was then added and the reaction product was quantified
by measuring the absorbance. The results were then normalized to
the number of total viable cells.
Cell migration and invasion assay
For Transwell migration assays, 2.5×104
cells were plated in the top chamber with the non-coated membrane
(24-well insert; pore size, 8 mm; BD Biosciences, San Jose, CA,
USA). For cell invasion assays, 1.25×105 cells were
plated in the top chamber with Matrigel-coated membrane (24-well
insert; pore size, 8 mm; BD Biosciences). In both assays, the cells
were plated in medium without serum or growth factors, and the
medium supplemented with serum was used as a chemoattractant in the
lower chamber. The cells were incubated for 24 h and the cells that
did not migrate or invade through the pores were removed using a
cotton swab. The cells on the lower surface of the membrane were
stained with the Diff-Quick stain set (Dade Behring, Newark, NJ,
USA) and counted under a Zeiss microscope (Zeiss, New York, NY,
USA).
Bioinformatics analysis
The analysis of potential miRNA target sites was
performed using the following 3 commonly used prediction
algorithms: TargetScan (http://www.targetscan.org), miRanda (http://www.microrna.org/microrna/home.do) and PicTar
(http://pictar.mdc-berlin.de/).
Immunofluorescence staining
To perform immunofluorescence staining, the A172
cells were plated on glass coverslips in 6-well plates and
transfected with 50 nM pre-miR-143 or control miR. At 36 h
following transfection, the coverslips were stained with the
above-mentioned anti-NUAK2 antibody. Alexa Fluor 488 goat
anti-rabbit IgG antibody (ab150077; Abcam) was used as a secondary
antibody. The coverslips were counterstained with DAPI
(Invitrogen-Molecular Probes, Eugene, OR, USA) for visualization of
the nuclei. Microscopic analysis was performed with a confocal
laser-scanning microscope (Leica Microsystems, Bensheim, Germany).
The fluorescence intensities were measured in a few viewing areas
for 200–300 cells/coverslip and analyzed using ImageJ 1.37v
software (http://rsb.info.nih.gov/ij/index.html).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) for NUAK2
Total RNA was isolated from the cells using TRIzol
reagent (Invitrogen). First-strand cDNA was synthesized from the
total RNA using M-MLV reverse transcriptase (Promega, Madison, WI,
USA) and random hexamer primers (Sangon, Shanghai, China). The
thermal cycling conditions were as follows: denaturation for 30 sec
at 95°C, annealing for 45 sec at 52–58°C depending on the primers
used, and extension for 45 sec at 72°C. The PCR products were
visualized on 2% agarose gels stained with ethidium bromide under
UV transillumination. qPCR was performed with a Power SYBR-Green
PCR Master Mix (Applied Biosystems, Carlsbad, CA, USA) according to
the manufacturer's instructions. The primer sequences were as
follows: NUAK2 forward, 5′-CTGAGACTGATAACGAGGAT-3′ and reverse,
5′-GAGGTGTTTCTGCTTGAC-3′.
RT-qPCR for miRNAs
Total RNA from the cultured cells, with the
efficient recovery of small RNA, was isolated using the mirVana
miRNA isolation kit (Ambion, Inc.). The detection of the mature
form of miRNAs was performed using the mirVana qRT-PCR miRNA
detection kit, according to the manufacturer's instructions
(Ambion, Inc.). U6 small nuclear RNA was used as an internal
control.
Statistical analysis
Data are expressed as the means ± SE, with the
number of independent experiments (n=3), and were analyzed using
the Student's t-test. A p<0.05 was considered to indicate a
statistically significant difference.
Results
Aberrant NUAK2 expression in glioma
tissues
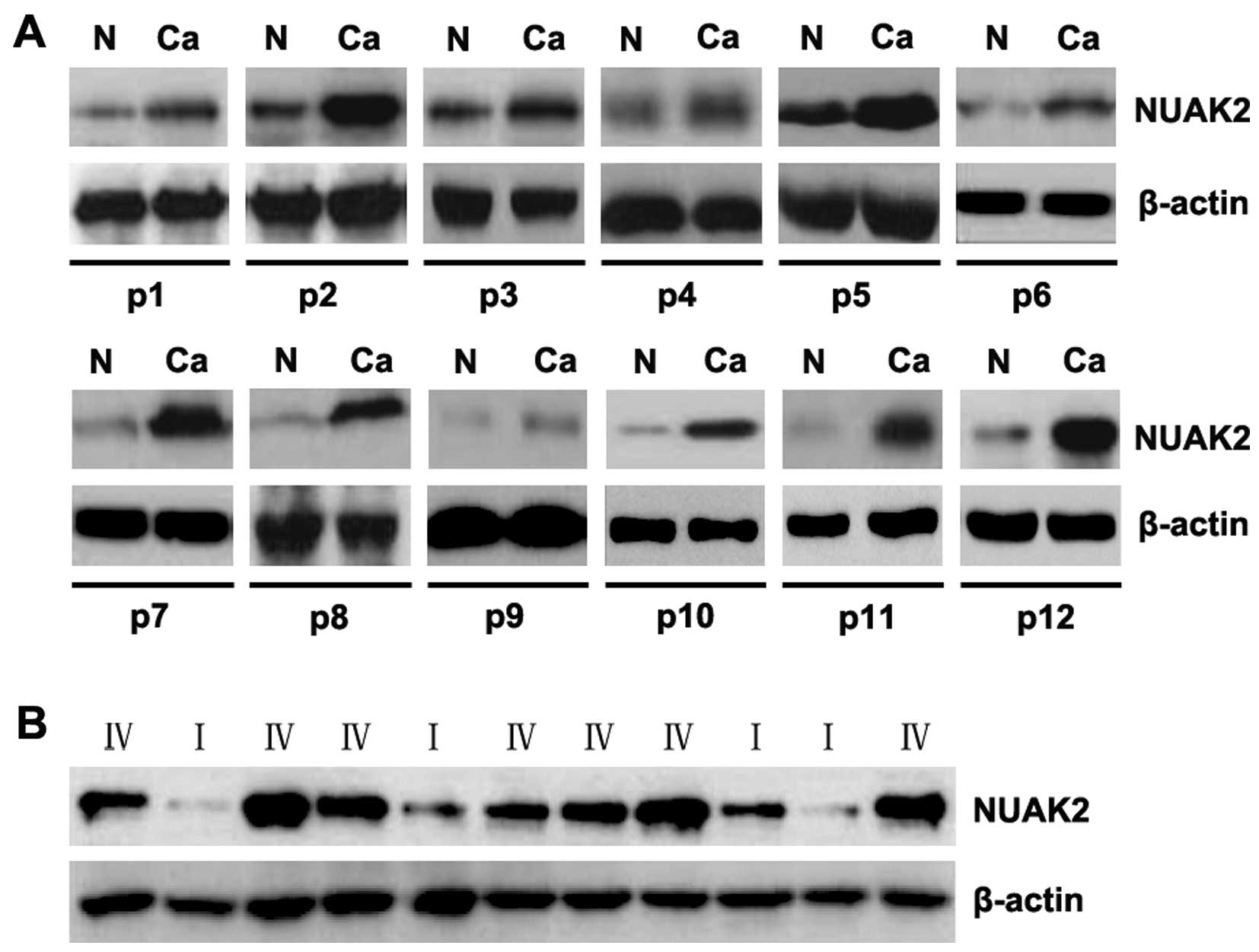
To assess the expression of NUAK2 in glioma tissues,
western blot analysis was conducted on 12 pairs of glioblastoma
tissues and matched adjacent normal tissue samples. The expression
of NUAK2 was consistently higher in the glioblastoma tissues than
in the normal tissues (Fig. 1A).
Moreover, the analysis of NUAK2 expression in the tissues from
patinets with high-grade (WHO grade IV) gliomas (namely
glioblastoma) and low-grade (WHO grade I) gliomas revealed that
NUAK2 was upregulated in the advanced stages of the disease
(Fig. 1B). These data support the
notion that NUAK2 functions as an oncogene in glioma tissues.
The overexpression of NUAK2 promotes the
proliferation, migration and invasion of A172 glioblastoma
cells
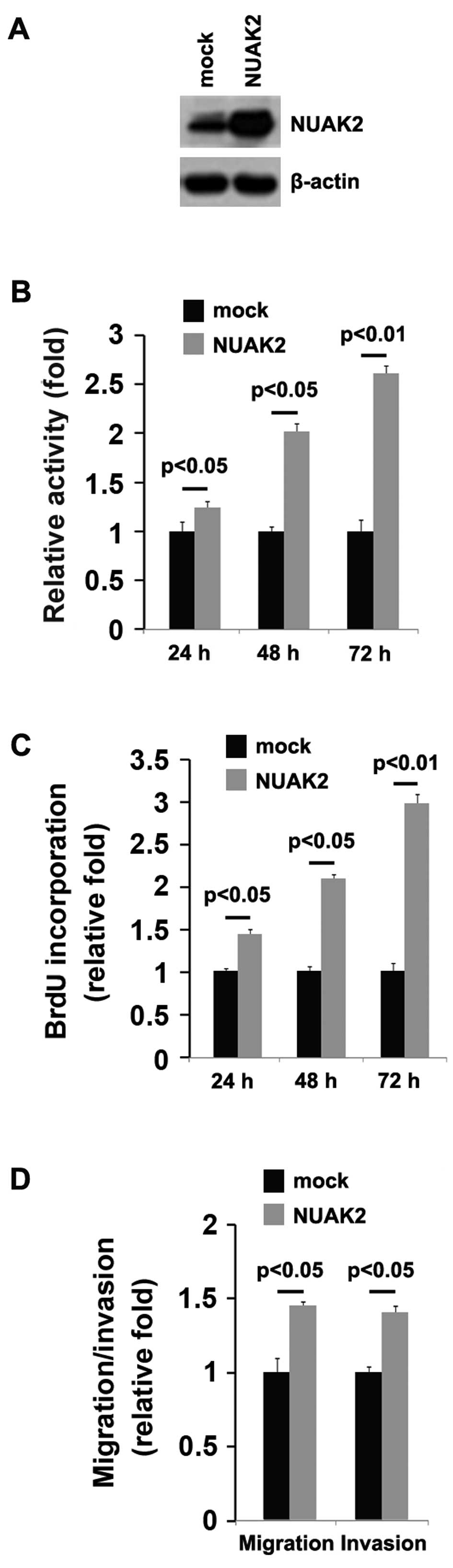
In an attempt to determine the role of NUAK2 in
regulating the proliferation of A172 cells, the cells were
transfected with a NUAK2 expression plasmid. We found that the
NUAK2 protein levels were significantly increased in the cells
transfectd with the NUAK2 expression plasmid compared to the cells
trans-fected with the emtpy vector (mock; Fig. 2A). Following stable transfection,
the proliferation rates of the A172 cells were determined by MTT
assay. The results revealed that the overexpression of NUAK2
significantly increased the proliferation rate of the A172 cells
and that the increase in cell proliferation was time-dependent
(Fig. 2B). This was further
confirmed by Brdu incorporation assay, which indicated that
transfection of the cells with NUAK2 resulted in increased DNA
synthesis activity per viable cell in the A172 cells in a
time-dependent manner (Fig.
2C).
Given that NUAK2 markedly promoted A172 cell
proliferation, we then sought to determine whether NUAK2 would have
an impact on the migration and invasion of A172 cells. The
migration and invasion assay of the A172 cells revealed that NUAK2
overexpression not only promoted the migration, but also promoted
the invasion of the A172 cells (Fig.
2D).
The silencing of NUAK2 inhibits the
proliferation, migration and invasion of A172 glioblastoma
cells
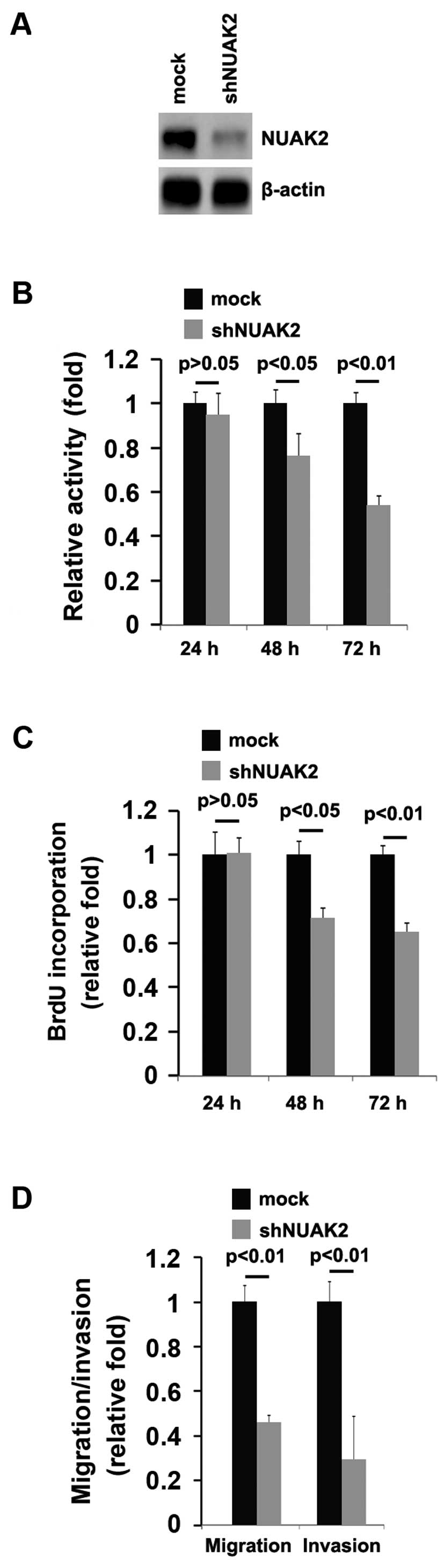
In order to further determine the role of NUAK2 in
regulating the proliferation of A172 cells, the cells were
transfected with a plasmid carrying shRNA targeting NUAK2
(shNUAK2). We found that the NUAK2 protein levels were
significantly decreased following transfection with shNUAK2
(Fig. 3A). Following stable
transfection, the proliferation rates of the A172 cells were
determined by MTT assay. The results revealed that the silencing of
NUAK2 significantly suppressed the proliferation rate of the A172
cells and that the decrease in cell proliferation was
time-dependent (Fig. 3B). This
was further confirmed by Brdu incorporation assy, which indicated
that transfection with shNUAK2 resulted in decreased DNA synthesis
activity per viable cell in the A172 cells, in a time-dependent
manner (Fig. 3C).
Given that the silencing of NUAK2 markedly inhibited
A172 cell proliferation, we then sought to determine whether
silencing NUAK2 would have an impact on the migration and invasion
of the A172 cells. The migration and invasion assay of the A172
cells revealed that the silencing of NUAK2 not only suppressed the
migration, but also inhibited the invasion of the A172 cells
(Fig. 3D).
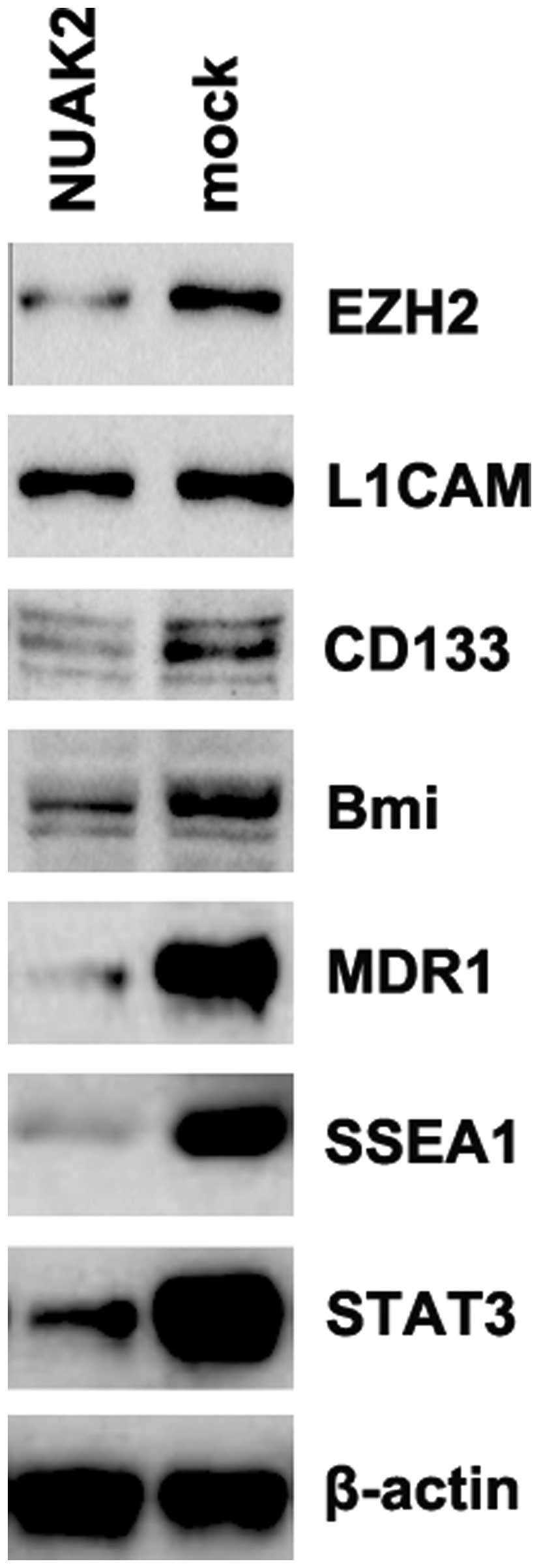
NUAK2 regulates CSC-related protein
expression in A172 glioblastoma cells
We demonstrated the aberrant expression of NUAK2 was
found in glioblastoma tissues, and that NUAK2 promoted the
proliferation, migration and invasion of A172 cells. It has been
reported that there is an association between proliferation,
migration and invasion, and CSCs (17–19). Thus, we performed western blot
analysis to measure the expression levels of CSC-related proteins
(EZH2, L1CAM, CD133, Bmi, MDR1, SSEA1 and STAT3) (20–26). Our results revealed that NUAK2
promoted the expression of EZH2, CD133, Bmi, MDR1, SSEA1 and STAT3
(Fig. 4). The results revealed
that NUAK2 overexpression was associated with the traits of CSCs in
the A172 cells.
miR-143 degrades NUAK2 in A172
glioblastoma cells
Having demonstrated that NUAK2 expression was
specifically upregulated and that it promoted the proliferation,
migration and invasion of the A172 cells, we then examined the
mechanisms promoting NUAK2 expression. miRNAs are a new class of
small (~22 nucleotide) non-coding RNAs that negatively regulate
protein-coding gene expression by targeting mRNAs for degradation
or translational inhibition (27–29). It has been previously reported
that oncogenes are upregulated in cancer, due to a lack of specific
miRNAs (30,31).
We hypothesized that NUAK2 was upregulated in glioma
due to defects in specific miRNAs in glioblastoma. To further
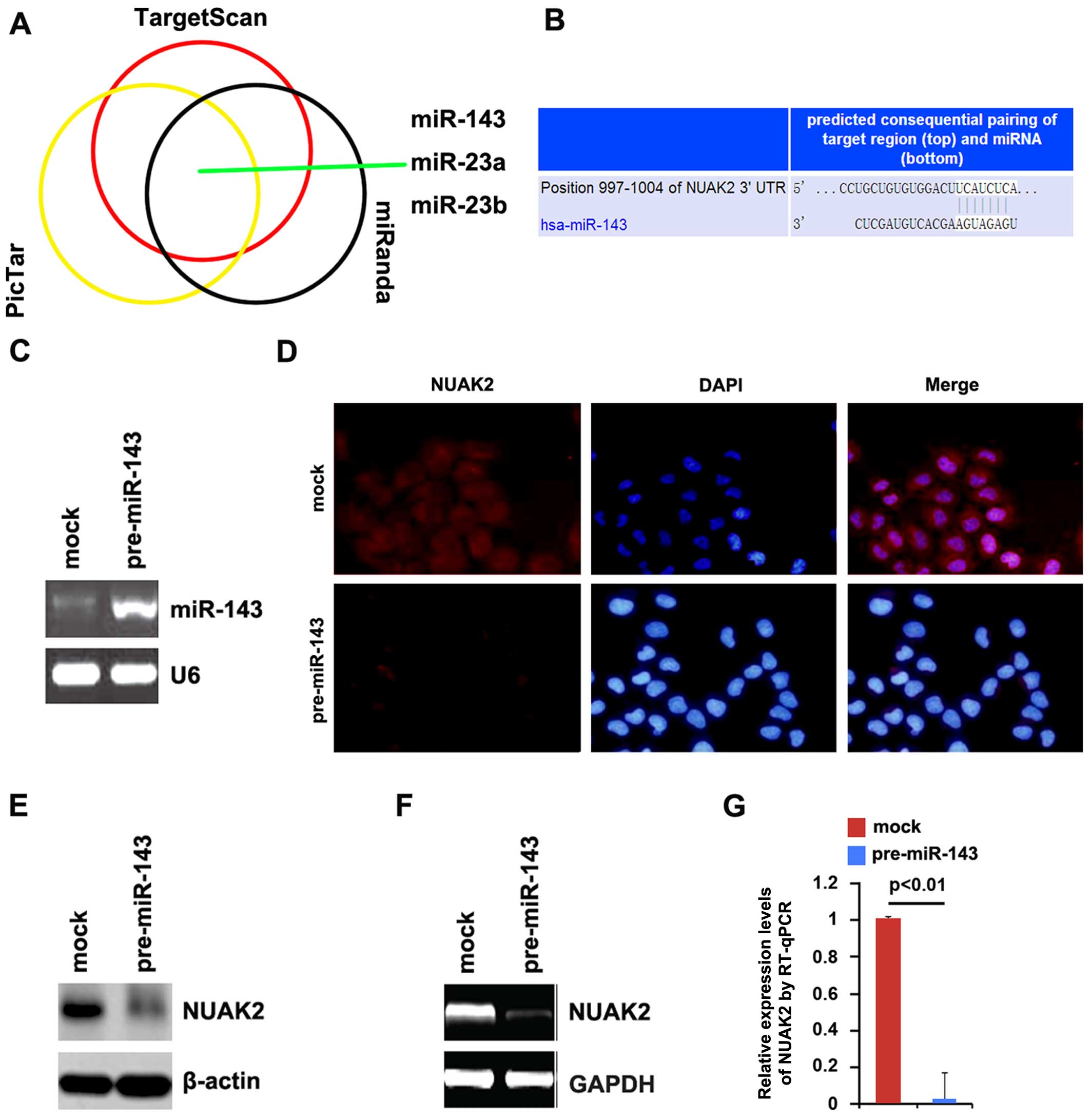
confirm this hypothesis, we used 3 commonly used prediction
algorithms, miRanda (http://www.microrna.org/), TargetScan (http://www.targetscan.org) and PicTar (http://pictar.mdc-berlin.de/) to analyze the 3′UTR of
NUAK2. All 3 algorithms predicted that miR-143, miR-23a and miR-23b
targeted the 3′UTR of NUAK2 (Fig.
5A).
Recently, Wang et al reported that miR-143
functions as a tumor suppressor by targeting N-RAS and enhances
TMZ-induced apoptosis in glioma (15). Thus, we hypothesized that the
upregulation of NUAK2 in glioblastoma is the result of a defect in
miR-143. The predicted target sites of miR-143 are illustrated in
Fig. 5B.
To determine whether NUAK2 can be downregulated by
miR-143, we transfected the A172 cells with pre-miR-143 and RT-qPCR
was then performed to detect miR-143 expression in the cells. The
results revealed that transfection with pre-miR-143 significantly
increased miR-143 expression (Fig.
5C). To determine whether NUAK2 protein expression was affected
by miR-143, we performed immunofluorescence staining. The results
revealed that NUAK2 protein expression was significantly
downregulated following transfection of the A172 cells with
pre-miR-143 compared to the cells transfected with control miR
(mock; Fig. 5D). Moreover,
western blot analysis was performed to measure the protein
expression levels of NUAK2 in the A172 cells transfected with
pre-miR-143. Consistent with the results of immunofluorescence
staining, we found that NUAK2 protein expression was significantly
downregulated following transfection of the A172 cells with
pre-miR-143 (Fig. 5E).
As miRNAs can suppress mRNA translation without
degrading the mRNA, we also performed RT-qPCR to determine whether
miR-143 affects NUAK2 mRNA expression. We transfected the A172
cells with pre-miR-143 and we found that pre-miR-143 evidently
degraded NUAK2 mRNA in the A172 cells (Fig. 5F and G). All the results confirmed
that miR-143 degrades NUAK2 and suggest that the overexpression of
NUAK2 is associated with a low expression of miR-143 in
gliomas.
miR-143 functions as a tumor
suppressorsuppressive gene in A172 glioblastoma cells
We demonstrated that NUAK2 was upregulated in the
glioblastoma tissues, and that it promoted the proliferation,
migration and invasion of the A172 glioblastoma cells, and that
miR-143 degraded NUAK2 in the A172 cells. Moreover, it has been
previously demonstrated that miR-143 functions as a tumor
suppressor by targeting N-RAS and enhances TMZ-induced apoptosis in
glioma (15). Thus, we
hypothesized that contrary to NUAK2, miR-203 may inhibit the
proliferation, migration and invasion of A172 cells. We
demonstrated that miR-143 expression was significantly increased
following transfection with pre-miR-143 (Fig. 5C).
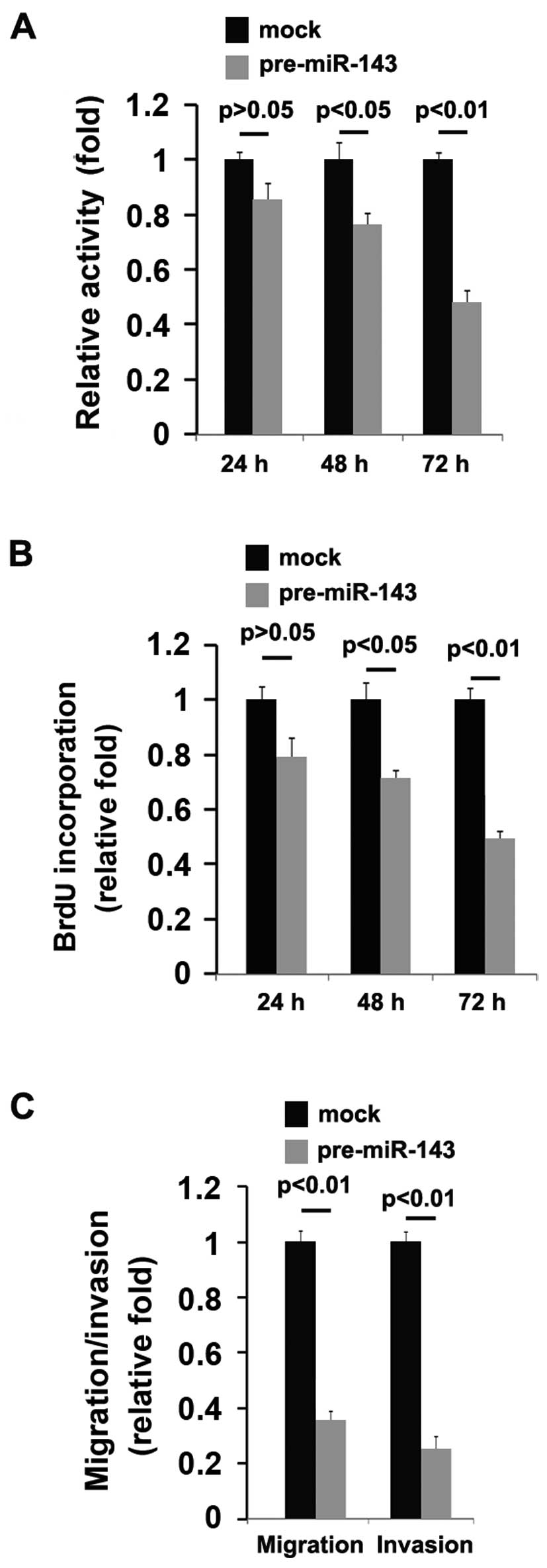
Subsequently, we observed the effects of miR-143 on
the proliferation, migration and invasion of the A172 cells. An MTT
assay, a migration assay and an invasion assay were performed on
the A172 cells. We demonstrated that miR-143 significantly
suppressed the proliferation rate of the A172 cells and that the
decrease in cell proliferation was time-dependent (Fig. 6A). This was further confirmed by
BrdU incorporation assay, which indicated that transfection with
pre-miR-143 resulted in decreased DNA synthesis activity per viable
cell in the A172 cells, also in a time-dependent manner (Fig. 6B).
Given that miR-143 inhibited A172 cell
proliferation, we then sought to determine whether miR-143 would
have an impact on the migration and invasion of A172 cells. The
migration and invasion assay of A172 cells revealed that miR-143
not only suppressed the migration, but also and inhibited the
invasion of the A172 cells (Fig.
6C).
Discussion
Gains or amplifications of the long arm of
chromosome 1 are among the frequent chromosomal abnormalities in
various types of cancer, and gains in the region spanning 1q31–1q32
are the most frequent abnormalities in these loci. The observation
that the gain of the 1q32 locus is shared by various types of
cancer emphasizes the importance of this locus for cancer
development and tumor progression in general (32). NUAK2 resides at 1q32 and public
databases, such as GeneCards (www.genecards.org) indicate that NUAK2 is highly
expressed in various types of cancer, including cancers of the
lymphoid tissues, lungs and breasts, implying that it may be
associated with cancer development and tumor progression (6,11,12). Recently, Namiki et al
(33) reported that the
AMPK-related kinase, NUAK2, affects tumor growth, migration and the
clinical outcome of human melanoma cells, which further confirms
the role of NUAK2 in cancer. However, to the best of our knowledge,
there is no study available to date on the role of NUAK2 in
glioma.
In this study, we demonstrated that NUAK2 expression
was upregulated in glioma tissues compared with matched adjacent
normal tissues and that its expression was increased in the
advanced stages of the disease, suggesting that it is associated
with the development and progression of glioma. NUAK2
overexpression promoted the proliferation, migration and invasion
of the glioma cells, while knockdown in vitro experiments
using plasmids containing shRNA targeting NUAK2 revealed that NUAK2
has a significant impact on the proliferation and migration of
glioblastoma cells.
A subset of cancer cells within some tumors, CSCs,
may drive the growth, multiple drug resistance and metastasis of
these tumors (34–36). Understanding the pathways that
regulate the proliferation, self-renewal, survival and
differentiation of malignant stem cells may shed light on the
mechanisms that lead to cancer development and may provide better
modes of treatment.
We found that NUAK2 downregulated the expression of
CSC-associated genes. It has been demonstrated that EZH2 is
essential for glioblastoma CSC maintenance (20); the stem cell marker, CD133, has
been found to affect clinical outcomes in glioma patients (21); Bmi-1 has also been shown to
promote stem cell self-renewal (22); it has been demonstrated that the
gene expression of MDR1 in glioblastoma stem cells is increased
(23); SSEA-1 has been
demonstrated to be an enrichment marker for tumor-initiating cells
in human glioblastoma (24);
evidence suggests that STAT3 is required for the maintenance of
multipotency in glioblastoma stem cells (25). We demonstrated that NUAK2
upregulated EZH2, CD133, Bmi-1, MDR1, SSEA-1 and STAT3 protein
expression, implying that NUAK2 overexpression may promote the
production of CSCs.
Consistent with the results of a previous study
demonstrating that miR-143 functions as a tumor suppressor by
targeting N-RAS (15), in this
study, we demonstrated that miR-143 degraded NUAK2 mRNA and
contrary to the role of NUAK2, it inhibited the proliferation,
migration and invasion of glioblastoma cells. Thus, from our data,
it can be concluded that miR-143 inhibits oncogenic traits by
degrading NUAK2 in glioblastoma cells.
References
|
1
|
Deorah S, Lynch CF, Sibenaller ZA and
Ryken TC: Trends in brain cancer incidence and survival in the
United States: surveillance, epidemiology, and end results program,
1973 to 2001. Neurosurg Focus. 20:E12006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Stupp R, Mason WP, van den Bent MJ, Weller
M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn
U, et al European Organisation for Research and Treatment of Cancer
Brain Tumor and Radiotherapy Groups; National Cancer Institute of
Canada Clinical Trials Group: Radiotherapy plus concomitant and
adjuvant temozolomide for glioblastoma. N Engl J Med. 352:987–996.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Stupp R, Hegi ME, Mason WP, van den Bent
MJ, Taphoorn MJ, Janzer RC, Ludwin SK, Allgeier A, Fisher B,
Belanger K, et al European Organisation for Research and Treatment
of Cancer Brain Tumour and Radiation Oncology Groups; National
Cancer Institute of Canada Clinical Trials Group: Effects of
radiotherapy with concomitant and adjuvant temozolomide versus
radiotherapy alone on survival in glioblastoma in a randomised
phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet
Oncol. 10:459–466. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hardie DG: AMP-activated/SNF1 protein
kinases: conserved guardians of cellular energy. Nat Rev Mol Cell
Biol. 8:774–785. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kato K1, Ogura T, Kishimoto A, Minegishi
Y, Nakajima N, Miyazaki M and Esumi H: Critical roles of
AMP-activated protein kinase in constitutive tolerance of cancer
cells to nutrient deprivation and tumor formation. Oncogene.
21:6082–6090. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lizcano JM, Göransson O, Toth R, Deak M,
Morrice NA, Boudeau J, Hawley SA, Udd L, Mäkelä TP, Hardie DG and
Alessi DR: LKB1 is a master kinase that activates 13 kinases of the
AMPK subfamily, including MARK/PAR-1. EMBO J. 23:833–843. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Manning G, Whyte DB, Martinez R, Hunter T
and Sudarsanam S: The protein kinase complement of the human
genome. Science. 298:1912–1934. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Martin MJ, Carling D and Marais R: Taking
the stress out of melanoma. Cancer Cell. 15:163–164. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ashrafian H: Cancer's sweet tooth: the
Janus effect of glucose metabolism in tumorigenesis. Lancet.
367:618–621. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Legembre P, Schickel R, Barnhart BC and
Peter ME: Identification of SNF1/AMP kinase-related kinase as an
NF-kappaB-regulated anti-apoptotic kinase involved in CD95-induced
motility and invasiveness. J Biol Chem. 279:46742–46747. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lefebvre DL, Bai Y, Shahmolky N, Sharma M,
Poon R, Drucker DJ and Rosen CF: Identification and
characterization of a novel sucrose-non-fermenting protein
kinase/AMP-activated protein kinase-related protein kinase, SNARK.
Biochem J. 355:297–305. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zagórska A, Deak M, Campbell DG, Banerjee
S, Hirano M, Aizawa S, Prescott AR and Alessi DR: New roles for the
LKB1-NUAK pathway in controlling myosin phosphatase complexes and
cell adhesion. Sci Signal. 3:ra252010.PubMed/NCBI
|
|
13
|
Bartel DP: MicroRNAs: genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Baek D, Villén J, Shin C, Camargo FD, Gygi
SP and Bartel DP: The impact of microRNAs on protein output.
Nature. 455:64–71. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang L, Shi ZM, Jiang CF, Liu X, Chen QD,
Qian X, Li DM, Ge X, Wang XF, Liu LZ, et al: MiR-143 acts as a
tumor suppressor by targeting N-RAS and enhances
temozolomide-induced apoptosis in glioma. Oncotarget. 5:5416–5427.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Heck S, Rom J, Thewes V, Becker N, Blume
B, Sinn HP, Deuschle U, Sohn C, Schneeweiss A and Lichter P:
Estrogen-related receptor alpha expression and function is
associated with the transcriptional coregulator AIB1 in breast
carcinoma. Cancer Res. 69:5186–5193. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bao B, Wang Z, Ali S, Ahmad A, Azmi AS,
Sarkar SH, Banerjee S, Kong D, Li Y, Thakur S and Sarkar FH:
Metformin inhibits cell proliferation, migration and invasion by
attenuating CSC function mediated by deregulating miRNAs in
pancreatic cancer cells. Cancer Prev Res (Phila). 5:355–364. 2012.
View Article : Google Scholar
|
|
18
|
Yu F, Li J, Chen H, Fu J, Ray S, Huang S,
Zheng H and Ai W: Kruppel-like factor 4 (KLF4) is required for
maintenance of breast cancer stem cells and for cell migration and
invasion. Oncogene. 30:2161–2172. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tang SN, Singh C, Nall D, Meeker D,
Shankar S and Srivastava RK: The dietary bioflavonoid quercetin
synergizes with epigallocathechin gallate (EGCG) to inhibit
prostate cancer stem cell characteristics, invasion, migration and
epithelial-mesenchymal transition. J Mol Signal. 5:142010.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Suvà ML, Riggi N, Janiszewska M,
Radovanovic I, Provero P, Stehle JC, Baumer K, Le Bitoux MA, Marino
D, Cironi L, et al: EZH2 is essential for glioblastoma cancer stem
cell maintenance. Cancer Res. 69:9211–9218. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zeppernick F, Ahmadi R, Campos B, Dictus
C, Helmke BM, Becker N, Lichter P, Unterberg A, Radlwimmer B and
Herold-Mende CC: Stem cell marker CD133 affects clinical outcome in
glioma patients. Clin Cancer Res. 14:123–129. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Godlewski J, Nowicki MO, Bronisz A,
Williams S, Otsuki A, Nuovo G, Raychaudhury A, Newton HB, Chiocca
EA and Lawler S: Targeting of the Bmi-1 oncogene/stem cell renewal
factor by microRNA-128 inhibits glioma proliferation and
self-renewal. Cancer Res. 68:9125–9130. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Nakai E, Park K, Yawata T, Chihara T,
Kumazawa A, Nakabayashi H and Shimizu K: Enhanced MDR1 expression
and chemoresistance of cancer stem cells derived from glioblastoma.
Cancer Invest. 27:901–908. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Son MJ, Woolard K, Nam DH, Lee J and Fine
HA: SSEA-1 is an enrichment marker for tumor-initiating cells in
human glioblastoma. Cell Stem Cell. 4:440–452. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sherry MM, Reeves A, Wu JK and Cochran BH:
STAT3 is required for proliferation and maintenance of multipotency
in glioblastoma stem cells. Stem Cells. 27:2383–2392. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bao S, Wu Q, Li Z, Sathornsumetee S, Wang
H, McLendon RE, Hjelmeland AB and Rich JN: Targeting cancer stem
cells through L1CAM suppresses glioma growth. Cancer Res.
68:6043–6048. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lee RC, Feinbaum RL and Ambros V: The C.
elegans heterochronic gene lin-4 encodes small RNAs with antisense
complementarity to lin-14. Cell. 75:843–854. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Pasquinelli AE, Reinhart BJ, Slack F,
Martindale MQ, Kuroda MI, Maller B, Hayward DC, Ball EE, Degnan B,
Müller P, et al: Conservation of the sequence and temporal
expression of let-7 heterochronic regulatory RNA. Nature.
408:86–89. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Reinhart BJ, Slack FJ, Basson M,
Pasquinelli AE, Bettinger JC, Rougvie AE, Horvitz HR and Ruvkun G:
The 21-nucleotide let-7 RNA regulates developmental timing in
Caenorhabditis elegans. Nature. 403:901–906. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Saito Y, Liang G, Egger G, Friedman JM,
Chuang JC, Coetzee GA and Jones PA: Specific activation of
microRNA-127 with down-regulation of the proto-oncogene BCL6 by
chromatin-modifying drugs in human cancer cells. Cancer Cell.
9:435–443. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Varambally S, Cao Q, Mani RS, Shankar S,
Wang X, Ateeq B, Laxman B, Cao X, Jing X, Ramnarayanan K, et al:
Genomic loss of microRNA-101 leads to overexpression of histone
methyltransferase EZH2 in cancer. Science. 322:1695–1699. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Corson TW, Huang A, Tsao MS and Gallie BL:
KIF14 is a candidate oncogene in the 1q minimal region of genomic
gain in multiple cancers. Oncogene. 24:4741–4753. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Namiki T1, Tanemura A, Valencia JC, Coelho
SG, Passeron T, Kawaguchi M, Vieira WD, Ishikawa M, Nishijima W,
Izumo T, et al: AMP kinase-related kinase NUAK2 affects tumor
growth, migration, and clinical outcome of human melanoma. Proc
Natl Acad Sci USA. 108:6597–6602. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Bjerkvig R, Tysnes BB, Aboody KS, Najbauer
J and Terzis AJ: Opinion: the origin of the cancer stem cell:
current controversies and new insights. Nat Rev Cancer. 5:899–904.
2005. View
Article : Google Scholar : PubMed/NCBI
|
|
35
|
Reya T, Morrison SJ, Clarke MF and
Weissman IL: Stem cells, cancer, and cancer stem cells. Nature.
414:105–111. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
36
|
Donnenberg VS and Donnenberg AD: Multiple
drug resistance in cancer revisited: the cancer stem cell
hypothesis. J Clin Pharmacol. 45:872–877. 2005. View Article : Google Scholar : PubMed/NCBI
|