Introduction
Nuclear factor-κB (NF-κB) is a dimeric transcription
factor that controls the expression of genes involved in multiple
processes, including immunity, inflammation, apoptosis and cell
cycle progression (1). Briefly,
in resting cells, NF-κB is kept inactive in the cytoplasm by its
association with the inhibitor of κBα (IκBα). Stimulating cells
with an agonist, such as tumor necrosis factor α (TNFα) or
interleukin (IL)-1β) activates the IκB kinase (IKK) complex, which
is composed of two catalytic subunits, IKKα and IKKβ, and a
regulatory subunit, NEMO. This signal-induced phosphorylation
targets IκBα for Lys48-linked polyubiquitination and subsequent
degradation by the ubiquitin-proteasome system (2,3).
In this way, NF-κB is released from IκBα, and translocates into the
nucleus to trigger the expression of its target genes. Recent
studies have revealed several enzymes involved in the
ubiquitination and deubiquitination of signaling proteins that
mediate IKK activation through a degradation-independent mechanism
(4). TNFR-associated factor
(TRAF) proteins, ubiquitin E3 ligases (5) have a pivotal role in signaling
pathways that are involved in the activation of NF-κB by many
cell-surface receptors, including the TNFR superfamily, the IL-1
receptor (IL-1R) and Toll-like receptors (TLRs) (6). Following ligand binding to IL-1R or
TLRs, TRAF6 is recruited to the receptor complexes and forms
oligomers. TRAF6 oligomerization activates its ligase activity,
leading to the Lys63-linked polyubiquitination of target proteins,
including TRAF6 and NEMO. It is also known that
receptor-interacting protein (RIP) is ubiquitinated by TRAF2
through the Lys63 linkage immediately following TNFα stimulation
(7). Both the Lys63-linked
polyubiquitylation of TRAF6 and RIP recruits transforming growth
factor β (TGFβ)-activated kinase (TAK1) and two adaptor proteins,
TAK1-binding protein (TAB)1 and TAB2. TAB2 contains a highly
conserved novel zinc-finger domain that binds preferentially to
polyubiquitin chains that are linked by Lys63 (8). TAB2-associated TAK1 phosphorylates
IKKβ at two serine residues in the activation loop, thereby
activating the IKK complex (9,10).
Ubiquitination has been shown to play important
roles in the regulation of NF-κB signaling pathways, as well as in
endoplasmic reticulum (ER) stress. There are two branches of ER
quality control that address the situation. One is the unfolded
protein response (UPR) pathway (11). The activation of UPR results in
attenuation of protein synthesis, and upregulation of genes
encoding chaperones that facilitate the protein folding process in
the ER. Thus, UPR reduces the accumulation and aggregation of
malfolded proteins, giving the cell the possibility of correcting
the environment inside the ER (11,12). A second major response is known as
the ER-associated degradation (ERAD) pathway to reduce the
misfolded proteins in the ER, which involves retro-translocation,
polyubiquitination and degradation in the cytosol through the 26S
proteasome (13,14). Synoviolin, a representative
ERAD-associated E3 ubiquitin ligase, is regulated by ER stress
through the ER stress-response element (ERSE) which is a regulatory
element located in many of these ER stress-response genes (15). Synoviolin, a mammalian homolog of
Hrd1p/Der3p, was identified from the cDNA of rheumatoid synovial
cells and is involved in the development of obesity, rheumatoid
arthritis, fibrosis, limb girdle muscular dystrophy and liver
cirrhosis (16–20).
In the present study, we searched for E3 ligases
which are associated with ER stress and regulate NF-κB signaling.
We identified an E3 ligase, mitochondrial ubiquitin ligase
activator of NF-κB (MULAN). Previous studies have reported that
MULAN is one of the NF-κB-activating genes by large scale screening
(21), and regulates
mitochondrial trafficking and morphology (22). In addition, mitochondrial
hyperfusion promotes NF-κB activation in a TAK1- and IKK-dependent
manner via MULAN (23). In this
study, we found that the expression of MULAN is upregulated by ER
stress. MULAN associates with TAK1 and activates NF-κB signaling in
an E3 ligase activity-dependent manner. The knockdown of MULAN by
siRNA resulted in the downregu-lation of NF-κB signaling in cells
subjected to ER stress. Our results suggest that MULAN is an E3
ligase which regulates NF-κB signaling under conditions of ER
stress.
Materials and methods
Plasmids
The coding sequences for full-length MULAN genes
were PCR-amplified (primers, 5′-acagaattcATGGAGAGCGGAGGGC-3′ and
5′-tgtgtcgacGCTGTTGTACAGGGGTATCA-3′) from reversed-transcribed HeLa
cell RNA. PCR products were digested with EcoRI and
SalI, and ligated into pGEX-5X-1 (Amersham Pharmacia
Biotechnology, Piscataway, NJ, USA) for in vitro ubiquitin
(Ub) assays. For expression in mammalian cells, the fragments of
MULAN and mutants were inserted into pcDNA3 hemagglutinin antigen
(HA) or pcDNA3 FLAG, which were constructed by inserting the HA
sequence or FLAG sequence into pcDNA3 (Invitrogen, Carlsbad, CA,
USA), respectively. Fragments of deletion mutants, termed MULAN-A,
MULAN-B, MULAN-N and MULAN-C were obtained by PCR amplification.
Fragments of additional deletion mutants, termed MULAN delta
transmembrane (dTM) and MULAN 3C were generated by PCR-based
methods. The sequences of all plasmids generated by PCR were
confirmed by sequence analysis. The IKKβ, IKKβ K44A, TRAF6, TRAF6
dominant negative (DN; delta RING), TRAF2, TRAF2 DN (delta RING)
and RIP1 plasmids used in this study were obtained by a PCR-based
method. The ERSE-Luc reporter plasmid was also obtained by a
PCR-based method and possesses 4 copies of synoviolin ERSE. The
TAK1, TAK1 K63W and TAB1 plasmids were a kindly gif from Dr
Kunihiro Matsumoto. The pcDNA3 HA-Ub expression plasmid, NF-κB-Luc
reporter plasmids, Som-Luc and CMV-β-gal plasmid have been
described in previous studies (16,24–26).
Cell culture and antibodies
HeLa cells and 293 cells [provided by RIKEN BRC
through the National Bio-Resource Project of MEXT (Ibaraki, Japan)]
were maintained in Dulbecco's modified Eagle's medium supplemented
with 10% fetal bovine serum plus penicillin and streptomycin at
37°C in 5% CO2. The following antibodies were used:
anti-FLAG (M2; F3165) and anti-β-actin (A5441) from Sigma Chemical
Co. (St. Louis, MO, USA), anti-HA [12CA5 (1583816) and 3F10
(1867423); Boehringer Mannheim GmbH, Mannheim, Germany], anti-IκB
(sc-203) and anti-p65 (sc-372) were purchased from Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA.
Induction of ER stress
To induce ER stress, the cells were stimulated with
tunicamycin at concentrations of 2 μg/ml for 0, 3, 6 and 9 h
and total RNA was collected following stimulation. ER stress was
also induced by thapsigargin in luciferase assay (Fig. 5). The cells were stimulated with
thapsigargin at concentrations of 2 μM for 4 h.
TNFα and IL-1β treatment
TNFα is known to activate NF-κB signaling by
promoting the degradation of IκBα and the nuclear translocation of
p65. We treated the cells with TNFα at 10 nM for 4 h as a positive
control (Fig. 4) and an inducer
of NF-κB signaling (Fig. 5).
IL-1β was also used at 10 ng/ml for 4 h.
Transfection and immunofluorescence
For immunofluorescence experiments, trypsinized
cells were seeded on a cover glass and incubated for 24 h prior to
transfection. Transfection was performed using Lipofectamine 2000
(Invitrogen) with 0.5 μg of MULAN expression plasmid,
according to the manufacturer's instructions. The cells were
incubated for a further 12 h after transfection, washed 3 times
with phosphate-buffered saline (PBS), and fixed with 3.7%
formaldehyde in PBS for 30 min, followed by permeabilization with
0.1% Triton X-100 for 30 min. The cells then were incubated with
the primary antibody followed by staining with Alexa Fluor 488
anti-mouse second antibody or Alexa Fluor 594 anti-rabbit second
antibody (Molecular Probes, Eugene, OR, USA). Samples were scanned
with a Zeiss LSM 510 confocal laser scanning microscopy (Carl Zeiss
Microscopy, Jena, Germany). Co-localization scatter diagrams were
generated using LSM 510 software (27).
Transient transfection assay
Transient transfection assays were performed using
the 293 cells, as previously described (24). The cells were lysed with cell
lysis buffer (Toyo Ink, Tokyo, Japan) 24 h following transfection
and luciferase activities were measured. The recorded activity was
normalized to the β-gal activity from CMV-β-gal. All experiments
were performed in triplicate. The 293 cells were transfected with
100 ng of NF-κB-Luc, 50 ng of CMV-β-gal control plasmid, and 0, 50
and 100 ng of MULAN or mutants, and/or DN-mutants plasmid, or
truncated forms of MULAN expression vector. To ensure an equal
amount of DNA, empty plasmids were added in each transfection.
Immunoprecipitation assay
The HeLa cells were transfected with HA TAK1, HA
TAB1, HA RIP1, HA IKKβ, and/or MULAN/FLAG expression vector. After
12 h of transfection, the cells were lysed in 1 ml of lysis buffer
(20 mM HEPES, pH 7.5, 100 mM NaCl, 1 mM EDTA, 1 mM DTT, 0.1% NP-40,
5% glycerol and protease inhibitors). The lysates were mixed with 1
μg of anti-FLAG antibody (M2) conjugated to protein
G-sepharose beads (GE Healthcare Bio-Sciences, Uppsala, Sweden).
Following 4 h of incubation at 4°C, the beads were washed 3 times
with lysis buffer. Bound proteins were fractionated by sodium
dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and
examined by western blot analysis.
Western blot analysis
The protein extracts were resolved by 10% SDS-PAGE,
transferred onto a PVDF membrane, and incubated with primary
antibodies [anti-FLAG M2 (F3165), anti-β-actin (A5441), anti-HA
3F10 (1867423), anti-IκB (sc-203) and anti-p65 (sc-372)] followed
by horseradish peroxidase-conjugated secondary antibodies. The
antigen-antibody complexes were visualized using an ECL detection
system (Promega, Madison, WI, USA).
RNA interference assay, RT-PCR and
real-time PCR
siRNAs (Stealth RNAi) for MULAN and GFP were
purchased from Invitrogen. The sequence of the MULAN siRNA-1 was
5′-UUUCCACAAACUGGCUGUUAAGCGU-3′, and that of control siRNAwas
5′-AAGAAGUCGUGCUGCUUCAUGUGGU-3′. Transfection with siRNAs was
carried out using Lipofectamine 2000 (Invitrogen) according to the
manufacturer's instructions. Isogen was used for total RNA
isolation and RT-PCR was performed with ReverTra Ace (Toyobo,
Tokyo, Japan) according to the manufacturer's instructions. Total
RNA (1 μg) was used for cDNA preparation. Real-time PCR was
carried out using the Universal ProbeLibrary system (Roche
Diagnostic, Mannheim, Germany). Signals from each sample were
normalized to values obtained for the human glyceraldehyde
3-phosphate dehydrogenase (GAPDH) gene, which was assayed
simultaneously with the experimental samples. Analyses were
performed with a sequence detector (model 7500; Applied Biosystems,
Foster City, CA, USA).
Statistical analysis
The non-paired Student's t-test was used to analyze
mean differences. A P-value <0.05 was considered to indicate a
statistically significant difference.
Results
MULAN is regulated by ER stress
Recent studies have revealed that ER stress induces
the activation of NF-κB (36,37). It is well known that
ubiquitination is critical in both the response to ER stress and
NF-κB activation. Therefore, we wished to determine whether E3
ligases, which are related to ER stress, regulate NF-κB signaling.
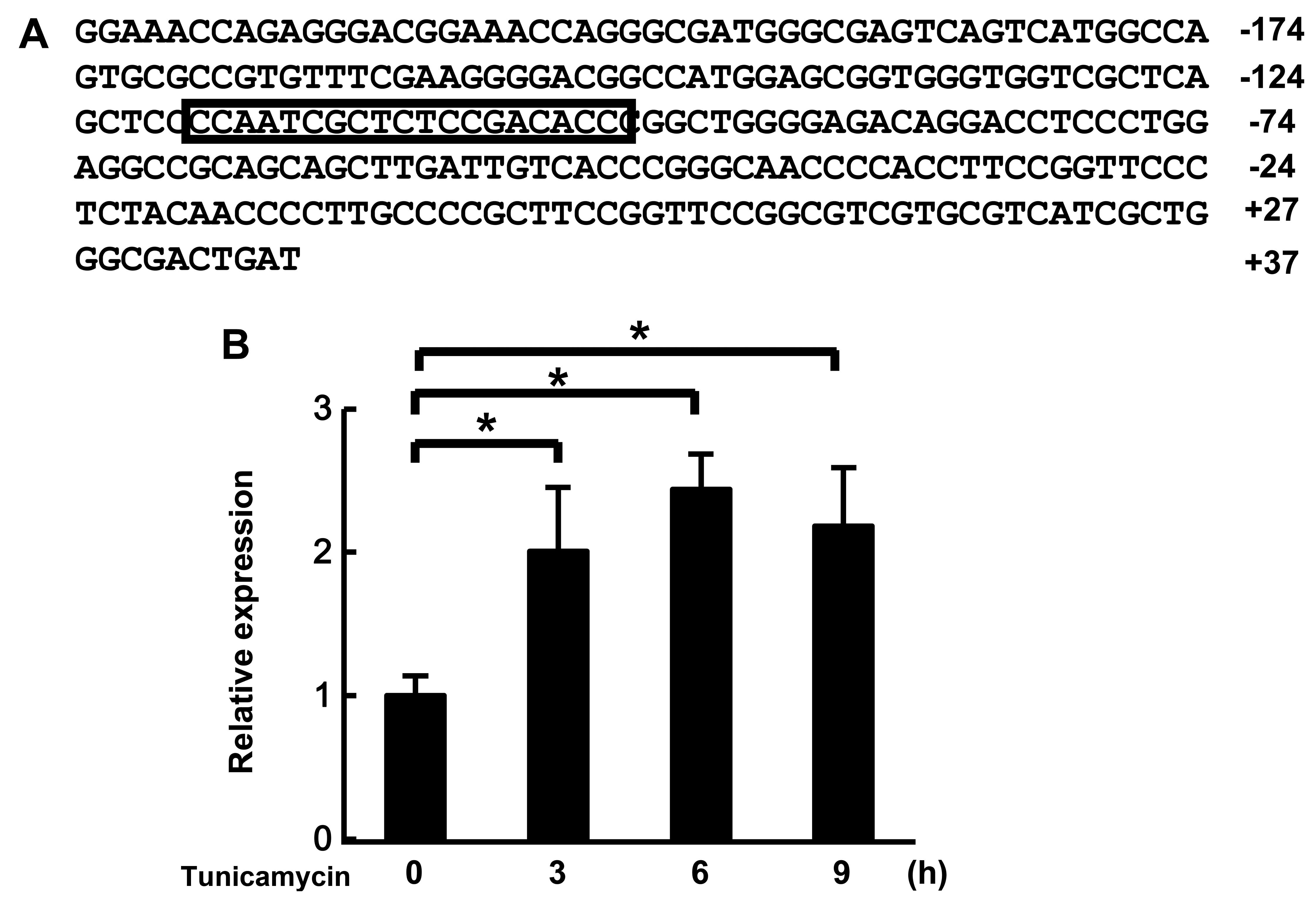
We found the putative ERSE located at positions −118 to −100 on the
promoter region of MULAN, which is one of the NF-κB-activating
genes (Fig. 1A). First, we
analyzed the mRNA expression of MULAN in cells subjected to ER
stress by real-time PCR. MULAN mRNA expression was increased by the
induction of ER stress by stimulation with tunicamycin (Fig. 1B). These results suggest that the
expression of MULAN is regulated by ER stress.
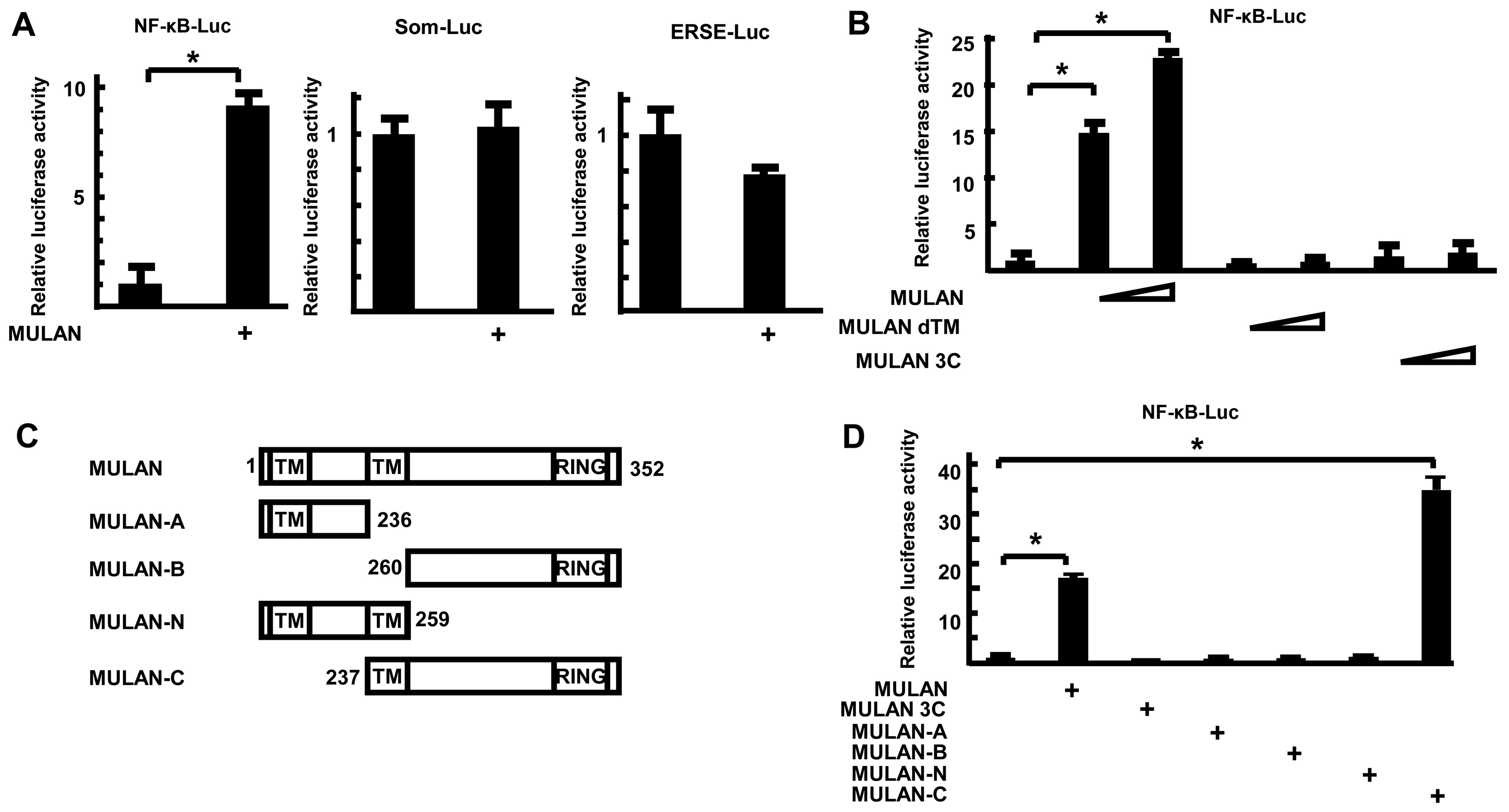
MULAN activates NF-κB gene expression in
an E3 ligase activity-dependent manner
To determine whether MULAN specifically activates
NF-κB-Luc, the luciferase reporter plasmids were co-transfected
with MULAN/HA-expressing plasmid into the 293 cells. Twenty-four
hours after transfection, the luciferase activities were measured.
MULAN enhanced the reporter activity of NF-κB-Luc, but not that of
Som-Luc and ERSE-Luc (Fig. 2A).
To further examine the mechanism of induction of NF-κB-dependent
gene expression by MULAN, we utilized several mutants of MULAN and
performed reporter assays. MULAN increased NF-κB-dependent gene
expression by 21.9-fold in a dose-dependent manner (Fig. 2B). By contrast, dTM, which lacks
the second transmembrane domain, and MULAN 3C, which includes a
cysteine to serine substitution in the RING finger domain and lacks
E3 ubiquitin ligase activity, did not stimulate the reporter
activity. To determine which domains contributes to the activation
of NF-κB-Luc, several truncations of MULAN were expressed as HA
fusion proteins (Fig. 2C) and
examined by reporter assay. MULAN induced the reporter activity
16.4-fold. Among these 4 mutants, only MULAN-C markedly activated
transcription 33.4-fold, whereas MULAN-A, MULAN-B and MULAN-N did
not (Fig. 2D). These results
suggest that both the transmembrane domain (TM) and RING finger
domain are important for the MULAN-dependent activation of
NF-κB.
The mechanism of MULAN-dependent NF-κB
activation
To further investigate the target of MULAN, we
utilized dominant negative (DN) mutants of major factors in NF-κB
signals, such as IKKβ, TAK1, TRAF6 and TRAF2. First, we examined
the inhibitory effects of these DN mutants in TNFα-treated cells,
and we found that they suppressed the reporter activity of
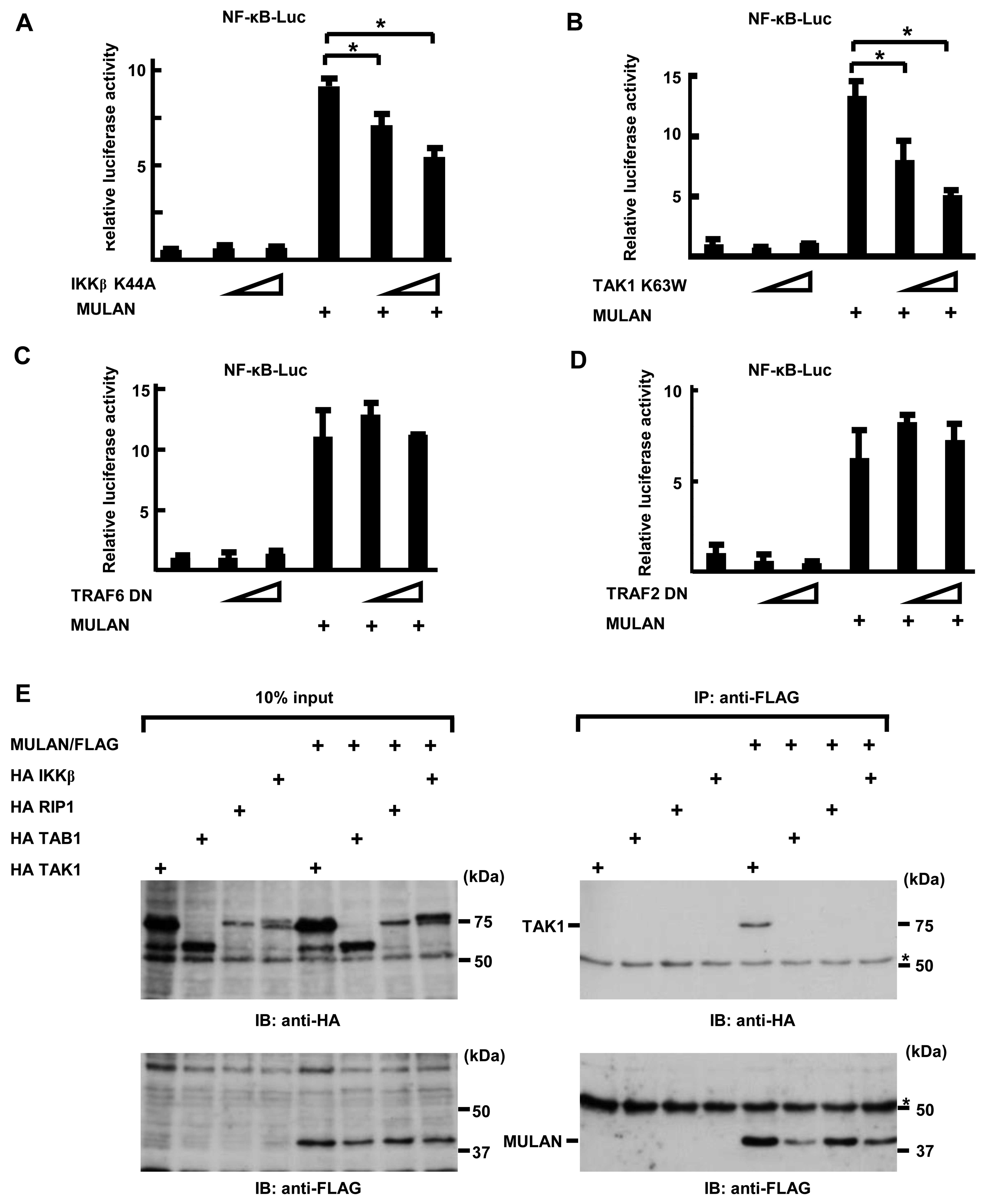
NF-κB-Luc in a dose-dependent manner. As shown in Fig. 3A, the introduction of DN-IKKβ
plasmids markedly inhibited the MULAN-induced activation of NF-κB
in a dose-dependent manner. In addition, transfection with DN-TAK1
also yielded the same result (Fig.
3B). Conversely, DN-TRAF2 and DN-TRAF6 failed to suppress the
MULAN-dependent activation of NF-κB (Fig. 3C and D). These results suggest
that MULAN plays a role in NF-κB activation upstream of TAK1 and
downstream of TRAF2 and TRAF6. These results prompted us to search
the interactants of MULAN. To determine whether MULAN interacts
with TAK1 TAB1, RIP1 and IKKβ, we transiently transfected HeLa
cells with HA TAK1, HA TAB1, HA RIP1, HA IKKβ expression plasmids
and/or MULAN/FLAG expression plasmids. We performed an
immunoprecipitation assay following western blot analysis. As shown
in Fig. 3E (upper panel), HA TAK1
was detected in the immunoprecipitate with anti-FLAG antibody, but
HA TAB1, HA RIP1 and HA IKKβ were not. These results suggest that
TAK1 may be one of the targets of MULAN through which it induces
the activation of NF-κB.
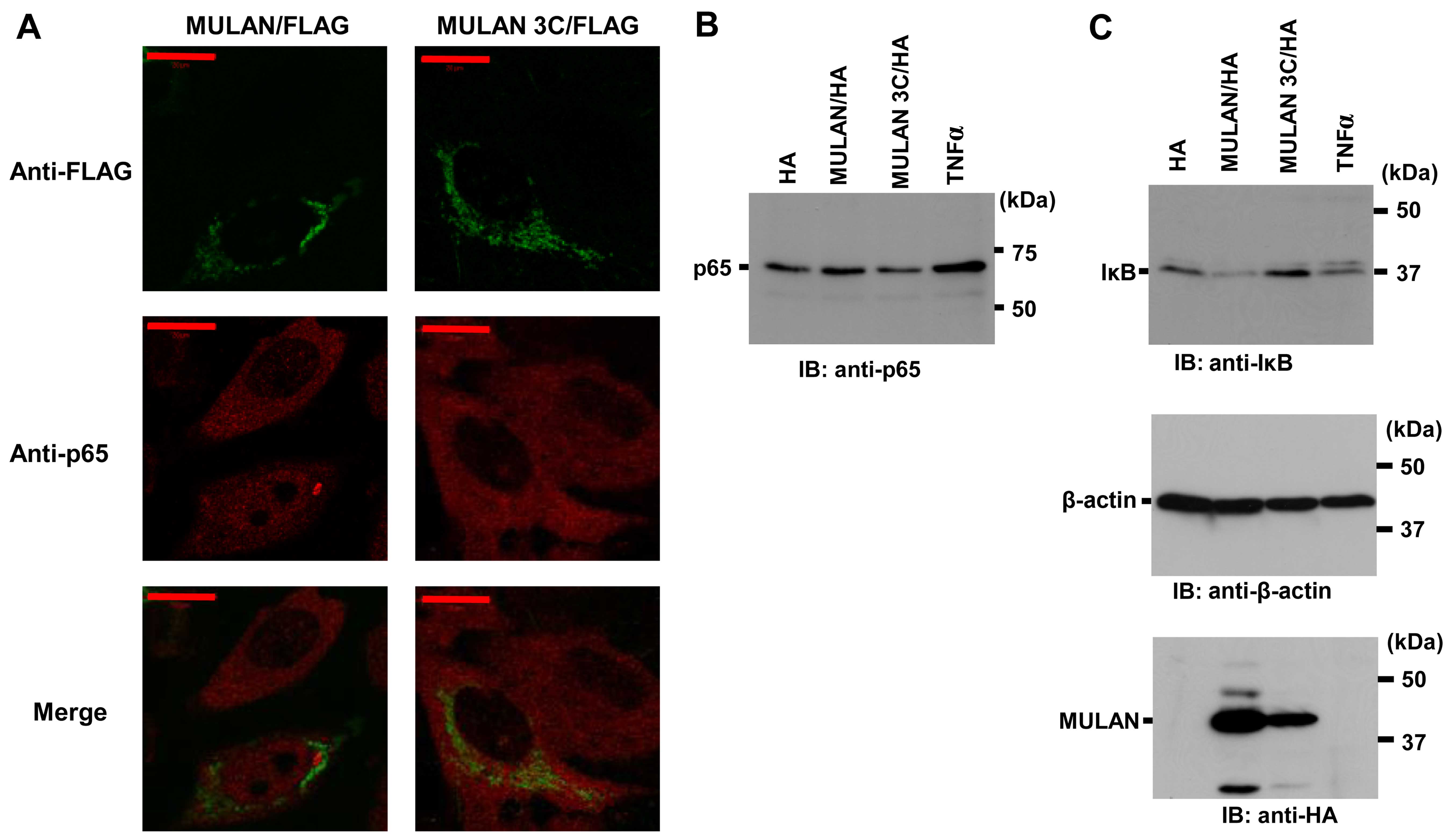
MULAN activates endogenous NF-κB
signals
It is well known that the NF-κB signaling pathway
can be activated through various processes; however, the release of
cytoplasmic NF-κB proteins from IκB proteins and nuclear
translocation are pivotal for signal transduction (28). To determine whether MULAN induces
endogenous NF-κB activation, we observed the translocation of
endogenous p65 into the nucleus. MULAN/FLAG or MULAN 3C/FLAG were
transiently transfected into the HeLa cells. The nuclear
localization of endogenous p65 was only observed in the cells that
expressed MULAN/FLAG (Fig. 4A).
To further confirm the nuclear localization of p65, we used nuclear
extraction and western blot analysis with anti-p65 specific
antibody. In the cells transfected with the pcDNA3 HA vector or
MULAN 3C/HA expression vector, a small amount of p65 was found in
the nuclear extracts. On the other hand, stimulation with TNFα and
the expression of MULAN led to an increase in p65 levels in the
nucleus (Fig. 4B). We then
examined the degradation of endogenous IκBα. We transfected
MULAN/HA or MULAN 3C/HA into the HeLa cells, and cell lysates were
prepared 12 h following transfection. In the cells transfected with
pcDNA3 HA vector or MULAN 3C/HA, hardly any IκBα degradation was
observed, whereas noticeable degradation was detected in the cells
expressing wild-type MULAN (Fig.
4C). These results indicate that MULAN activates
NF-κB-dependent gene expression in an enzymatic activity-dependent
manner.
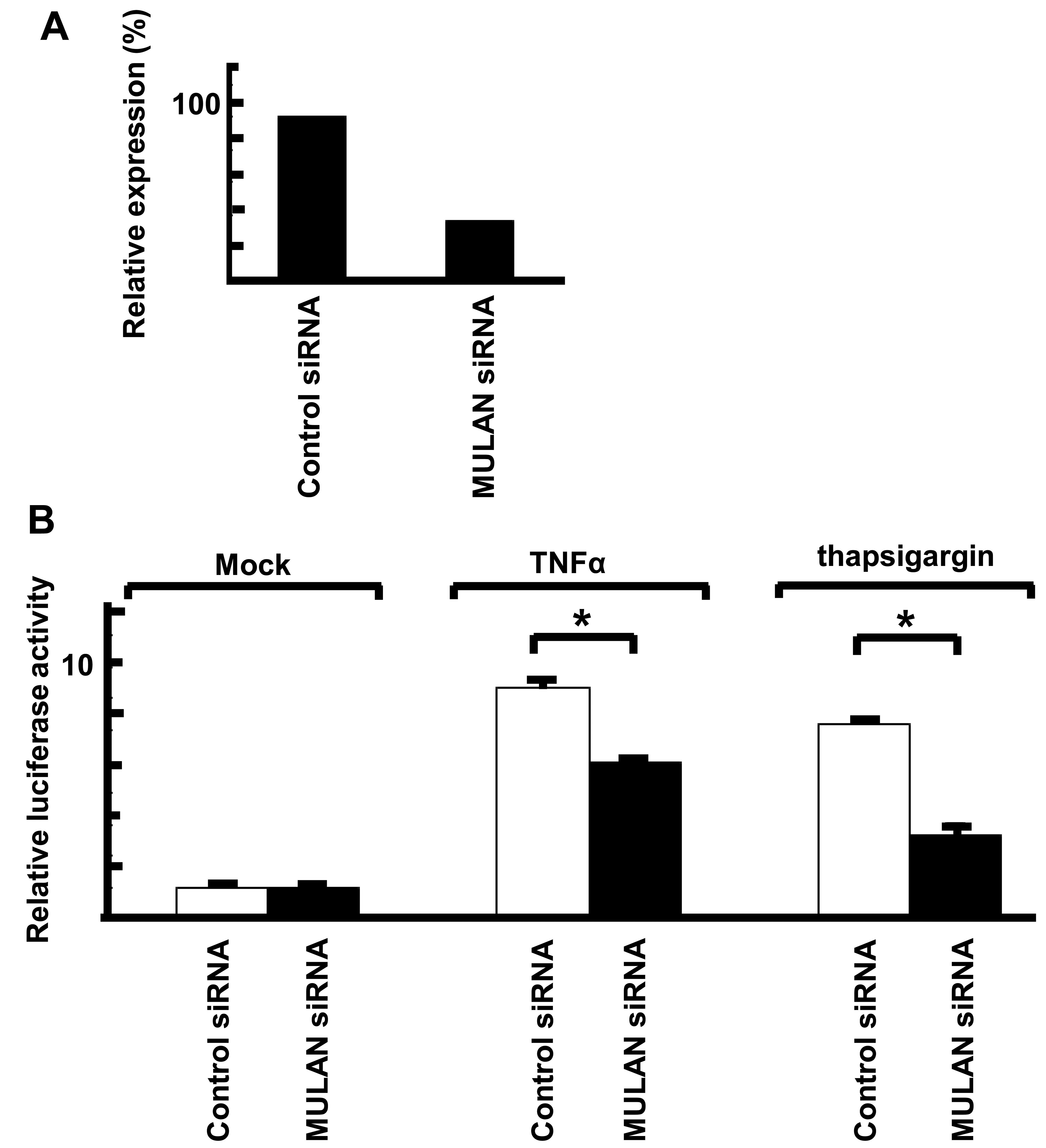
Knockdown of MULAN induces a decrease in
ER stress-dependent NF-κB activity
To examine the upper signal of MULAN-dependent NF-κB
activation, we performed reporter assay with siRNA against MULAN.
To confirm the effects of siRNAs, MULAN siRNA and control siRNA
were transiently transfected into the HeLa cells. After 48 h, total
RNA was isolated and the mRNA levels were measured by real-time
PCR. MULAN siRNA induced the downregulation of MULAN (Fig. 5A). We then performed a reporter
assay with NF-κB-Luc. The cells stimulated with TNFα and
transfected with control siRNA exhibited an enhanced reporter
activity by 7.3-fold and the MULAN siRNA-transfected cells
exhibited a low reporter activity (Fig. 5B). We also observed no difference
in reporter activity when the cells were stimulated with IL-1β
(data not shown). When the cells were treated with thapsigargin, a
decrease in reporter activity of approximately 50% was observed in
the MULAN siRNA-transfected cells compared with the cells
transfected with control siRNA. These results indicate that the
activation of NF-κB by MULAN plays an important in cells subjected
to ER stress.
Discussion
MULAN is regulated by ER stress
We found putative ERSE on the promoter region of
MULAN gene (Fig. 1). ERSE is
found in many of these ER stress-response genes and is regulated by
the ER responsive transcriptional factors, activating transcription
factor 6 (ATF6) and X-box binding protein 1 (XBP1). ERSE, with a
consensus sequence of CCAAT-N9-CCACG, is a cis-acting
element that is necessary and sufficient for transcriptional
induction of ER chaperone genes (29–31). The putative ERSE of MULAN,
CCAAT-N9-ACACC, differs slighlty from the consensus sequence of
ERSE. Many ER stress-responsive genes also have a different ERSE
from a consensus sequence; for example, Bip/Grp78, CCAAT-N9-CCAAC
and PDI, CCAGT-N9-ACAGC. In addition, the expression of MULAN was
induced by ER stress (Fig. 1).
However, we cannot not rule out possibility of the contribution of
other elements on the MULAN promoter. These results suggest that
the putative ERSE of MULAN may function as an ERSE and it is
certain that MULAN plays a role in the response to ER stress.
The mechanism of MULAN-induced NF-κB
activation
Ubiquitination is important in NF-κB activation. It
has been demonstrated that TRAF2, which is recruited to the TNFR
complex, ubiquitinates RIP, and polyubiquitinated RIP associates
with TNFR as well as with TAB2 (8,32,33). These studies suggest that the
polyubiquitination of RIP recruits the TAK1-TAB2 complex, which
subsequently activates the IKK complex. It has been shown that
TRAF6 recruited to the receptor complexes polyubiquitinates these
complexes, and polyubiquitinated TRAF6 associates with the
TAB2/TAB1/TAK1 complex (8). In
this study, we revealed that MULAN activated NF-κB dependent
transcription via E3 ubiquitin ligase activity. The activation of
NF-κB via MULAN was inhibited by IKK DN and TAK1 DN, but not by
TRAF2 and TRAF6. In addition, our binding assay revealed that MULAN
associated with TAK1. TAK1 has been reported to be induced by
polyubiquitination. TAK1 has Lys63-linked ubiquitination sites and
the polyubiquitination of TAK1 induces IK-dependent NF-κB
activation (34,35). Therefore, it is possible that
MULAN may induce the polyubiqutination of TAK1 to activate NF-κB
signaling.
MULAN-induced NF-κB activation in cells
subjected to ER stress
The ER stress-induced activation of NF-κB has been
demonstrated to be involved in both the protection of cells from ER
stress-induced apoptosis (36)
and in the induction of apoptosis (37). TRAF2 mediates the activation of
both the c-Jun N-terminal kinase (JNK)/stress-activated protein
kinase (SAPK) and the NF-κB pathways following ER stress (38,39). Therefore, TRAF2 simultaneously
mediates the activation of the NF-κB survival pathway and the
pro-apoptotic JNK pathway, and the fate of the cell would be
determined by the interplay between these opposing signals. TAK1
also activates both the NF-κB survival pathway and the
pro-apoptotic JNK pathway. The activation of TAK1 is induced by
K63-linked polyubiquitination reactions by TRAF2 or TRAF6 at the
K158, K34 and K209 residues (35). Recently, Lys562 of TAK1 was
identified as a novel Lys63-linked ubiquitination site. The
accumulation of polyubiquitination at Lys562 induces IKK-dependent
NF-κB activation, but not JNK/p38 pathway activation (34). In this study, MULAN activated
NF-κB dependent gene expression, and the expression of MULAN was
regulated by ER stress. In addition, the MULAN-dependent NF-κB
activation was TAK1-dependent and MULAN interacted with TAK1. We
thus hypothesized that MULAN may induce NF-κB activation via TAK1
for cell survival following ER stress; MULAN may activate
NF-κB-dependent gene expression by the induction of de novo
MULAN by ER stress.
In conclusion, in the present study we provide
evidence that MULAN is an E3 ligase that mediates NF-κB-dependent
gene expression in cells subjected to ER stress. Further analysis
of MULAN will be helpful in order to broaden our understanding of
the physiological significance of MULAN and of the mechanisms of
NF-κB activation by MULAN.
Abbreviations:
|
TAK1
|
transforming growth factor β-activated
kinase
|
|
MULAN
|
mitochondrial ubiquitin ligase
activator of nuclear factor-κB
|
|
ERSE
|
endoplasmic reticulum stress-response
element
|
|
TRAF
|
tumor necrosis factor
receptor-associated factor
|
|
IκBα
|
inhibitor of κBα
|
|
IKK
|
IκB kinase
|
Acknowledgments
We would like to thank Dr Kunihiro Matsumoto for
kindly providing the TAK1, TAK1 K63W and TAB1 expression vectors.
We would also like to thank Ms. Yukari Nakagawa and Mrs. Sanae
Shinkawa for their technical assistance and the members of the
Nakajima Laboratory for their helpful comments on our manuscript.
This study was funded in part by grants from the Naito Foundation,
Inoue Foundation for Science, Natural Science Scholarship
Daiichi-Sankyo Foundation of Life Science, Mitsubishi Tanabe Pharma
Corporation, Bureau of Social Welfare and Public Health, Academic
contribution of Pfizer, Eisai, Asahi Kasei Pharma, Abbvie, Santen,
Takeda Science Foundation and AstraZeneca (R&D grant 2013).
This study was supported in part by funds provided through an
MEXT-Supported program of the Strategic Research Foundation at
Private Universities (nos. S1411011 and 2014–2018) from the
Ministry of Education, Culture, Sports, Science and Technology of
Japan. This study was also supported in part by the Japan Society
for the Promotion of Science KAKENHI and Industry-university
cooperation (BioMimetics Sympathies Inc., Tokyo, Japan).
References
|
1
|
Karin M and Lin A: NF-kappaB at the
crossroads of life and death. Nat Immunol. 3:221–227. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chen Z, Hagler J, Palombella VJ, Melandri
F, Scherer D, Ballard D and Maniatis T: Signal-induced
site-specific phosphorylation targets I kappa B alpha to the
ubiquitin-proteasome pathway. Genes Dev. 9:1586–1597. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Karin M and Ben-Neriah Y: Phosphorylation
meets ubiquitination: The control of NF-[kappa]B activity. Annu Rev
Immunol. 18:621–663. 2000. View Article : Google Scholar
|
|
4
|
Chen ZJ: Ubiquitin signalling in the
NF-kappaB pathway. Nat Cell Biol. 7:758–765. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Deng L, Wang C, Spencer E, Yang L, Braun
A, You J, Slaughter C, Pickart C and Chen ZJ: Activation of the
IkappaB kinase complex by TRAF6 requires a dimeric
ubiquitin-conjugating enzyme complex and a unique polyubiquitin
chain. Cell. 103:351–361. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chung JY, Park YC, Ye H and Wu H: All
TRAFs are not created equal: Common and distinct molecular
mechanisms of TRAF-mediated signal transduction. J Cell Sci.
115:679–688. 2002.PubMed/NCBI
|
|
7
|
Wertz IE, O'Rourke KM, Zhou H, Eby M,
Aravind L, Seshagiri S, Wu P, Wiesmann C, Baker R, Boone DL, et al:
De-ubiquitination and ubiquitin ligase domains of A20 downregulate
NF-kappaB signalling. Nature. 430:694–699. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kanayama A, Seth RB, Sun L, Ea CK, Hong M,
Shaito A, Chiu YH, Deng L and Chen ZJ: TAB2 and TAB3 activate the
NF-kappaB pathway through binding to polyubiquitin chains. Mol
Cell. 15:535–548. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang C, Deng L, Hong M, Akkaraju GR, Inoue
J and Chen ZJ: TAK1 is a ubiquitin-dependent kinase of MKK and IKK.
Nature. 412:346–351. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ninomiya-Tsuji J, Kishimoto K, Hiyama A,
Inoue J, Cao Z and Matsumoto K: The kinase TAK1 can activate the
NIK-I kappaB as well as the MAP kinase cascade in the IL-1
signalling pathway. Nature. 398:252–256. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kaufman RJ: Stress signaling from the
lumen of the endoplasmic reticulum: Coordination of gene
transcriptional and translational controls. Genes Dev.
13:1211–1233. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sitia R and Braakman I: Quality control in
the endoplasmic reticulum protein factory. Nature. 426:891–894.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kopito RR: ER quality control: The
cytoplasmic connection. Cell. 88:427–430. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Plemper RK and Wolf DH: Retrograde protein
translocation: ERADication of secretory proteins in health and
disease. Trends Biochem Sci. 24:266–270. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hu MC, Gong HY, Lin GH, Hu SY, Chen MH,
Huang SJ, Liao CF and Wu JL: XBP-1, a key regulator of unfolded
protein response, activates transcription of IGF1 and Akt
phosphorylation in zebrafish embryonic cell line. Biochem Biophys
Res Commun. 359:778–783. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Amano T, Yamasaki S, Yagishita N,
Tsuchimochi K, Shin H, Kawahara K, Aratani S, Fujita H, Zhang L,
Ikeda R, et al: Synoviolin/Hrd1, an E3 ubiquitin ligase, as a novel
pathogenic factor for arthropathy. Genes Dev. 17:2436–2449. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li L, Shen Y, Ding Y, Liu Y, Su D and
Liang X: Hrd1 participates in the regulation of collagen I
synthesis in renal fibrosis. Mol Cell Biochem. 386:35–44. 2014.
View Article : Google Scholar
|
|
18
|
Bianchini E, Fanin M, Mamchaoui K, Betto R
and Sandonà D: Unveiling the degradative route of the V247M
α-sarcoglycan mutant responsible for LGMD-2D. Hum Mol Genet.
23:3746–3758. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fujita H, Yagishita N, Aratani S,
Saito-Fujita T, Morota S, Yamano Y, Hansson MJ, Inazu M, Kokuba H,
Sudo K, et al: The E3 ligase synoviolin controls body weight and
mitochondrial biogenesis through negative regulation of PGC-1β.
EMBO J. 34:1042–1055. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hasegawa D, Fujii R, Yagishita N,
Matsumoto N, Aratani S, Izumi T, Azakami K, Nakazawa M, Fujita H,
Sato T, et al: E3 ubiquitin ligase synoviolin is involved in liver
fibrogenesis. PLoS One. 5:e135902010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Matsuda A, Suzuki Y, Honda G, Muramatsu S,
Matsuzaki O, Nagano Y, doi T, Shimotohno K, Harada T, Nishida E, et
al: Large-scale identification and characterization of human genes
that activate NF-kappaB and MAPK signaling pathways. Oncogene.
22:3307–3318. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li W, Bengtson MH, Ulbrich A, Matsuda A,
Reddy VA, Orth A, Chanda SK, Batalov S and Joazeiro CA: Genome-wide
and functional annotation of human E3 ubiquitin ligases identifies
MULAN, a mitochondrial E3 that regulates the organelle's dynamics
and signaling. PLoS One. 3:e14872008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zemirli N, Pourcelot M, Ambroise G, Hatchi
E, Vazquez A and Arnoult D: Mitochondrial hyperfusion promotes
NF-κB activation via the mitochondrial E3 ligase MULAN. FEBS J.
281:3095–3112. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Fujita H, Fujii R, Aratani S, Amano T,
Fukamizu A and Nakajima T: Antithetic effects of MBD2a on gene
regulation. Mol Cell Biol. 23:2645–2657. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Nakajima T, Fukamizu A, Takahashi J, Gage
FH, Fisher T, Blenis J and Montminy MR: The signal-dependent
coactivator CBP is a nuclear target for pp90RSK. Cell. 86:465–474.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Nakajima T, Uchida C, Anderson SF, Lee CG,
Hurwitz J, Parvin JD and Montminy M: RNA helicase A mediates
association of CBP with RNA polymerase II. Cell. 90:1107–1112.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Fujita H, Ohshima T, Oishi T, Aratani S,
Fujii R, Fukamizu A and Nakajima T: Relevance of nuclear
localization and functions of RNA helicase. A Int J Mol Med.
15:555–560. 2005.
|
|
28
|
Hoffmann A and Baltimore D: Circuitry of
nuclear factor kappaB signaling. Immunol Rev. 210:171–186. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Roy B and Lee AS: The mammalian
endoplasmic reticulum stress response element consists of an
evolutionarily conserved tripartite structure and interacts with a
novel stress-inducible complex. Nucleic Acids Res. 27:1437–1443.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yoshida H, Haze K, Yanagi H, Yura T and
Mori K: Identification of the cis-acting endoplasmic reticulum
stress response element responsible for transcriptional induction
of mammalian glucose-regulated proteins. Involvement of basic
leucine zipper transcription factors. J Biol Chem. 273:33741–33749.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yoshida H, Okada T, Haze K, Yanagi H, Yura
T, Negishi M and Mori K: ATF6 activated by proteolysis binds in the
presence of NF-Y (CBF) directly to the cis-acting element
responsible for the mammalian unfolded protein response. Mol Cell
Biol. 20:6755–6767. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lee TH, Shank J, Cusson N and Kelliher MA:
The kinase activity of Rip1 is not required for tumor necrosis
factor-alpha-induced IkappaB kinase or p38 MAP kinase activation or
for the ubiqui-tination of Rip1 by Traf2. J Biol Chem.
279:33185–33191. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Legler DF, Micheau O, Doucey MA, Tschopp J
and Bron C: Recruitment of TNF receptor 1 to lipid rafts is
essential for TNFalpha-mediated NF-kappaB activation. Immunity.
18:655–664. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chen IT, Hsu PH, Hsu WC, Chen NJ and Tseng
PH: Polyubiquitination of transforming growth factor β-activated
kinase 1 (TAK1) atlysine 562 residue regulates TLR4-mediated JNK
and p38 MAPK activation. Sci Rep. 5:123002015. View Article : Google Scholar
|
|
35
|
Ajibade AA, Wang HY and Wang RF: Cell
type-specific function of TAK1 in innate immune signaling. Trends
Immunol. 34:307–316. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Mauro C, Crescenzi E, De Mattia R,
Pacifico F, Mellone S, Salzano S, de Luca C, D'Adamio L, Palumbo G,
Formisano S, et al: Central role of the scaffold protein tumor
necrosis factor receptor-associated factor 2 in regulating
endoplasmic reticulum stress-induced apoptosis. J Biol Chem.
281:2631–2638. 2006. View Article : Google Scholar
|
|
37
|
Hu P, Han Z, Couvillon AD, Kaufman RJ and
Exton JH: Autocrine tumor necrosis factor alpha links endoplasmic
reticulum stress to the membrane death receptor pathway through
IRE1alpha-mediated NF-kappaB activation and downregulation of TRAF2
expression. Mol Cell Biol. 26:3071–3084. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Leonardi A, Vito P, Mauro C, Pacifico F,
Ulianich L, Consiglio E, Formisano S and Di Jeso B: Endoplasmic
reticulum stress causes thyroglobulin retention in this organelle
and triggers activation of nuclear factor-kappa B via tumor
necrosis factor receptor-associated factor 2. Endocrinology.
143:2169–2177. 2002.PubMed/NCBI
|
|
39
|
Urano F, Wang X, Bertolotti A, Zhang Y,
Chung P, Harding HP and Ron D: Coupling of stress in the ER to
activation of JNK protein kinases by transmembrane protein kinase
IRE1. Science. 287:664–666. 2000. View Article : Google Scholar : PubMed/NCBI
|