Introduction
Multidrug resistance is a major cause of cancer
chemotherapy failure, and reversing this resistance is an emerging
area of interest in cancer treatment. MicroRNAs (miRNAs or miRs)
have the potential to act as mediators of multidrug resistance
reversal. miRNAs are a class of small non-coding RNAs of about
19–22 nucleotides that play an important role in cell occurrence,
development, differentiation, proliferation, aging and apoptosis
(1). miRNAs are evolutionarily
conserved and are located in the introns or exons of protein-coding
genes, or in intergenic regions. Mature miRNAs regulate gene
expression by inhibiting protein translation and promoting mRNA
degradation.
Differential expression of miRNAs has been described
in multiple cancer cell types, and miRNAs commonly function as
either tumor suppressors or oncogenes (2,3).
The expression of miR-133a is low in the majority of types of tumor
cells, and particularly low in squamous cell carcinoma of the
maxillary sinus, renal cell carcinoma, and rhabdomyosarcoma
(4–6). High miR-133a expression induces the
death of multiple cancer cells, including squamous cell carcinomas
of the lungs (7), tongue
(8) and esophagus (9), as well as prostate carcinoma
(10) and bladder carcinoma
(11). In squamous cell carcinoma
of the head and neck, increased miR-133a expression has been shown
to exert anticancer effects through downregulation of the
scaffolding protein caveolin-1 (12).
The expression of copper transporting P-type
adenosine triphosphatase (ATP7B) has previously been implicated in
cisplatin resistance (13). ATP7B
transports copper to the Golgi complex to promote the synthesis of
copper-containing enzymes, or it mediates the direct elimination of
copper from cells. Furukawa et al and others (14,15) have noted that the presence of
ATP7B in the Golgi complex is associated with cisplatin resistance
and that copper transport is implicated in multidrug resistance.
Others have reported that ATP7B-positive esophageal and oral
carcinoma patients had poorer outcomes and a noticeably lower
long-term survival rate following chemotherapy with
cisplatin-containing drugs when compared with patients with
ATP7B-negative lesions (16).
Nakayama et al (17)
reported that ATP7B-transfected KB/WD cells exhibited reduced
accumulation of cisplatin compared with KB/CV control cells.
Additionally, a cisplatin-resistant oral squamous cell carcinoma
cell line established by cultivation of cells in gradually
increasing concentrations of cisplatin demonstrated high levels of
ATP7B and copper-transporting ATPase 1 (ATP7A) expression (18,19).
In the present study, we report on ATP7B expression
in the cell line Hep-2 and the vincristine-resistant cell line
Hep-2v. We investigated the influence of exogenous miR-133a on
ATP7B expression in both cell lines, and correlated ATP7B
expression with sensitivity to cisplatin chemotherapy in Hep-2
cells.
Materials and methods
Cell lines and reagents
The Hep-2 and NP69 cell lines were purchased from
Guangzhou Jinibio Biotechnology Co., Ltd. (Guangdong, China). The
vincristine-resistant l Hep-2v cells were developed in our
laboratory (Institute of Otolaryngology-Head and Neck Surgery,
Changchun, China). RPMI-1640, fetal calf serum, Alexa 555-labeled
goat anti-rabbit IgG (A-21428), and TRIzol reagent were purchased
from Invitrogen Life Technologies (Carlsbad, CA, USA). ATP7A
(bs-1572R) and ATP7B (bs-1718R) antibodies were purchased from
Bioss (Beijing, China). The Cell Counting Kit (CCK)-8 was obtained
from Signalway Antibody LLC (College Park, MD, USA). The miRcute
miRNA cDNA first-strand synthesis and miRcute miRNA isolation kits
were purchased from Tiangen Biotech (Beijing) Co., Ltd. (Beijing,
China). The SYBR-Green I PCR kit was supplied by Roche Biochemicals
(Indianapolis, IN, USA).
Cell culture
Hep-2 cells were recovered from frozen storage and
sub-cultured in RPMI-1640 medium supplemented with 10% fetal calf
serum at 37°C, in an atmosphere with 5% CO2 and 95%
relative humidity. Culture medium was refreshed daily. When cells
reached 90% confluence, they were detached with 0.25% trypsin and
seeded into a new culture flask at a ratio of 1:3. The
vincristine-resistant cell line Hep-2v was produced by incubation
with gradually increasing concentrations of vincristine (Sigma, St.
Louis, MO, USA) for 8 months, with cells developing a vincristine
resistance index of 45, as previously described (20).
Plasmids and transfection
pEZX-miR-133 and pEZX-control lentiviral vectors
were from Guangzhou FulenGen Co., Ltd. (Guangdong, China). The
sequence of mature miR-133a-1 was identified using the query
hsa-miR-133a in GenBank (http://www.ncbi.nlm.nih.gov). The gene ID is
MIMAT0000427 and the sequence is: 5′-UUU GGUCCCCUUCAACCAGCUG-3′.
Precursor miRNA expr ession constructs were prepared in a feline
immunodeficiency virus-based lentiviral plasmid vector system
(Guangzhou FulenGen Co., Ltd.). H1 RNA polymerase III
promoter-driven pre-miRNA has a stem loop of
5′-ACAAUGCUUUGCUAGAGCUGGUAAAAUGGAACC
AAAUCGCCUCUUCAAUGGAUUUGGUCCCCUUCAAC CAGCUGUAGCUAUGCAUUGA-3′ which
is processed into mature miR-133a-1 by the RNAi enzyme system
(Guangzhou FulenGen Co., Ltd.). The vector co-expresses reporter
gene eGFP under the control of cytomegalovirus (CMV) promoter
(Guangzhou FulenGen Co., Ltd.).
Hep-2v cells were detached using 0.25% trypsin and
collected by centrifugation at 800 × g for 5 min at room
temperature. Sediments were re-suspended in RPMI-1640 culture
medium and then seeded onto a 24-well plate (2×105
cells/well). Upon reaching 70–80% confluence, cells were cultured
with antibiotic-free serum-containing RPMI-1640 medium overnight.
For transfection, 3 µg pEZX-miR-133a plasmid was diluted in
50 µl serum-free culture medium (both self-prepared with FCS
and RPMI-1640), centrifuged on a short spin, and slightly mixed.
Subsequently, 3 µl GenEscort™ transfection reagent (Wisegen
Biotechnology Co., Nanjing, China) was diluted in 50 µl
serum-free culture medium and then lightly shaken. Transfection
reagent-containing- and pEZX-miR-133a plasmid-containing solutions
were then combined. The resulting mixture was lightly shaken and
then incubated at room temperature for 15 min. Cells seeded onto
24-well plates were cultured with 100 µl of the resulting
mixture at 37°C. Culture medium was refreshed with serum-containing
complete culture medium after 4 h, and cells were further cultured
for 48–72 h.
Immunofluorescence staining
After removal of the culture medium, glass
coverslips with cultured cells were washed with 0.1 M
phosphate-buffered saline (PBS), treated with 4% paraformaldehyde
at room temperature for 30 min, washed three times with PBS for 10
min each wash, treated with 0.1% Triton X-100 for 10 min, and
washed a further three times with PBS. After the addition of 5%
goat serum, cells were incubated at room temperature for 1 h, and
then incubated with primary antibody (ATP7A or ATP7B) at 4°C
overnight. Cells were then washed three times with PBS, incubated
with Alexa-555 goat anti-rabbit IgG secondary antibody (1:200
dilution) at room temperature for 1 h in the dark, and washed three
times with PBS for 10 min each wash. Hoechst 33342 was used for
staining. Cells were mounted in glycerol and observed by laser
confocal microscopy (FluoView FV1000; Olympus, Tokyo, Japan) to
determine protein expression.
CCK-8 assay
Hep-2 and Hep-2v cells were seeded onto a 96-well
plate at 1×104 cells/well. Cells were then treated with
indicated concentrations of cisplatin for indicated time periods
followed by addition of 100 µl culture medium and 10
µl/well CCK-8 solution. Cells were incubated at 37°C for 1–4
h, and optical density (OD) at 450 nm was determined. Cell
viability was calculated as OD value in the treated group/OD value
in the control group × 100. The control group was Hep-2v or Hep-2
cells which were not treated with cisplatin.
Reverse transcription-quantitative PCR
(RT-qPCR)
miRNA was isolated from Hep-2, Hep-2v, and NP69
cells using the miRcute miRNA isolation kit from Tiangen Biotech
(Beijing) Co., Ltd.. Poly(A) tails were added to the 3′ ends of
miRNA, and reverse transcription was performed using
oligo(dT)-universal tag and the miRcute miRNA cDNA first-strand
synthesis kit following the manufacturer's instructions [Tiangen
Biotech (Beijing) Co., Ltd.]. Briefly, 5 µl total RNA, 2
µl 10X poly(A) polymerase buffer, 4 µl 5X rATP
solution, 8.6 µl RNase-free ddH2O, and 0.4
µl E. coli poly(A) polymerase were combined in an
RNase-free reaction tube, which was pre-cooled on ice. Next, 2
µl poly(A) reaction solution was used for first-strand cDNA
synthesis in a 20-µl reaction containing 2 µl 10X RT
primer, 2 µl 10X RT buffer, 1 µl ultrapure dNTP
mixture (2.5 mM each), 1 µl RNasin (40 U/µl), 0.5
µl Quant KTase, and 11.5 µl RNase-free
ddH2O. After thorough mixing, the mixture was incubated
at 37°C for 60 min. qPCR was performed using the miRcute miRNA
fluorescent quantitative detection kit [Tiangen Biotech (Beijing)
Co., Ltd.] with an upstream primer for hsa-miR-133a,
5′-TTTGGTCCCCTTCAACCAG-3′, and the generic downstream primer
provided in the kit. A 20-µl reaction containing 10
µl 2X miRcute miRNA Premix, 0.4 µl hsa-miR-133a
upstream primer (10 µmol/l), 0.4 µl downstream
primer, 2 µl cDNA, and 7.2 µl RNase-free
ddH2O was established. qPCR involved denaturation at
94°C for 2 min and 40 cycles of 94°C for 30 sec and 60°C for 45
sec. The fluorescence value for each cycle was recorded during
extension at 75°C. Relative changes in miRNA levels were calculated
using 5S RNA [Tiangen Biotech (Beijing) Co., Ltd.] as an internal
control.
RT-qPCR-based analysis of mRNA
expression
Cell culture medium was removed, cells were washed
twice with PBS and total RNA was extracted using TRIzol. RNA
concentration was determined using a NanoDrop 2000
spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA).
cDNA synthesis was then performed using a cDNA first-strand
synthesis kit [Tiangen Biotech (Beijing) Co., Ltd.] in 20-µl
reactions, each with 2 µl 5X avian myeloblastosis virus
(AMV) buffer, 2 µl dNTP mixture, 1 µl AMV, 1
µl Oligo dT, 1 µl RNase Inhibitor, and 13 µl
total RNA (2 µg). Reverse transcription was performed by
incubation at 42°C for 1 h, followed by 75°C for 10 min to
inactivate AMV. PCR primers for ATP7B were designed based on the
GenBank sequence (http://www.ncbi.nlm.nih.gov) using Primer 3.0
software: 5′-GGTGTTC TCTCCGTGTTGGT-3 and 5′-GGCTGCACAGGAAAGA
CTTC-3′. qPCR was performed using FastStart Essential DNA Green
Master (Roche). Each reaction contained 10 µl Master
SYBR-Green I mix, 5 µl primer mixture (1.25 nmol/l), 2
µl cDNA, and 3 µl H2O. qPCR involved
denaturation at 95°C for 10 min and 40 cycles of 94°C for 1 min,
60°C for 50 sec, and 72°C for 50 sec. The fluorescence value for
each cycle was recorded during the extension at 75°C. Values for
ATP7B were normalized against β-actin. Relative values for mRNA
levels were calculated using the ΔΔCt method: 2−Δ(ΔCt),
where ΔCt = Ct(target) − Ct(actin) and Δ(ΔCt) = ΔCt(treated) − ΔCt
(untreated).
Western blot analysis
Cells were collected and lysed in buffer containing
50 mM HEPES, 250 mM NaCl, 5 mM EDTA, 0.1% NP-40, 1 mM PMSF, 1 mM
DTT and supplemented with protease inhibitor cocktail). Following
centrifugation (10,000 × g for 10 min at 4°C), the supernatant was
discarded and protein concentration was determined using BCA
protein assay. Protein samples (4:1 in SDS loading buffer) were
denatured at 95°C for 5 min, subjected to SDS-PAGE, electrically
transferred onto PVDF membranes using a semi-dry transfer system,
and blocked in 5% dry milk for 1 h. Protein samples were incubated
with anti-ATP7B primary antibody (dilution 1:200) at 4°C overnight
and then with HRP-labeled goat anti-rabbit IgG secondary antibody
(sc-2004; dilution 1:2,000; Santa Cruz Biotechnology, Santa Cruz,
CA, USA). The membranes were washed three times with TBST for 15
min after each incubation. Blots were developed using ECL reagent
(Thermo Fisher Scientific) and images obtained using a
Dolphin-Chemi Mini image system (Wealtec Corp., Sparks, NV, USA).
Gray-scale values for each band were analyzed, and relative protein
levels were calculated using β-actin as an internal control.
Statistical analysis
All data were collected from at least three
independent experiments. SPSS 17.0 software was used for
statistical analysis. One-way ANOVA was used for analysis of cell
viability and mRNA or protein expression, and Tukey's test was used
for multiple pairwise comparisons. A p-value <0.05 was
considered to indicate a statistically significant difference.
Results
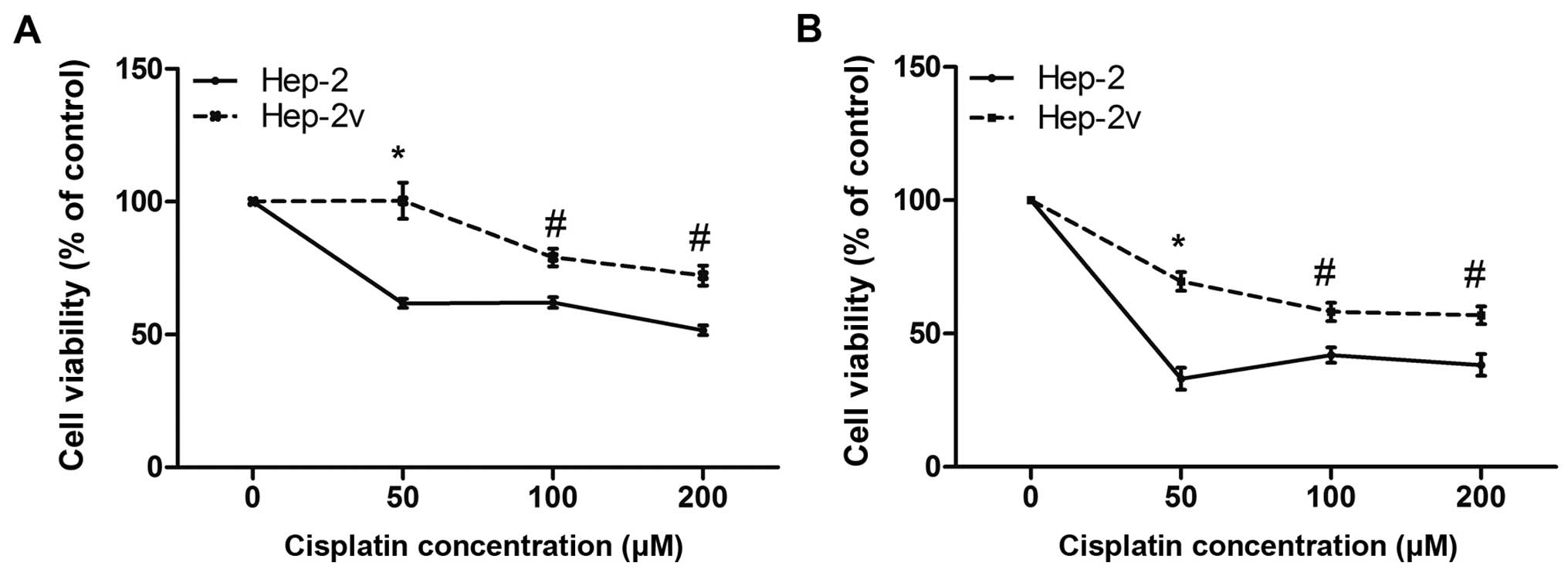
Cisplatin resistance in Hep-2v cells
A CCK-8 assay was performed to investigate the
effects of cisplatin on Hep-2 and Hep-2v cell proliferation
(Fig. 1). Following treatment
with 50 µM cisplatin for 24 h, the survival rate of Hep-2
cells was 61.7%, while that of Hep-2v had not changed. The
difference between the two cell lines was statistically significant
(p<0.01). Following treatment with 100 µM cisplatin for
24 h, the survival rate of Hep-2 cells was closer to that of those
treated with 50 µM cisplatin, while the survival rate of
Hep-2v cells was 80%. However, this difference was also
statistically significantly (p<0.05). Treatment with 200
µM cisplatin for 24 h resulted in survival rates of 51.2%
and 72% for Hep-2v and Hep-2 cells, respectively. Similar results
were obtained when 48-h cisplatin treatment was undertaken
(Fig. 1B). These findings suggest
that vincristine-treated Hep-2 cells exhibit cisplatin resistance,
particularly at 50 µM.
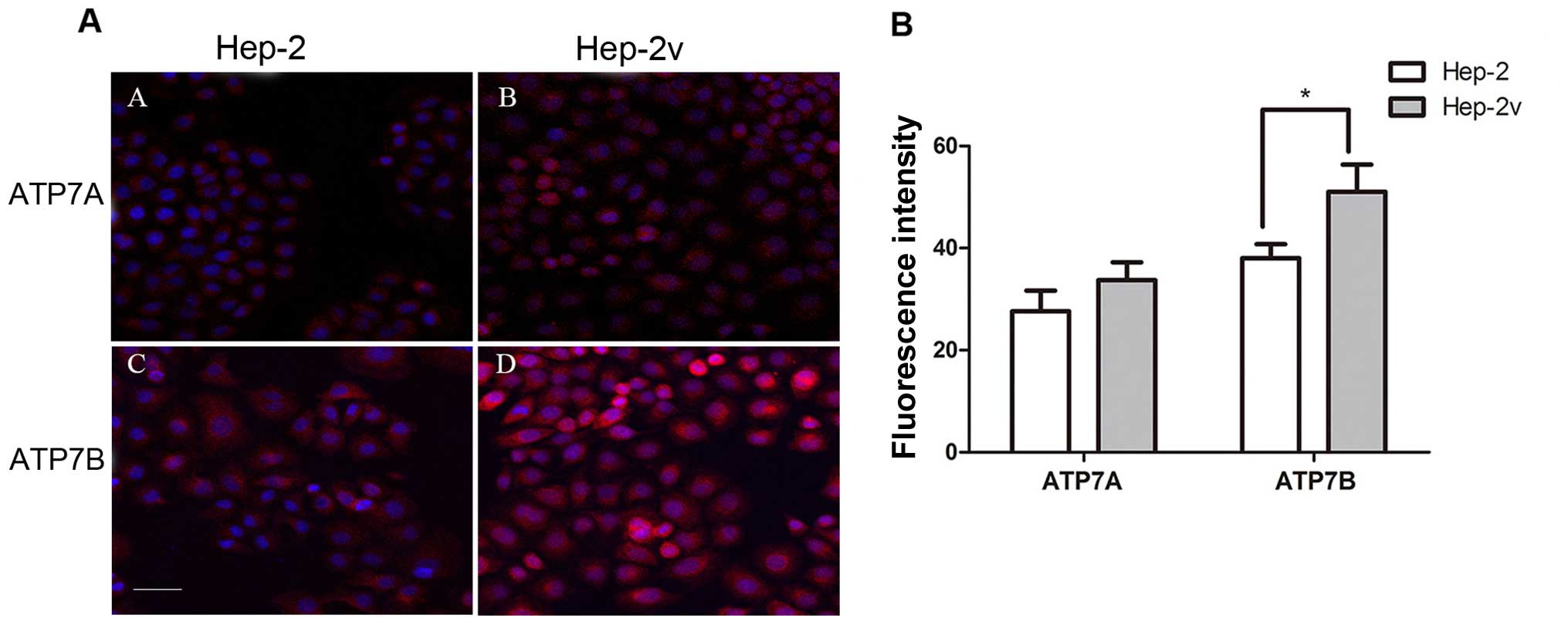
ATP7A and ATP7B expression in Hep-2v
cells
We subsequently assessed the expression of cisplatin
resistance-related molecules ATP7A and ATP7B by immunofluorescence
staining to determine the molecular mechanism underlying cisplatin
resistance of Hep-2v cells. Both Hep-2 and Hep-2v cells expressed
ATP7A and ATP7B protein. Quantitative fluorescence analysis showed
that ATP7B protein expression in Hep-2v cells was significantly
higher than that in the Hep-2 cells; however, there was no
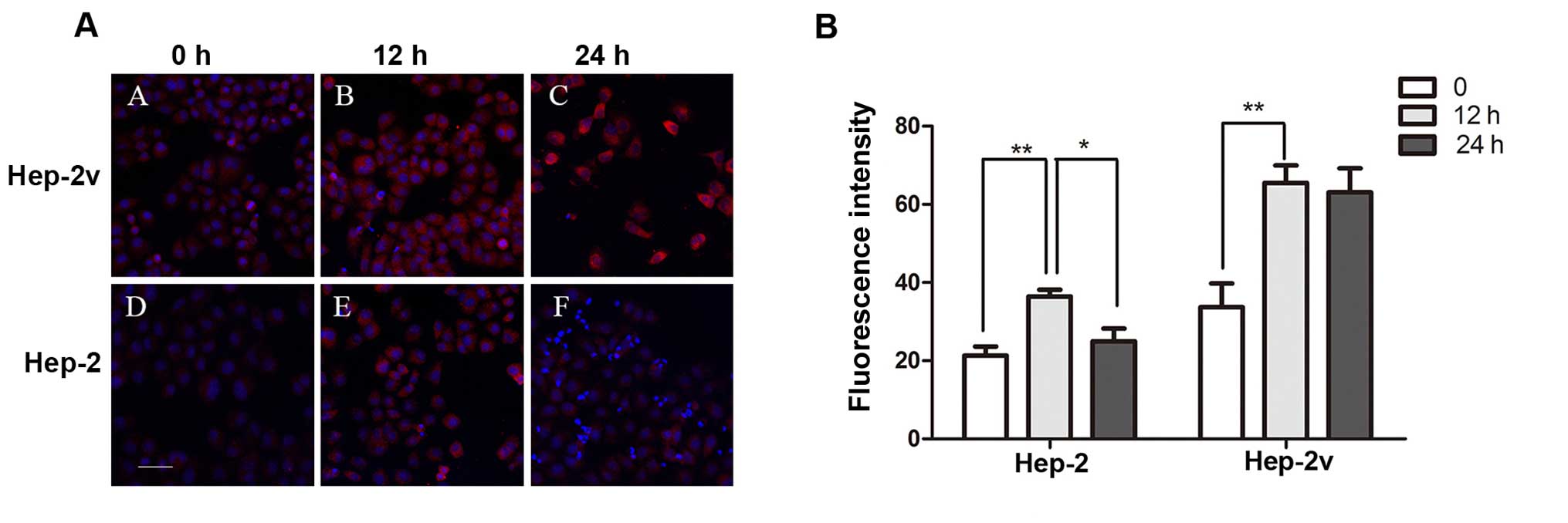
significant difference in ATP7A expression (Fig. 2). Furthermore, treatment with 50
µM cisplatin increased levels of ATP7B protein expression in
both Hep-2 and Hep-2v cells (Fig.
3). In Hep-2v cells, ATP7B protein levels reached a peak at 12
h and began to decrease at 24 h following treatment. Hep-2v cells
expressed significantly higher levels of ATP7B than Hep-2 cells at
all time points observed (Fig.
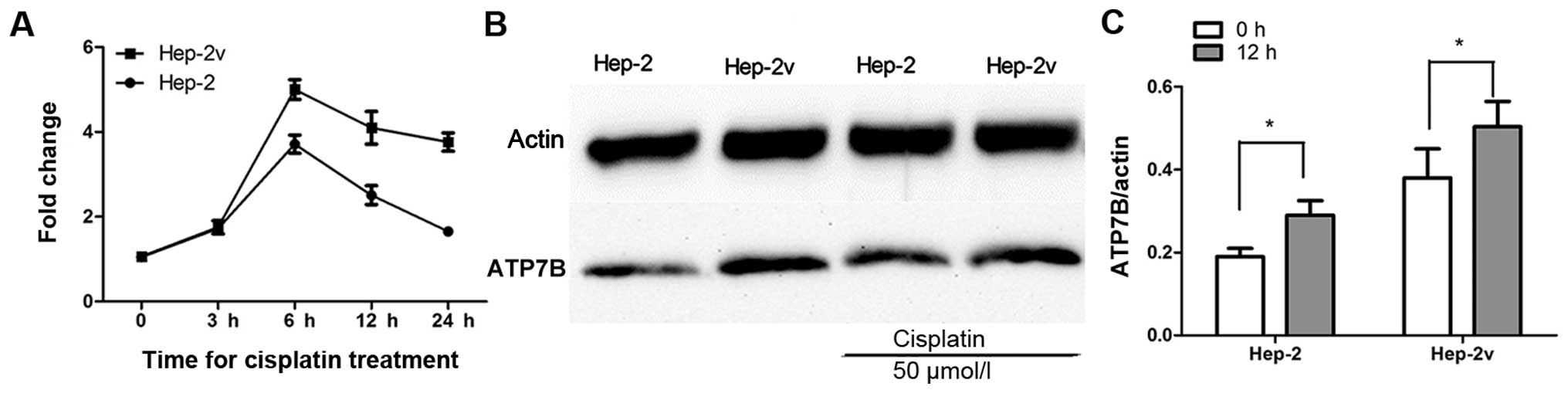
3). RT-qPCR revealed that 50 µM cisplatin increased
ATP7B gene expression in both Hep-2 and Hep-2v cells. ATP7B
expression in Hep-2 cells began to increase at 3 h, peaked at 6 h,
and began to decrease at 12 h after cisplatin treatment. Moreover,
ATP7B expression in Hep-2v cells followed a similar trend but
remained at peak level until 24 h after cisplatin treatment
(Fig. 4A). Western blot analysis
revealed that ATP7B protein expression in both Hep-2 and Hep-2v
cells was significantly greater than that in control cells at 12 h
after cisplatin treatment (P<0.05; Fig. 4B and C).
ATP7B expression in
miR-133a-overexpressing Hep-2v cells
We subsequently used RT-qPCR, western blot analysis
and immunofluorescence staining to assess ATP7B expression in
Hep-2v cells overexpressing miR-133a. Hep-2v cells were transfected
with either pEZX-miR-133 or pEZX-control plasmid vectors.
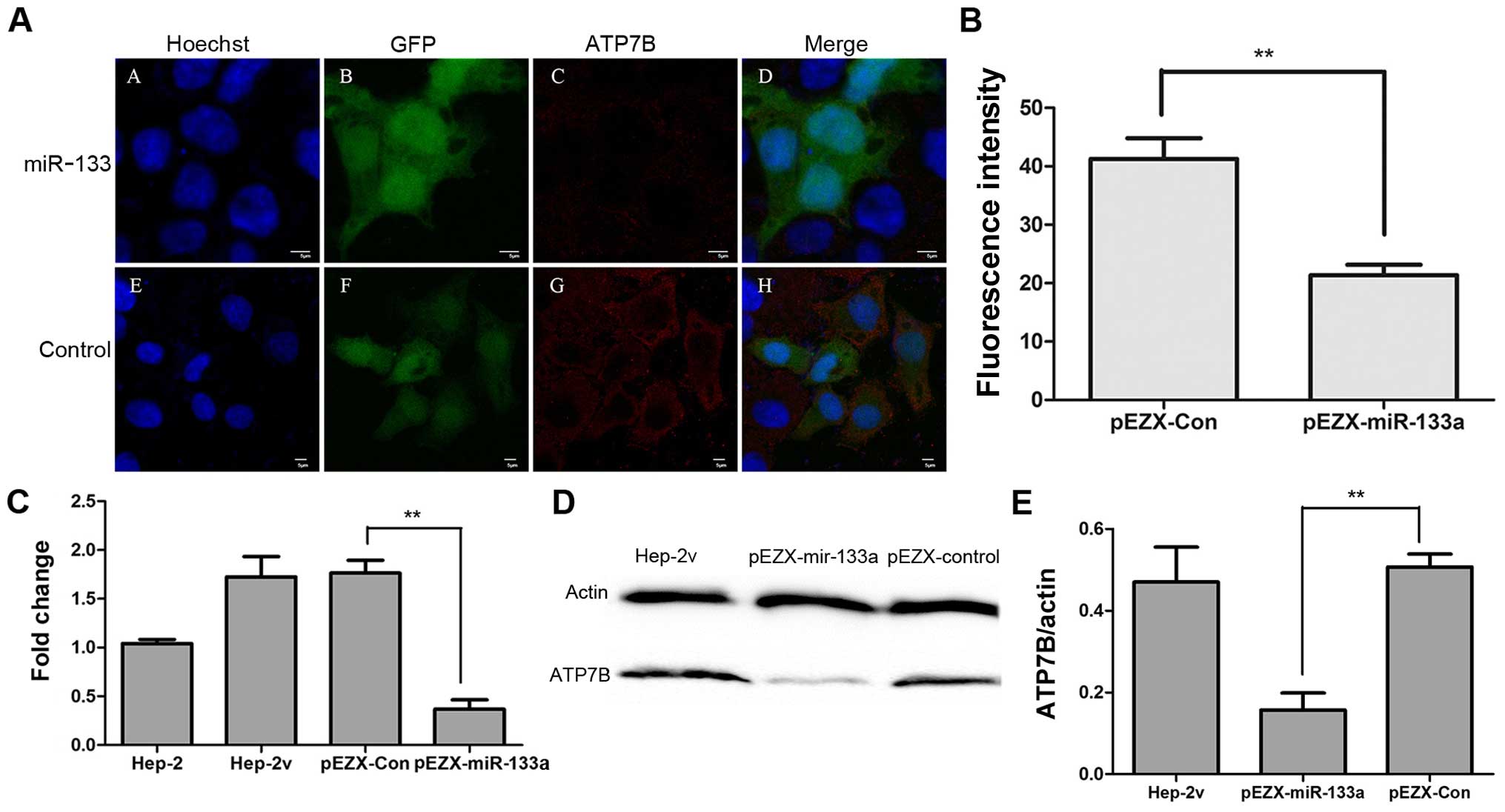
Transfected cells exhibit reporter gene GFP expression, while ATP7B
expression is indicated by red fluorescence following
immunostaining (Fig. 5A). ATP7B
staining in pEZX-miR-133a-transfected cells was significantly
weaker than that in pEZX-control transfected cells (Fig. 5B). Western blot analysis and
RT-qPCR results showed that exogenous expression of miR-133a
reduced ATP7B mRNA and protein expression in Hep-2v cells (Fig. 5C–E).
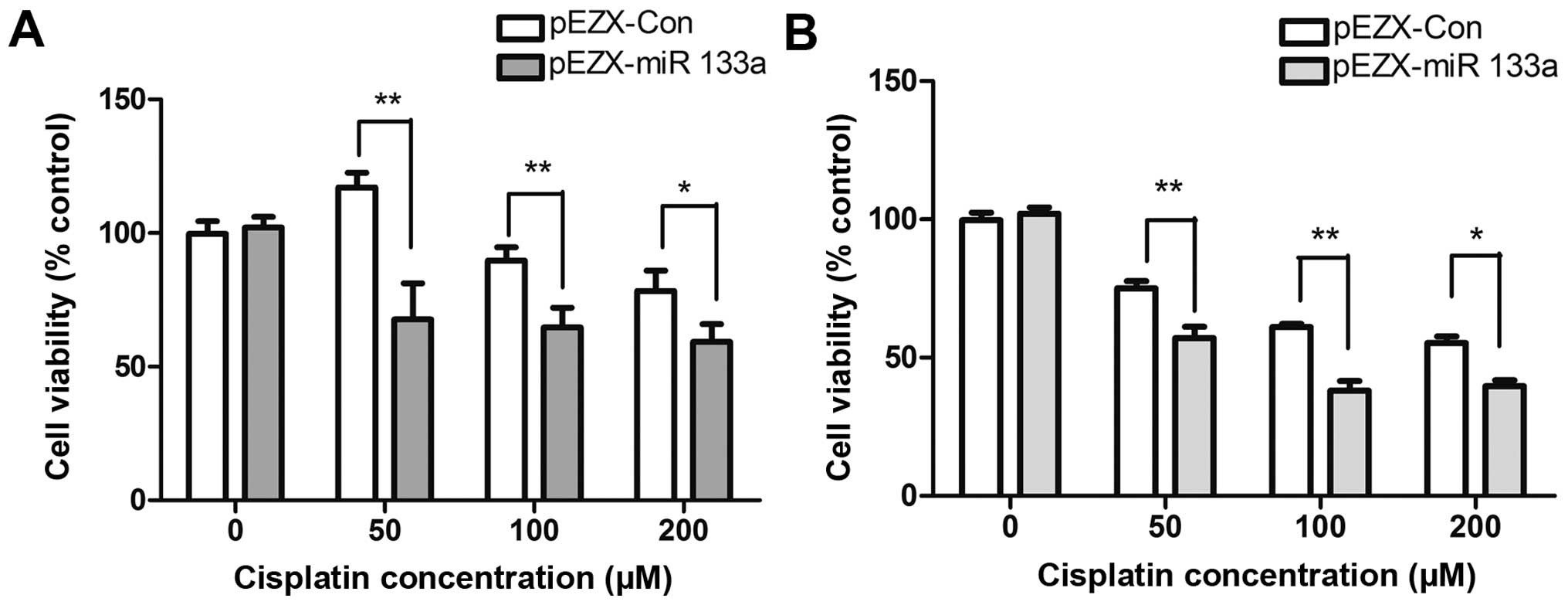
miR-133a overexpression increases the
sensitivity of drug-resistant Hep-2v cells to cisplatin
Hep-2v cells were transfected with a recombinant
lentivirus vector expressing miR-133a and then treated with 50, 100
or 200 µM cisplatin 48 h later. After a further 24-48 h,
cell viability was evaluated by CCK-8 assay. Compared with control
cells, Hep-2v cells expressing exogenous miR-133a had significantly
significantly reduced survival following treatment with 50 or 100
µM cisplatin (Fig. 6).
These results suggest that upregulation of miR-133a enhances the
cytotoxic effects of cisplatin on Hep-2v cells, particularly at
50–100 µM concentrations.
Discussion
We previously created a vincristine-resistant cell
line and found that these cells were resistant to cisplatin.
Therefore, Hep-2v cells are multidrug-resistant.
The dynamic balance of heavy metals in living
organisms is maintained by a series of proteins: e.g., in
vivo copper metabolism involves the membrane protein copper
transporter receptor 1, which selectively imports copper into
cells. Following importation, at least three further proteins,
homolog of Ace1 activator (HAA1), ATOX1, copper chaperone for
superoxide dismutase (CCS), are involved in transporting copper to
the Golgi complex and mitochondria (21). HAA1 transports copper to ATP7B
through a unique copper-binding site (22). ATP7B transports copper and other
heavy metal molecules such as cisplatin outside cells, and ATP7B
has been implicated in cellular resistance to stibium and arsenite
(23,24). Schilsky et al (25) found that chromium resistance in
hepatoblastoma cell lines involves ATP7B upregulation. Cisplatin
functions by inhibiting DNA synthesis through a platinum-DNA
reaction (26), and intracellular
accumulation of platinum-based compounds is closely related to the
degree of cell sensitivity to these drugs.
In the present study, we demonstrated that ATP7B
expression was low in Hep-2 cells, and increased in
vincristine-resistant Hep-2v cells. Following cisplatin treatment,
ATP7B expression in these two cell lines was increased. While ATP7B
expression in Hep-2 cells began to decrease at 12 h, expression in
Hep-2v cells remained higher. This suggests that the resistance of
Hep-2v cells to cisplatin is mediated by persistent high expression
of ATP7B. Cisplatin-resistant cells exhibited reduced intracellular
accumulation of cisplatin and increased extracellular outflow of
cisplatin, and cisplatin resistance is associated with increased
ATP7B expression in ovarian carcinoma cell lines (27,28).
miR-133a functions as a tumor inhibitor and is
expressed at low levels in a variety of tumor tissues; exogenous
miR-133a induces tumor cell apoptosis and inhibits tumor cell
metastasis (29); however,
whether miR-133a is involved in chemotherapeutic drug resistance
remains unknown. Yuan et al (30) and others (31) have found that decreased miR-133a
expression in adriamycin-resistant MCF-7 cells led to an increase
in target gene UCP-2 expression. Previously (32), we compared differential expression
levels of miRNAs in laryngeal carcinoma and normal tissues and
found that miR-133 expression was low in laryngeal carcinoma
tissues.
Investigations utilizing miRanda/miRBase and
miRNA.org identified ATP7B as a target of miR-133a.
Therefore, we questioned whether the high ATP7B expression in
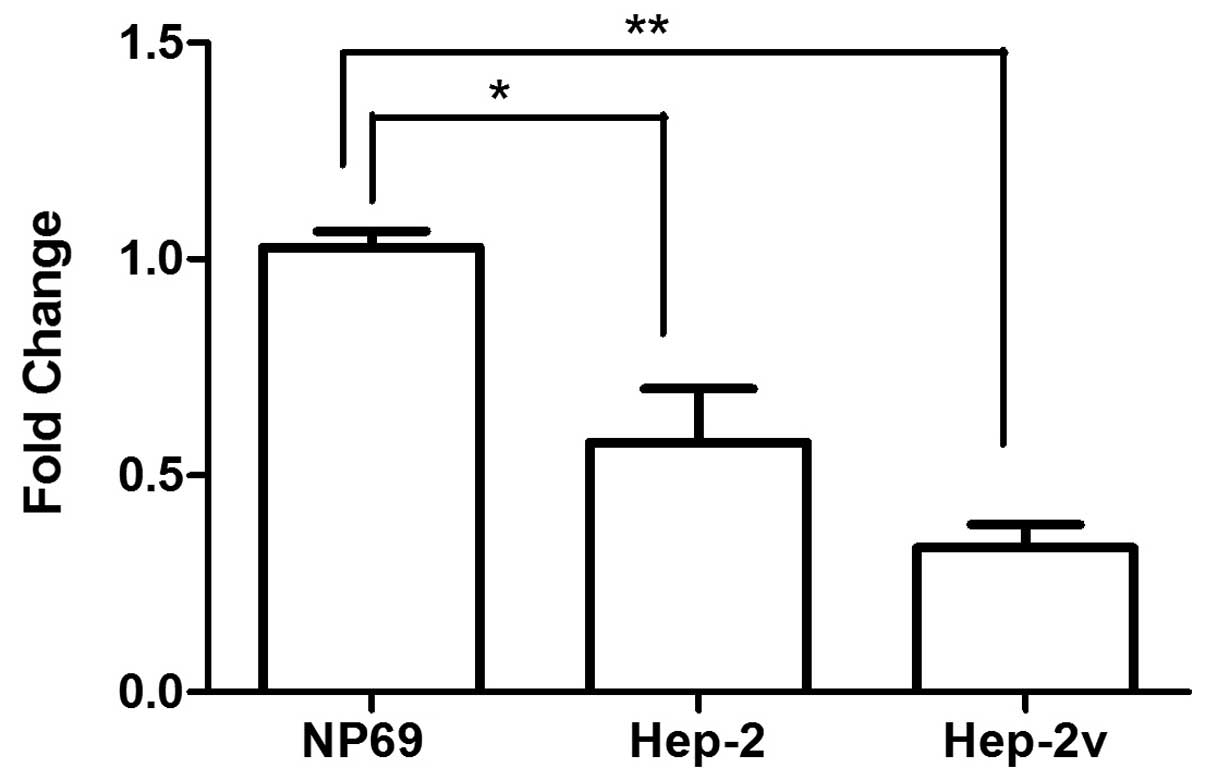
Hep-2v cells was influenced by miR-133a. In the present study, we
noted that miR-133a expression in Hep-2 and Hep-2v cells was
significantly lower than that in NP69 cells, but there was no
marked difference between expression in Hep-2 and Hep-2v cells
(Fig. 7). This suggests that high
ATP7B expression in Hep-2v cells involves factors additional to
miR-133a downregulation.
We transfected a recombinant lentiviral vector
expressing miR-133a into Hep-2v cells and detected intracellular
ATP7B expression using RT-qPCR and immunofluorescence staining.
ATP7B expression was significantly lower in the
miR-133a-transfected group compared with the control. These
findings indicate that exogenous miR-133a transfection negatively
regulates ATP7B expression.
Hep-2v cells expressing exogenous miR-133a exhibited
low ATP7B expression, and cell viability was significantly
decreased after cisplatin treatment. This provides further evidence
that ATP7B promotes multidrug resistance in Hep-2v cells. However,
there was no significant change in the viability of Hep-2 cells
overexpressing miR-133a. We suggest that this was due to the
increase in ATP7B expression, as it is transient in
non-drug-resistant Hep-2 cells, and ATP7B expression rapidly
returns to a low level. Therefore, decreased ATP7B expression after
miR-133a transfection was not obvious (data not shown).
Hep-2v cells persistently express high levels of
ATP7B, and cisplatin treatment stimulates increased ATP7B
expression of ATP7B in these cells. ATP7B contributes to the
removal of intracellular cisplatin to the extracellular space,
thereby promoting cell survival. However, ATP7B expression was
significantly decreased following exogenous expression of miR-133a.
Reduced levels of ATP7B likely impaired the transportation of
cisplatin to the extracellular space, thereby increasing the
sensitivity of Hep-2v cells to cisplatin. These findings suggest
that ATP7B is a useful target in cisplatin-resistant tumors.
Abbreviations:
|
miRNAs
|
microRNAs
|
|
ATP7B
|
copper transporting P-type adenosine
triphosphatase
|
Acknowledgments
This study was supported by grants from Jilin
Province Technique-Development Plan (200905197) and Jilin Province
Health Development (2009Z021).
References
|
1
|
Bartel DP: MicroRNAs: genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Calin GA, Dumitru CD, Shimizu M, Bichi R,
Zupo S, Noch E, Aldler H, Rattan S, Keating M, Rai K, et al:
Frequent deletions and down-regulation of micro-RNA genes miR15 and
miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci
USA. 99:15524–15529. 2002. View Article : Google Scholar
|
|
3
|
Calin GA, Sevignani C, Dumitru CD, Hyslop
T, Noch E, Yendamuri S, Shimizu M, Rattan S, Bullrich F, Negrini M
and Croce CM: Human microRNA genes are frequently located at
fragile sites and genomic regions involved in cancers. Proc Natl
Acad Sci USA. 101:2999–3004. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kawakami K, Enokida H, Chiyomaru T,
Tatarano S, Yoshino H, Kagara I, Gotanda T, Tachiwada T, Nishiyama
K, Nohata N, et al: The functional significance of miR-1 and
miR-133a in renal cell carcinoma. Eur J Cancer. 48:827–836. 2012.
View Article : Google Scholar
|
|
5
|
Nohata N, Hanazawa T, Kikkawa N, Sakurai
D, Sasaki K, Chiyomaru T, Kawakami K, Yoshino H, Enokida H,
Nakagawa M, et al: Identification of novel molecular targets
regulated by tumor suppressive miR-1/miR-133a in maxillary sinus
squamous cell carcinoma. Int J Oncol. 39:1099–1107. 2011.PubMed/NCBI
|
|
6
|
Rao PK, Missiaglia E, Shields L, Hyde G,
Yuan B, Shepherd CJ, Shipley J and Lodish HF: Distinct roles for
miR-1 and miR-133a in the proliferation and differentiation of
rhabdomyosarcoma cells. FASEB J. 24:3427–3437. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Moriya Y, Nohata N, Kinoshita T, Mutallip
M, Okamoto T, Yoshida S, Suzuki M, Yoshino I and Seki N: Tumor
suppressive microRNA-133a regulates novel molecular networks in
lung squamous cell carcinoma. J Hum Genet. 57:38–45. 2012.
View Article : Google Scholar
|
|
8
|
Wong TS, Liu XB, Chung-Wai Ho A, Po-Wing
Yuen A, Wai-Man Ng R and Ignace Wei W: Identification of pyruvate
kinase type M2 as potential oncoprotein in squamous cell carcinoma
of tongue through microRNA profiling. Int J Cancer. 123:251–257.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kano M, Seki N, Kikkawa N, Fujimura L,
Hoshino I, Akutsu Y, Chiyomaru T, Enokida H, Nakagawa M and
Matsubara H: miR-145, miR-133a and miR-133b: tumor-suppressive
miRNAs target FSCN1 in esophageal squamous cell carcinoma. Int J
Cancer. 127:2804–2814. 2010. View Article : Google Scholar
|
|
10
|
Kojima S, Chiyomaru T, Kawakami K, Yoshino
H, Enokida H, Nohata N, Fuse M, Ichikawa T, Naya Y, Nakagawa M and
Seki N: Tumour suppressors miR-1 and miR-133a target the oncogenic
function of purine nucleoside phosphorylase (PNP) in prostate
cancer. Br J Cancer. 106:405–413. 2012. View Article : Google Scholar :
|
|
11
|
Yoshino H, Chiyomaru T, Enokida H,
Kawakami K, Tatarano S, Nishiyama K, Nohata N, Seki N and Nakagawa
M: The tumour-suppressive function of miR-1 and miR-133a targeting
TAGLN2 in bladder cancer. Br J Cancer. 104:808–818. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nohata N, Hanazawa T, Kikkawa N, Mutallip
M, Fujimura L, Yoshino H, Kawakami K, Chiyomaru T, Enokida H,
Nakagawa M, et al: Caveolin-1 mediates tumor cell migration and
invasion and its regulation by miR-133a in head and neck squamous
cell carcinoma. Int J Oncol. 38:209–217. 2011.
|
|
13
|
Yoshizawa K, Nozaki S, Kitahara H, Ohara
T, Kato K, Kawashiri S and Yamamoto E: Copper efflux transporter
(ATP7B) contributes to the acquisition of cisplatin-resistance in
human oral squamous cell lines. Oncol Rep. 18:987–991.
2007.PubMed/NCBI
|
|
14
|
Furukawa T, Komatsu M, Ikeda R, Tsujikawa
K and Akiyama S: Copper transport systems are involved in multidrug
resistance and drug transport. Curr Med Chem. 15:3268–3278. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Komatsu M, Sumizawa T, Mutoh M, Chen ZS,
Terada K, Furukawa T, Yang XL, Gao H, Miura N, Sugiyama T and
Akiyama S: Copper-transporting P-type adenosine triphosphatase
(ATP7B) is associated with cisplatin resistance. Cancer Res.
60:1312–1316. 2000.PubMed/NCBI
|
|
16
|
Nakayama K, Kanzaki A, Ogawa K, Miyazaki
K, Neamati N and Takebayashi Y: Copper-transporting P-type
adenosine triphosphatase (ATP7B) as a cisplatin based
chemoresistance marker in ovarian carcinoma: comparative analysis
with expression of MDR1, MRP1, MRP2, LRP and BCRP. Int J Cancer.
101:488–495. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Nakayama K, Miyazaki K, Kanzaki A,
Fukumoto M and Takebayashi Y: Expression and cisplatin sensitivity
of copper-transporting P-type adenosine triphosphatase (ATP7B) in
human solid carcinoma cell lines. Oncol Rep. 8:1285–1287.
2001.PubMed/NCBI
|
|
18
|
Nakamura M, Nakatani K, Uzawa K, Ono K,
Uesugi H, Ogawara K, Shiiba M, Bukawa H, Yokoe H, Wada T, et al:
Establishment and characterization of a cisplatin-resistant oral
squamous cell carcinoma cell line, H-1R. Oncol Rep. 14:1281–1286.
2005.PubMed/NCBI
|
|
19
|
Nakatani K, Nakamura M, Uzawa K, Wada T,
Seki N, Tanzawa H and Fujita S: Establishment and gene analysis of
a cisplatin-resistant cell line, Sa-3R, derived from oral squamous
cell carcinoma. Oncol Rep. 13:709–714. 2005.PubMed/NCBI
|
|
20
|
Yin W, Wang P, Wang X, Song W, Cui X, Yu H
and Zhu W: Identification of microRNAs and mRNAs associated with
multidrug resistance of human laryngeal cancer Hep-2 cells. Braz J
Med Biol Res. 46:546–554. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Petzoldt S, Kahra D, Kovermann M,
Dingeldein AP, Niemiec MS, Ådén J and Wittung-Stafshede P: Human
cytoplasmic copper chaperones Atox1 and CCS exchange copper ions in
vitro. Biometals. 28:577–585. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wernimont AK, Huffman DL, Lamb AL,
O'Halloran TV and Rosenzweig AC: Structural basis for copper
transfer by the metallochaperone for the Menkes/Wilson disease
proteins. Nat Struct Biol. 7:766–771. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Palm-Espling ME, Lundin C, Björn E, Naredi
P and Wittung-Stafshede P: Interaction between the anticancer drug
Cisplatin and the copper chaperone Atox1 in human melanoma cells.
Protein Pept Lett. 21:63–68. 2014. View Article : Google Scholar
|
|
24
|
Chen ZS, Mutoh M, Sumizawa T, Furukawa T,
Haraguchi M, Tani A, Saijo N, Kondo T and Akiyama S: An active
efflux system for heavy metals in cisplatin-resistant human KB
carcinoma cells. Exp Cell Res. 240:312–320. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Schilsky ML, Stockert RJ, Kesner A, Gorla
GR, Gagliardi GS, Terada K, Miura N and Czaja MJ: Copper resistant
human hepatoblastoma mutant cell lines without metallothionein
induction overexpress ATP7B. Hepatology. 28:1347–1356. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang D and Lippard SJ: Cellular processing
of platinum anticancer drugs. Nat Rev Drug Discov. 4:307–320. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Parker RJ, Eastman A, Bostick-Bruton F and
Reed E: Acquired cisplatin resistance in human ovarian cancer cells
is associated with enhanced repair of cisplatin-DNA lesions and
reduced drug accumulation. J Clin Invest. 87:772–777. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Loh SY, Mistry P, Kelland LR, Abel G and
Harrap KR: Reduced drug accumulation as a major mechanism of
acquired resistance to cisplatin in a human ovarian carcinoma cell
line: circumvention studies using novel platinum (II) and (IV)
ammine/amine complexes. Br J Cancer. 66:1109–1115. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lai C, Chen Z and Li R: MicroRNA-133a
inhibits proliferation and invasion, and induces apoptosis in
gastric carcinoma cells via targeting fascin actin-bundling protein
1. Mol Med Rep. 12:1473–1478. 2015.PubMed/NCBI
|
|
30
|
Yuan Y, Yao YF, Hu SN, Gao J and Zhang LL:
MiR-133a is functionally involved in doxorubicin-resistance in
breast cancer cells MCF-7 via its regulation of the expression of
uncoupling protein 2. PLoS One. 10:e01298432015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chekhun VF, Lukyanova NY, Burlaka CA,
Bezdenezhnykh NA, Shpyleva SI, Tryndyak VP, Beland FA and Pogribny
IP: Iron metabolism disturbances in the MCF-7 human breast cancer
cells with acquired resistance to doxorubicin and cisplatin. Int J
Oncol. 43:1481–1486. 2013.PubMed/NCBI
|
|
32
|
Wang P, Fu T, Wang X and Zhu W: Primary,
study of miRNA expression patterns in laryngeal carcinoma by
microarray. Lin Chung Er Bi Yan Hou Tou Jing Wai Ke Za Zhi.
24:535–538. 2010.In Chinese. PubMed/NCBI
|