Introduction
The control of human fertility is an important
global issue (1,2). In contrast to the range of
contraceptives available to women, contraceptive options for men
are limited to condoms or a vasectomy (3). Thus, the discovery of effective,
safe and reversible male contraceptives remains a challenge.
Currently, the only drugs in clinical trials are testosterone
analogs (4,5), although there are other possible
targets (e.g., GAPDHS) (6) and
drugs (e.g., gamendazole) (7). To
provide alternative male contraceptives, the identification of
novel protein targets that may be exploited to develop novel
chemical structures as male contraceptive agents is required. One
such contraceptive target is the testis-specific
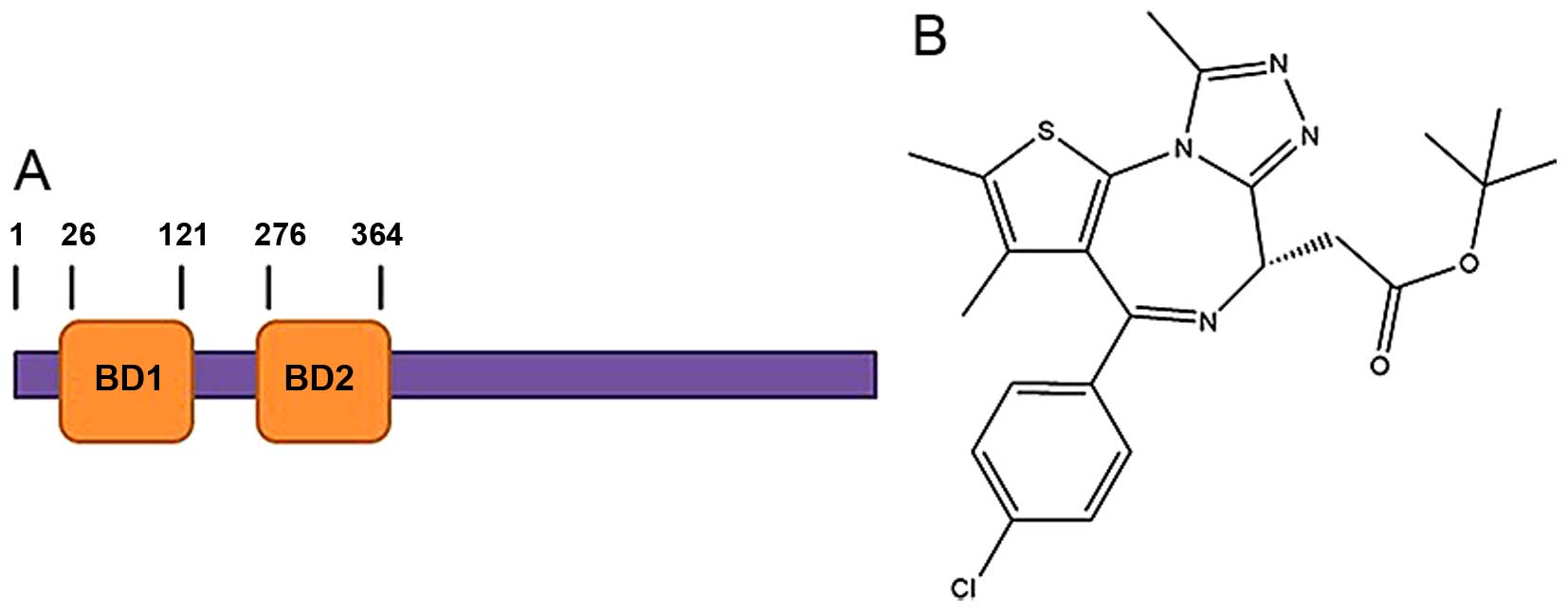
bromodomain-containing protein (BRDT), which is a member of the
bromo and extra-terminal (BET) family. The BET family of BRD
proteins is composed of 4 members in mammals (BRD2, BRD3, BRD4 and
BRDT), each containing 2 conserved N-terminal bromodomains
(8). BET proteins play critical
roles in cellular proliferation and cell cycle progression
(9). BRDT is a tissue-specific,
chromatin-associated protein expressed throughout the pachytene
stage in diplotene spermatocytes (10). BRDT is a key protein that
participates in chromatin remodeling during spermatogenesis. The
essential role of BRDT in spermatogenesis is mediated by the first
bromodomain (BD1; Fig. 1A), which
specifically binds the hyperacetylated histone 4 (H4Kac4) with
moderate potency (20 µM) (11). The loss of the BD1 of BRDT results
in improper/incomplete spermatid elongation and sperm that exhibit
severe morphological defects (12). The genetic analysis of BRDT has
demonstrated that the selective deletion of the BD1 encoding region
is sufficient to confer sterility in homozygous hypomorphic male
mice (10). The lack of
full-length BRDT leads to changes in transcription and alternative
splicing in spermatocytes and spermatids (12). Thus, a small molecule that
specifically inhibits BRDT function represents a suitable choice
for a male contraceptive. Furthermore, BRDT is not expressed in
mitotically dividing spermatogonia, and thus a BRDT-specific
inhibitor would not affect the spermatogonial stem cell population,
theoretically making any spermatogenic impairment reversible
(12).
Currently, several potent pan-BET inhibitors, such
as (+)-JQ1 (13), GSK1210151A
(I-BET151) (14) and PFI-1
(15) have been found. Among
these, only (+)-JQ1 (Fig. 1B) has
been reported to be an effective acetyl-lysine competitive
inhibitor of BRDT. In addition, there are currently no specific
BRDT inhibitors undergoing clinical trials, at least to the best of
our knowledge. Therefore, pursuing and discovering more potent and
selective BRDT inhibitors with new scaffolds is mandatory.
High-throughput screening methods, particularly computer-aided
virtual screening may provide economical approaches for the rapid
discovery of lead compounds with new scaffolds of specified targets
from large chemical databases. Docking-based virtual screening and
pharmacophore-based virtual screening are classical methods that
have been widely used to identify hit compounds in drug discovery.
However, previous studies have shown that the individual use of
these methods generally leads to a low hit rate and low enrichment
factor, as well as to a high false-positive rate (16,17). On the other hand, the combined use
of virtual screening methods in a hybrid protocol would overcome
these drawbacks. In 2012, Matzuk et al solved the crystal
structure of human BRDT (hBRDT) in complex with (+)-JQ1 (18). This structure provides a good
basis for the structure-based discovery of drugs targeting
hBRDT.
Thus, in this study, we have taken advantage of the
crystal structure of hBRDT-JQ1 to discover novel hit compounds
targeting hBRDT. Both the structure-based pharmacophore modeling
and molecular docking methods were adopted for virtual screening,
and the hit compounds were evaluated by a protein-based assay. The
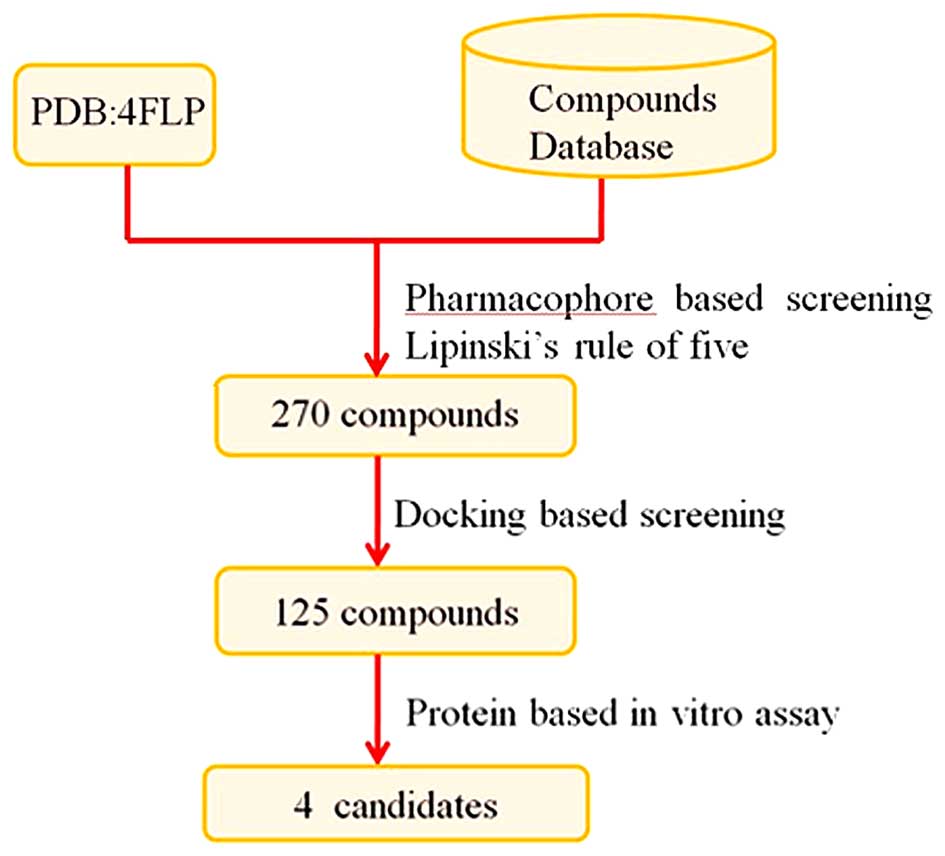
virtual screening protocol is illustrated in Fig. 2. To the very best of our
knowledge, this is the first example of a successful application of
virtual screening to discover novel hBRDT inhibitors.
Data collection methods
Preparation of compound database
In this study, the in-house chemical database used
for virtual screening was developed by the Institute of Medicinal
Biotechnology, Chinese Academy of Medical Sciences, Beijing, China.
This database contains the structural information of 80,000
compounds. All the compounds were energy minimized by applying the
CHARMM force field and subjected to a conformational analysis using
the Polling algorithm.
Structure-based pharmacophore
modeling
Pharmacophore-based methods have been widely used in
virtual screening (19).
Structure-based pharmacophore generation uses the spatial
information of the target protein for the topological description
of ligand-receptor interactions. It also provides an efficient
alternative to docking-based virtual screening, while continuing to
represent specific ligand-protein interactions. Moreover, it has
been demonstrated that the structure-based pharmacophore approach
provides more detailed information and accuracy in its description
of ligand binding than ligand-based methods (20). The information about the protein
structure is a good source to bring forth the structure-based
pharmacophore and its use as a first screening before docking
studies. As only a few hBRDT inhibitors targeting the BD1 of hBRDT
have been reported (18), in this
study, a structure-based pharmacophore modeling based on the
crystal structure of BD1 of hBRDT in complex with the inhibitor,
JQ1, was carried out using the 'Receptor-Ligand Pharmacophore
Generation' protocol in Discovery Studio 3.1 (DS; Accelrys, San
Diego, CA, USA) with default parameters. This protocol generates
selective pharmacophore models based on receptor-ligand
interactions. The crystal structure of the first bromodomain of
hBRDT was retrieved from the Protein Data Bank (PDB ID: 4FLP). As
the water molecule is very important in the binding site of the BET
family (13), the receptor
structure was prepared by retaining the water molecules and adding
hydrogen atoms, as previously described (21).
According to the interactions between ligand and
receptor, the features, including hydrogen acceptors (HA) and
hydrophobic regions (HP), were generated through the
'Receptor-Ligand Pharmacophore Generation' protocol. In addition,
the excluded volumes were involved in the pharmacophore models to
improve the effectiveness of virtual screening.
Docking-based virtual screening
Since pharmacophore-based virtual screening usually
suffers a higher 'false-positive' rate (22), the combined use of
pharmacophore-based virtual screening with docking should lead to a
reduction in the false-positive rate. In this study, a docking
analysis was carried out after the pharmacophore-based analysis to
filter the virtual screening results. All of the molecular docking
studies were carried out using the program genetic optimisation for
ligand docking (GOLD) 4.0 (23).
GOLD adopts the genetic algorithm to dock flexible ligands into the
binding site of a protein. The crystal structure of BRDT complexed
with JQ1 (PDB ID: 4FLP) was used as the receptor structure. The
binding site was defined as a sphere containing residues within 9 Å
of the co-ligand JQ1, which is large enough to cover the
acetyl-lysine binding pocket of the N-terminal bromodomain of BRDT
(w).
Subsequently, we adjusted the docking parameters
until the docked pose of JQ1 was as close as possible to the
original crystallized structure in the hydrophobic acetyl-lysine
binding pocket of hBRDT. The final optimized docking parameters
mainly included: i) the 'number of dockings' was set to 10 without
using the early termination option; ii) the 'detect cavity' was
turned on; iii) the optimized positions of the polar protein
hydrogen atoms were saved; iv) the GA parameter was set to 'gold
default'; v) the top 10 scoring poses were saved for each compound;
and vi) the default settings were used for the other parameters.
The scoring function ChemPLP was used.
In vitro assay
The in vitro bioactivities of the 125
selected hit compounds were performed by a time-resolved
fluorescence resonance energy transfer (TR-FRET) assay with the
Cayman BRDT bromodomain 1 TR-FRET assay kit (600650; Cayman
Chemical Co., Ann Arbor, MI, USA), according to the vendor's
instructions. The TR-FRET assay kit is a homogeneous, TR-FRET assay
method amenable to the rapid characterization of inhibitors of
bromodomain/acetylated peptide interactions in a high-throughput
format. The assay was performed in 384-well plates with a 20
µl final assay volume. Briefly, the control (JQ1) and sample
(positive compounds acquired from virtual screening) were incubated
in 10 µl of the BRDT europium chelate for 15 min at room
temperature. Following incubation, the reaction was initiated by
the addition of 5 µl of the reconstituted BRDT bromodomain 1
ligand/APC acceptor mixture. The plate was then sealed with an
adhesive aluminum seal and incubated at room temperature for 1 h.
The TR-FRET ratio (670 nm emission/620 nm emission) was measured
using an EnVision 2104 multilabel plate reader (PerkinElmer,
Waltham, MKA, USA) in our laboratory. A plot of the TR-FRET ratio
(670 nm emission/620 nm emission) vs. the inhibitor concentration
on semi-log axes resulted in a sigmoidal dose-response curve
typical of competitive binding assays. The data were fit to a
4-parameter logistic equation to calculate the half maximal
inhibitory concentration (IC50) values. Samples were
prepared in dimethyl sulfoxide (DMSO) and the final concentration
of DMSO in the assay was <2% (v/v).
Results and Discussion
Generation of structure-based
pharmacophore modeling
The 'Receptor-Ligand Pharmacophore Generation'
protocol of DS presents the chemical features which instigate key
interactions between protein and ligand, as well as some excluded
volume spheres corresponding to the 3D structure of protein. In
this study, the 3D structures of hBRDT bound with its inhibitor,
JQ1, were selected as input for structure-based pharmacophore
generation. In general, the excluded volumes attempt to penalize
molecules occupying steric regions that are not occupied by active
molecules. The filtering of the pharmacophore with these excluded
volume features provides a more selective model to reduce
false-positives and a better enrichment rate in virtual screening.
The parameters for the minimum features and the maximum features
were set to 4 and 6, respectively. The parameter for keeping water
molecules was set to false. The 'Receptor-Ligand Pharmacophore
Generation' module identified all the possible pharmacophore
features based on the interactions between the acetyl-lysine
binding pocket of hBRDT and JQ1. Finally, a pharmacophore model
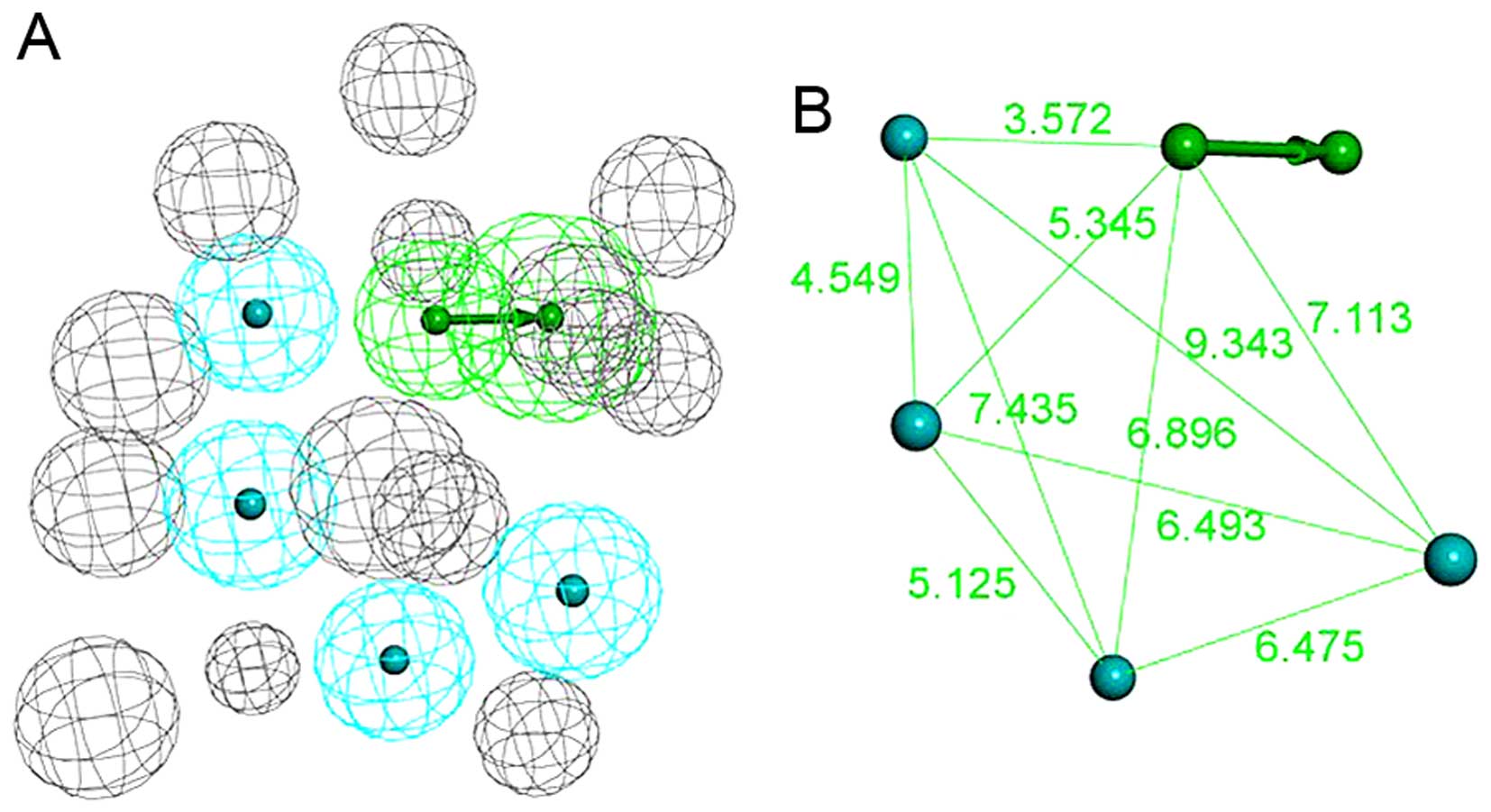
which involves 1 hydrogen bond acceptor, 4 hydrophobic features and
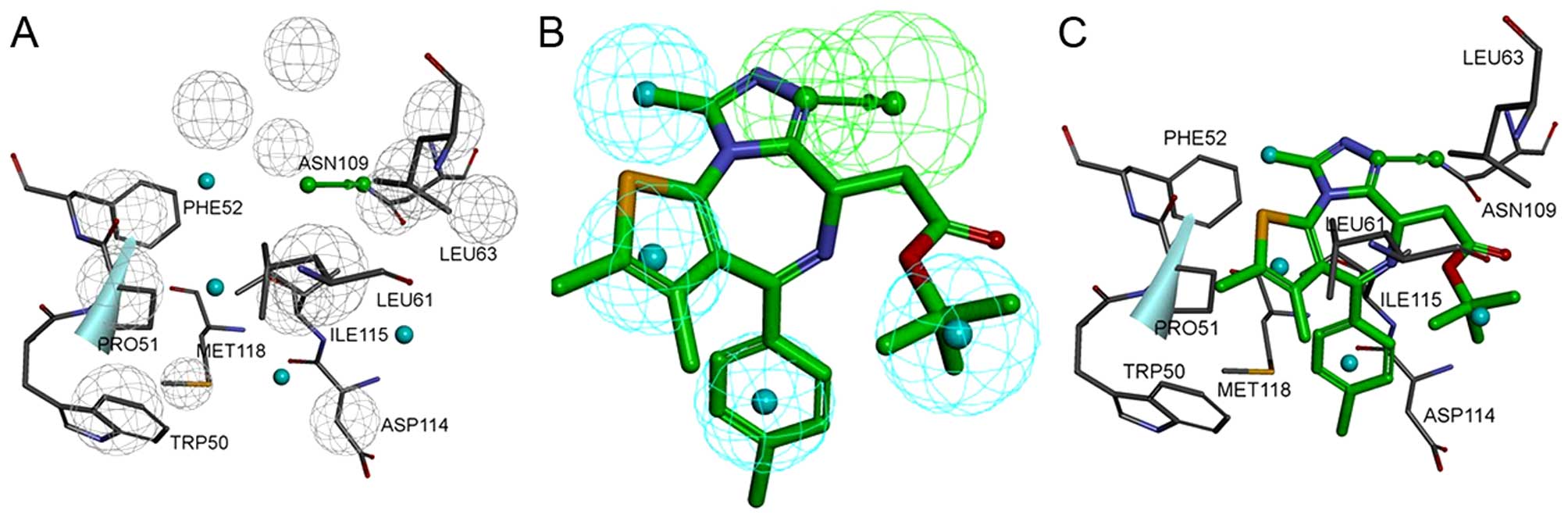
14 excluded volumes was obtained (Fig. 3A and B). Fig. 4A shows the pharmacophore model and
residues responsible for the pharmacophore features. Fig. 4B shows the inhibitor, JQ1, mapped
with the pharmacophore features. Clearly, inhibitor JQ1 is mapped
very well with these features. Fig.
4C shows that the hydrogen bond acceptor corresponds to the
hydrogen bond formed between the triazole ring of JQ1 and the
evolutionarily conserved asparagine (ASN109). Four hydrophobic
features map the different parts of JQ1; H1, H2, H3 and H4 map the
methyl, thiophene ring, benzene ring and tertiary butyl,
respectively. These hydrophobic features refer to the hydrophobic
regions at the active site of BRDT, formed mainly by TRP50, PRO51,
PHE52, LEU61, LEU63, ILE115 and MET118.
Pharmacophore-based virtual
screening
The setup of the pharmacophore model was complete.
Subsequently, the best pharmacophore model, including 5 chemical
features and 14 excluded volumes was used as a 3D search query for
retrieving potent BRDT inhibitors from an in-house chemical
database (80,000 molecules). Only those compounds that mapped at
least 3 of the 5 features were selected. These compounds were
further screened using Lipinski's rule of five to make them more
drug-like. A total of 270 drug-like molecules wa finally
selected.
Docking-based virtual screening
To further filter the retrieved hits, the remaining
270 compounds were docked into the inhibitor binding site of BRDT
using GOLD within the Discovery Studio 3.1 program package.
Compounds were ranked according to the scoring function ChemPLP
incorporated in GOLD; ChemPLP was used as the pre-evaluation of the
results of various scoring functions revealed that ChemPLP was more
effective than others (data not shown). Finally, 125 compounds were
selected based on the ranking order and visual examination. These
compounds were used in the subsequent in vitro assay.
In vitro assay
The in vitro inhibitory activities of the 125
compounds were evaluated by TR-FRET assay using JQ1 as a standard
reference drug. From this assay, 4 compounds were identified to
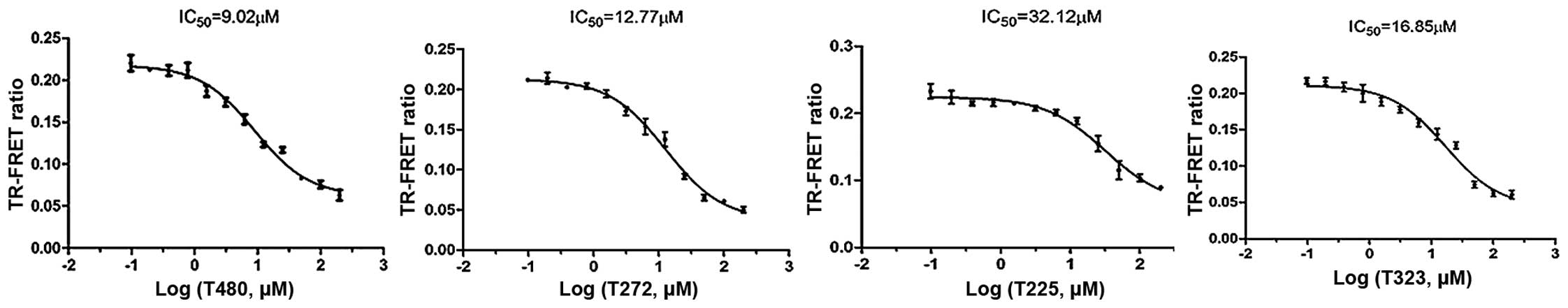
display obvious inhibitory activities with IC50 values
<33 µM (Table I and
Fig. 5). The fitting curves for
the IC50 data of the compounds are shown in Fig. 6. These hits, which were novel
small molecule inhibitors, showed structural diversity.
 | Table IInhibitory rates and inhibitory
activities against hBRDT. |
Table I
Inhibitory rates and inhibitory
activities against hBRDT.
| Compound | Inhibition
ratea (%) | IC50
(µM) |
|---|
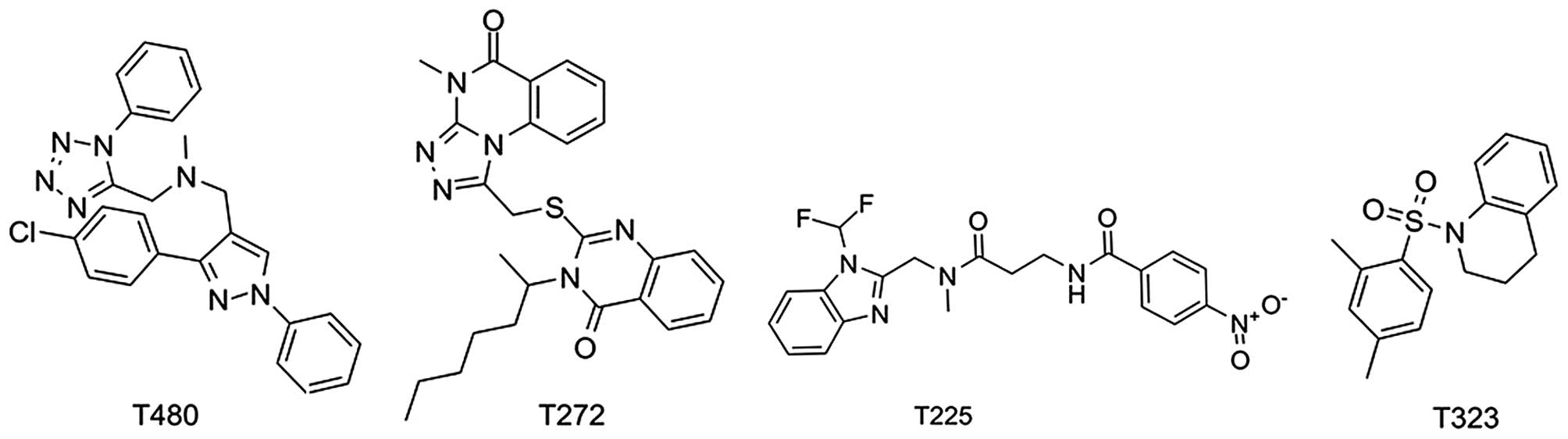
| T480 | 65.53 | 9.02±0.45 |
| T272 | 71.23 | 12.77±0.38 |
| T225 | 44.39 | 32.12±0.63 |
| T323 | 74.08 | 16.85±0.59 |
| JQ1 | 70.34b | 0.133 |
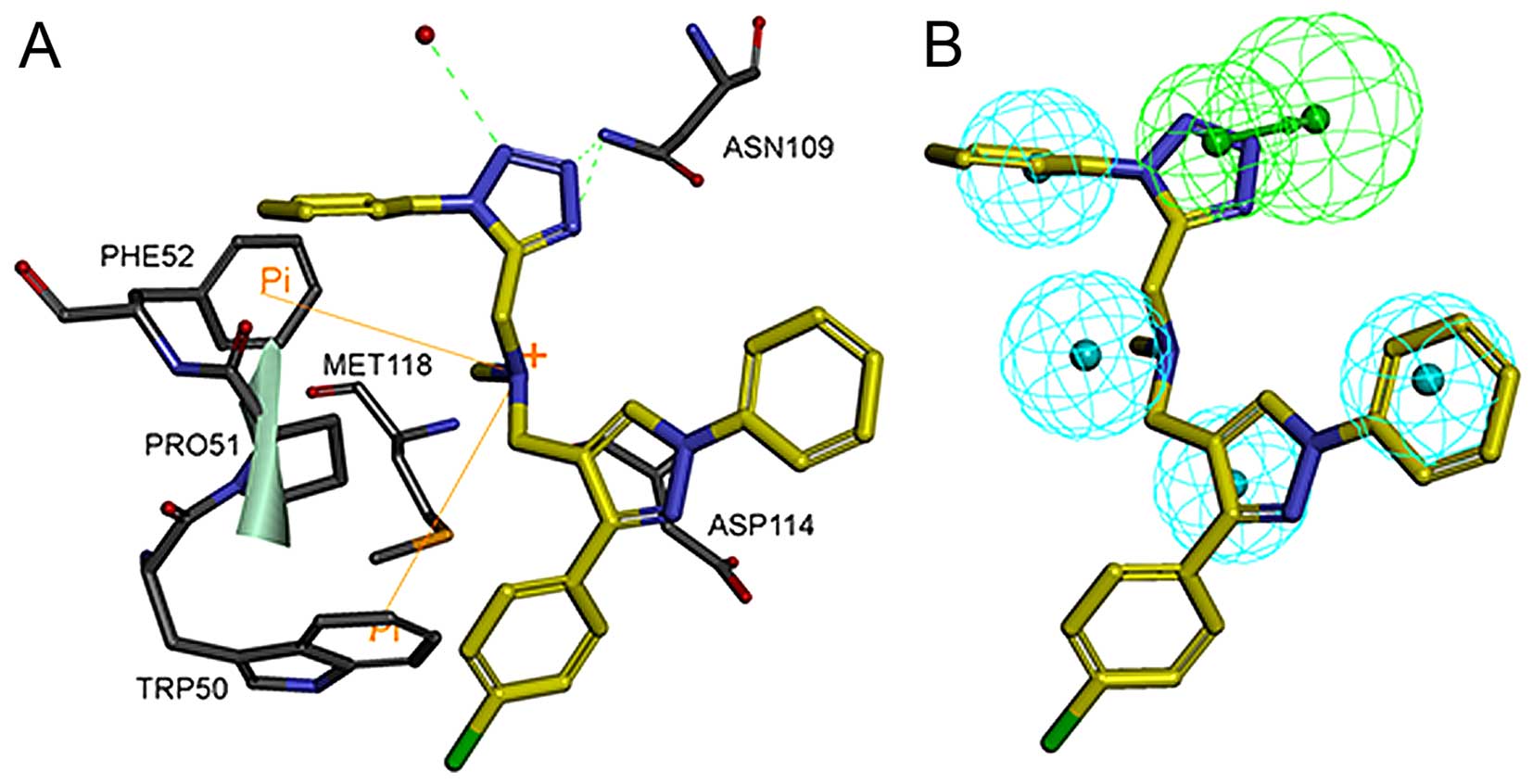
The detailed binding mode of the most active
compound, T480, and the mapping of compound T480 with the
pharmacophore model are shown in Fig.
7A and B, respectively. This compound will be analyzed in
subsequent investigations, including chemical structural
modification and further pharmacological studies. Fig. 7A shows that compound T480 docks
tightly into the active site of hBRDT. Two hydrogen-bond
interactions are formed between compound T480 and hBRDT: one is
between the tetrazole ring nitrogen atom and the amino group of the
amide of the conserved ASN109 residue, whereas the other is between
the other nitrogen atom on the tetrazole ring of compound T480 and
the same amino group of the conserved ASN109. One hydrogen-bond
interaction is formed between one nitrogen atom on the tetrazole
ring of T480 and the conservative water molecule. The phenyl
moieties of T480 form hydrophobic interactions with PHE52, PRO51,
TRP50 and ASP114. In addition, the nitrogen of methenamine forms
cation-π interactions with PHE52 and TRP50. Fig. 7B shows that the tetrazole ring and
2 phenyl moieties of compound T480 map the hydrogen-bond acceptor
feature and2 wo hydrophobic features, respectively. The good
binding pose and interactions of T480 with the active site of hBRDT
provide a solid basis for their bioactivity against hBRDT.
Virtual screening resulted in multiple novel
inhibitor classes targeting hBRDT. In the combined screening
method, ligand-protein X-ray structure pharmacophore was used to
search large databases and provided a much smaller set of compounds
for docking into the active site. This method provided an efficient
method of finding new leads.
In conclusion, our study demonstrates that hBRDT is
a promising target in the discovery of male contraceptive drugs. In
this study, both pharmacophore-based and docking-based virtual
screening methods were developed and applied in a virtual screening
of an in-house database for retrieving inhibitors of hBRDT. A total
of 125 compounds were selected based on the ranking order in the
virtual screening for the in vitro protein-based assays.
Four novel inhibitors were identified and revealed moderate
inhibitory potencies with IC50 values ranging from
9.02±0.45 to 32.12±0.63 µM. The detailed analysis of the
binding modes using the docking study for compound T480 showed that
the inhibitor was stabilized by the formation of 2 hydrogen bonds
with the key residue ASN109. Moreover, further development of these
inhibitors is currently ongoing.
Acknowledgments
This study was supported by grants from the China
Postdoctoral Science Foundation (no. 2014M550678 to N.G.), the
Special Research Funds from the Ministry of Science and Technology
of P.R. China (no. 2012GJSSJKB01 to H.W.).
References
|
1
|
Henshaw SK: Unintended pregnancy in the
United States. Fam Plann Perspect. 30:24–29. 461998. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Finer LB and Henshaw SK: Abortion
incidence and services in the United States in 2000. Perspect Sex
Reprod Health. 35:6–15. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Piccinino LJ and Mosher WD: Trends in
contraceptive use in the United States: 1982–1995. Fam Plann
Perspect. 30:4–10. 461998. View
Article : Google Scholar
|
|
4
|
Gu Y, Liang X, Wu W, Liu M, Song S, Cheng
L, Bo L, Xiong C, Wang X, Liu X, et al: Multicenter contraceptive
efficacy trial of injectable testosterone undecanoate in Chinese
men. J Clin Endocrinol Metab. 94:1910–1915. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hay CJ, Brady BM, Zitzmann M, Osmanagaoglu
K, Pollanen P, Apter D, Wu FC, Anderson RA, Nieschlag E, Devroey P,
et al: A multicenter Phase IIb study of a novel combination on
intramuscular androgen (testosterone decanoate) and oral
progestogen (etonogestrel) for male hormonal contraception. J Clin
Endocrinol Metab. 90:2042–2049. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Frayne J, Taylor A, Cameron G and Hadfield
AT: Structure of insoluble rat sperm glyceraldehyde-3-phosphate
dehydrogenase (GAPDH) via heterotetramer formation with Escherichia
coli GAPDH reveals target for contraceptive design. J Biol Chem.
284:22703–22712. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Aitken RJ, Baker MA, Doncel GF, Matzuk MM,
Mauck CK and Harper MJ: As the world grows: contraception in the
21st century. J Clin Invest. 118:1330–1343. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Picaud S, Da Costa D, Thanasopoulou A,
Filippakopoulos P, Fish PV, Philpott M, Fedorov O, Brennan P,
Bunnage ME, Owen DR, et al: PFI-1, a highly selective protein
interaction inhibitor, targeting BET Bromodomains. Cancer Res.
73:3336–3346. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dey A, Nishiyama A, Karpova T, McNally J
and Ozato K: Brd4 marks select genes on mitotic chromatin and
directs postmitotic transcription. Mol Biol Cell. 20:4899–4909.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Shang E, Nickerson HD, Wen D, Wang X and
Wolgemuth DJ: The first bromodomain of Brdt, a testis-specific
member of the BET sub-family of double-bromodomain-containing
proteins, is essential for male germ cell differentiation.
Development. 134:3507–3515. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Morinière J, Rousseaux S, Steuerwald U,
Soler-López M, Curtet S, Vitte AL, Govin J, Gaucher J, Sadoul K,
Hart DJ, et al: Cooperative binding of two acetylation marks on a
histone tail by a single bromodomain. Nature. 461:664–668. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Berkovits BD and Wolgemuth DJ: The role of
the double bromodomain-containing BET genes during mammalian
spermatogenesis. Curr Top Dev Biol. 102:293–326. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Filippakopoulos P, Qi J, Picaud S, Shen Y,
Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, et
al: Selective inhibition of BET bromodomains. Nature.
468:1067–1073. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dawson MA, Prinjha RK, Dittmann A,
Giotopoulos G, Bantscheff M, Chan WI, Robson SC, Chung CW, Hopf C,
Savitski MM, et al: Inhibition of BET recruitment to chromatin as
an effective treatment for MLL-fusion leukaemia. Nature.
478:529–533. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Fish PV, Filippakopoulos P, Bish G,
Brennan PE, Bunnage ME, Cook AS, Federov O, Gerstenberger BS, Jones
H, Knapp S, et al: Identification of a chemical probe for bromo and
extra C-terminal bromodomain inhibition through optimization of a
fragment-derived hit. J Med Chem. 55:9831–9837. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang H, Xiang ML, Liang JY, Zeng T, Zhang
XN, Zhang J and Yang SY: Combination of pharmacophore model,and
molecular docking to identify novel inhibitors of S6K1. Mol Divers.
17:767–772. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ren JX, Li LL, Zheng RL, Xie HZ, Cao ZX,
Feng S, Pan YL, Chen X, Wei YQ and Yang SY: Discovery of novel
Pim-1 kinase inhibitors by hierarchical multistage virtual
screening approach based on SVM model,pharmacophore, and molecular
docking. J Chem Inf Model. 51:1364–1375. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Matzuk MM, McKeown MR, Filippakopoulos P,
Li Q, Ma L, Agno JE, Lemieux ME, Picaud S, Yu RN, Qi J, et al:
Small-molecule inhibition of BRDT for male contraception. Cell.
150:673–684. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Leach AR, Gillet VJ, Lewis RA and Taylor
R: Three-dimensional pharmacophore methods in drug discovery. J Med
Chem. 53:539–558. 2010. View Article : Google Scholar
|
|
20
|
Steindl T and Langer T: Influenza virus
neuraminidase inhibitors: generation and comparison of
structure-based and common feature pharmacophore hypotheses and
their application in virtual screening. J Chem Inf Comput Sci.
44:1849–1856. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Smellie A, Stanton R, Henne R and Teig S:
Conformational analysis by intersection: CONAN. J Comput Chem.
24:10–20. 2003. View Article : Google Scholar
|
|
22
|
Yang SY: Pharmacophore modeling and
applications in drug discovery: challenges and recent advances.
Drug Discov Today. 15:444–450. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jones G, Willett P, Glen RC, Leach AR and
Taylor R: Development and validation of a genetic algorithm for
flexible docking. J Mol Biol. 267:727–748. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dhalluin C, Carlson JE, Zeng L, He C,
Aggarwal AK and Zhou MM: Structure and ligand of a histone
acetyltransferase bromodomain. Nature. 399:491–496. 1999.
View Article : Google Scholar : PubMed/NCBI
|