Introduction
Non-alcoholic fatty liver disease (NAFLD) is
characterized by the accumulation of fat in the livers of
individuals who are abstinent from alcohol or drink infrequently
and do not have other liver diseases such as hepatitis B and C,
alcoholic disease, metabolic liver disease and autoimmune diseases.
The histological pattern of NAFLD may progress from non-alcoholic
fatty liver into non-alcoholic steatohepatitis (NASH), liver
fibrosis, cirrhosis and even hepatocellular carcinoma (HCC)
(1). It is now one of the most
common liver diseases worldwide. Approximately 10 to 40% of adults
in the USA and 20% in other developed countries suffer from NAFLD
(2).
The pathogenesis and underlying mechanisms of NAFLD
remain to be elucidated. Accumulating evidence has verified that
NAFLD is associated with obesity, type 2 diabetes, hyperlipidemia
and other comorbid conditions. The most widely supported theory
proposes that insulin resistance is the key mechanism responsible
for the metabolic abnormalities and may cause NAFLD. Current
treatment relies on lifestyle modifications including long-term
weight management and regular physical exercise. Studies have been
performed for the purpose of developing novel treatments for NAFLD
including insulin-sensitizers, antioxidants, lipid-lowering drugs,
pentoxifylline and cannabinoid receptor antagonists. However, the
current pharmacological treatment options for the disease remain
unsatisfactory (3–6).
The endoplasmic reticulum (ER) is an organelle in
cells which is important for the folding of protein molecules and
the transport of synthesized proteins. Failure of the ER's adaptive
capacity results in the activation of the unfolded protein response
(UPR), which intersects with many different inflammatory and stress
signaling pathways. Increasing evidence has demonstrated that the
UPR is activated in NAFLD and may play an important role in the
development and progression of this disease (7–9).
4-Phenylbutyric acid (4-PBA), a chemical chaperone, is used to
suppress the activation of the UPR (10).
Previous studies have indicated that lipid
accumulation in the liver may regulate the hepatic production of
proinflammatory cytokines, including interleukin (IL)-6,
transforming growth factor (TGF)-β and basic fibroblast growth
factor (bFGF) through nuclear factor-κB (NF-κB) activation and
downstream cytokine production (11–13).
Magnesium isoglycyrrhizinate (MgIG), a traditional
herbal remedy extracted from the roots of the plant Glycyrrhiza
glabra, is a magnesium salt of 18α-glycyrrhizic acid
stereoisomer. Increasing evidence indicates that MgIG acts as an
anti-inflammatory and hepatoprotective agent as it protects hepatic
cells against tissue injury, improves liver function and suppresses
inflammatory responses in the liver (14–16). A previous study demonstrated that
MgIG maintained cell viability and markedly decreased oleic acid
(OA)-induced cell apoptosis and lipid accumulation (17). It has also been demonstrated that
MgIG provided protection against various organ injuries and
diseases, including alcoholic liver disease and lung injury induced
by paraquat poisoning as a clinical medication (18–20). However, the effect of MgIG in the
treatment of NAFLD, particularly with regard to the effect of MgIG
on lipid-overloaded hepatic cells as well as the underlying
molecular mechanisms remain unknown.
The aim of the study was to explore the the possible
molecular mechanisms responsible for the effects of MgIG including
activation of the UPR, NF-κB activation and associated inflammatory
factor expression in hepatic L02 cells, as well as to examine the
protective effects of MgIG in a model of NAFLD.
Materials and methods
Cell line and OA/MgIG treatment
The human hepatic cell line L02 was obtained from
the Peking University Health Science Center (Beijing, China). The
L02 cells were cultured in RPMI-1640 medium (Biological Industries,
Beit-Haemek, Israel), at 37°C in a 5% CO2 humidified
atmosphere. All media were supplemented with 10% fetal bovine serum
and 1% Pen-Strep solution (both from Biological Industries).
The cells were seeded at either 5×105 in
2 ml complete medium in a 6-well plate, or at 1×104 in
200 µl medium in a 96-well plate. The cells were grown in
fresh medium until 50–60% confluent and treated with medium alone
or supplemented with either OA (2 mM) (Sigma, St. Louis, MO, USA)
or OA coupled with MgIG (Chia-tai Tianqing Pharmaceutical Co. Ltd,
Lianyungang, China) or 4-PBA (Sigma) for 24 h. The cells were then
cultured for another 24 h after treatment as described above.
Specifically, the cells were cultured with the following
treatments: i) RPMI-1640 medium alone as the control group, ii)
RPMI-1640 with OA as the OA group, iii) RPMI-1640 with OA and MgIG
as the OA+MgIG group, and iv) RPMI-1640 with OA and 4-PBA as the
OA+PBA group.
Cell Counting Kit-8 (CCK-8) assay
After individual treatments, cell viability was
measured with CCK-8 according to the manufacturer's instructions
(Dojindo Molecular Technologies, Inc., Kumamoto, Japan). CCK-8 (10
µl) was added to each well and then the cells were incubated
for another 3 h in a incubator at 37°C and 5% CO2. The
OD values of the cells from the five different groups were measured
at 450 nm using an ELISA reader (Bio-Tek Instruments, Inc.,
Winooski, VT, USA). Each assay was performed in triplicate.
Oil red O staining
The L02 cells were seeded in 6-well plates and
cultured in RPMI-1640 medium or supplemented with either OA and/or
MgIG as described above. The culture medium was discarded and the
cells were washed with phosphate-buffered saline (PBS) twice and
then fixed with 4% paraformaldehyde for 20 min. Oil red O working
solution (Sigma-Aldrich, St. Louis, MO, USA) was added to each well
and incubated for 15 min at room temperature. The cells were then
rinsed with 60% isopropyl alcohol once and then rinsed with PBS
twice. Images were then captured under an upright microscope
(magnification, ×400; BX53; Olympus, Tokyo, Japan).
Hoechst 33258 staining
The effect of MgIG and OA on apoptosis were
determined by fluorescence microscopy following staining with the
DNA binding fluorophore Hoechst 33258. Briefly, the L02 cells were
seeded in 6-well plates for 24 h and treated with either growth
medium or medium containing OA with or without MgIG. The cells were
washed twice with PBS, fixed with 4% paraformaldehyde for 10 min,
and stained with 5 mM Hoechst 33258 (Dojindo Molecular
Technologies, Inc.) for 10 min at room temperature. The cells were
washed with PBS twice before images were captured under a
fluorescence microscope (Olympus) at 340 nm excitation
(magnification, ×400).
Annexin V-FITC/propidium iodide (PI)
staining and flow cytometric analysis
Following the OA and MgIG treatment for 24 h as
described above, apoptosis was detected using the Annexin V-FITC
Apoptosis Detection kit (Dojindo Molecular Technologies, Inc.)
according to the manufacturer's instructions. Individually treated
L02 cells were collected by removing the medium and washed twice
with PBS. The cells were gently resuspended in 100 µl
binding solution. Annexin V-FITC (5 µl) was added to the
cells, followed by 5 µl PI solution. The L02 cells were then
incubated at room temperature in the dark for 15 min. Subsequently,
400 µl 1X Annexin V binding solution was added. The cells
were analyzed by flow cytometry using a BD FACSCalibur flow
cytometer (Becton-Dickinson, Franklin Lakes, NJ, USA) within 1 h.
The number of apoptotic cells was then calculated using FlowJo
software.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Following a 24-h incubation, total RNA was extracted
from the L02 cells using TRIzol reagent (Sigma) and reverse
transcribed according to the manufacturer's instructions. Briefly,
the SuperReal PreMix RT-PCR kit (Tiangen Biotech Co., Ltd.,
Beijing, China) was used to perform Real-Time PCR and the Golden
Fast PCR kit (Tiangen Biotech Co., Ltd.) was used to perform PCR.
Isolated total RNA (2 µg) was reverse transcribed into cDNA
using the High Capacity cDNA Reverse Transcription kit (Applied
Biosystems, Foster City, CA, USA). The primers used for IL-6,
TGF-β, bFGF, NF-κB, BiP, C/EBP homologous protein (CHOP), X-box
binding protein 1 (XBP-1), activating transcription factor 6
(ATF-6) and β-actin are presented in Table I. The primers were selected using
the NCBI/Primer-BLAST program (www.ncbi.nlm.nih.gov/tools/primer-blast/) and were
synthesized by Sangon Biotech (Shanghai, China). In addition, the
PCR products were also separated in a 2% agarose gel and analyzed
using Image Quant software. Quantitative (real-time) PCR was
performed on a 7300 Real-Time PCR system (Applied Biosystems).
Cycle threshold (Ct) values were normalized to β-actin and PCR
system reactions were run and calculated in triplicates.
 | Table IPrimer sequences used for
RT-qPCR. |
Table I
Primer sequences used for
RT-qPCR.
| Gene | Primer sequence
(5′→3′) |
|---|
| BiP | Forward:
CTAATGGTGGAAACCCACAACG
Reverse: TATCGCCAGGAATTGTTGCTG |
| CHOP | Forward:
GCTCAGGAGGAAGAGGAGGA
Reverse: CCTGCTTGAGCCGTTCATT |
| XBP-1 | Forward:
CAGACTACGTGCACCTCTGC
Reverse: GGCTGGTAAGGAACTGGGTC |
| ATF-6 | Forward:
TCCTCGGTCAGTGGACTCTTA
Reverse: CTTGGGCTGAATTGAAGGTTTTG |
| NF-κB | Forward:
GAAGCACGAATGACAGAGGC
Reverse: GCTTGGCGGATTAGCTCTTTT |
| IL-6 | Forward:
GGCACTGGCAGAAAACAACC
Reverse: TGGCATTTGTGGTTGGGTCA |
| TGF-β | Forward:
CTAATGGTGGAAACCCACAACG
Reverse: ATATCGCCAGGAATTGTTGCTG |
| bFGF | Forward:
AGTGTGTGCTAACCGTTACCT
Reverse: ACTGCCCAGTTCGTTTCAGTG |
| β-actin | Forward:
GTCACCAACTGGGACGACAT
Reverse: AGGGATAGCACAGCCTGGAT |
Protein extraction and western blot
analysis
After individual treatments, the L02 cells were
harvested, rinsed with PBS twice and lysed in RIPA buffer
containing 10 mM phosphate buffer, 150 mM NaCl, 2 mM EDTA, 0.1%
SDS, 1% sodium deoxycholate, 1% Triton X-100, 1 mM sodium
orthovanadate and protease inhibitors for 30 min on ice. Protein
concentrations were determined using a BCA Protein Assay kit
(Beyotime Institute of Biotechnology, Shanghai, China). The samples
were boiled for 10 min at 100°C and insoluble material was removed
by centrifugation. Total protein (50 µg) was separated on
12% SDS-PAGE gels and transferred to polyvinylidene difluoride
(PVDF) membranes. The membranes were blocked with 7% fat-free milk
in 0.5% Triton X-100-TBS (TBST) and incubated overnight at 4°C in
blocking solution containing primary antibodies. The membranes were
then washed with TBST 3 times and incubated with secondary
HRP-conjugated goat anti-rabbit IgG diluted in TBST with 7% milk
for 2 h at room temperature. Protein bands were developed using
chemiluminescence detection reagents and then visualized on rabbit
polyclonal antibodies against XBP-1 (SAB3500381), CHOP (SAB4500631)
or NF-κB protein (SAB4502610) were purchased from Sigma. The rabbit
polyclonal antibodies against BiP (sc-33757) or ATF-6 protein
(sc-22799) were purchased from Santa Cruz Biotechnology, Inc.
Enzyme-linked immunosorbent assay
(ELISA)
The expression of IL-6, TGF-β and bFGF in the L02
cells was detected by ELISA. After a 24-h treatment period, the
supernatants were collected and added into the human ELISA kit
(Abcam, Cambridge, MA, USA) according to the manufacturer's
instructions. The OD value was measured at 450 nm using an ELISA
reader (Bio-Tek Instruments). The expression values were calculated
using a standard curve and ELISA was performed with supernatants
with each independent culture run in triplicate.
Statistical analysis
All quantitative data are presented as the means ±
SD. Statistical analysis was performed using the ANOVA and
Student's t-test with SPSS 17.0 software. A P-value <0.05 was
considered to indicate a statistically significant difference.
Results
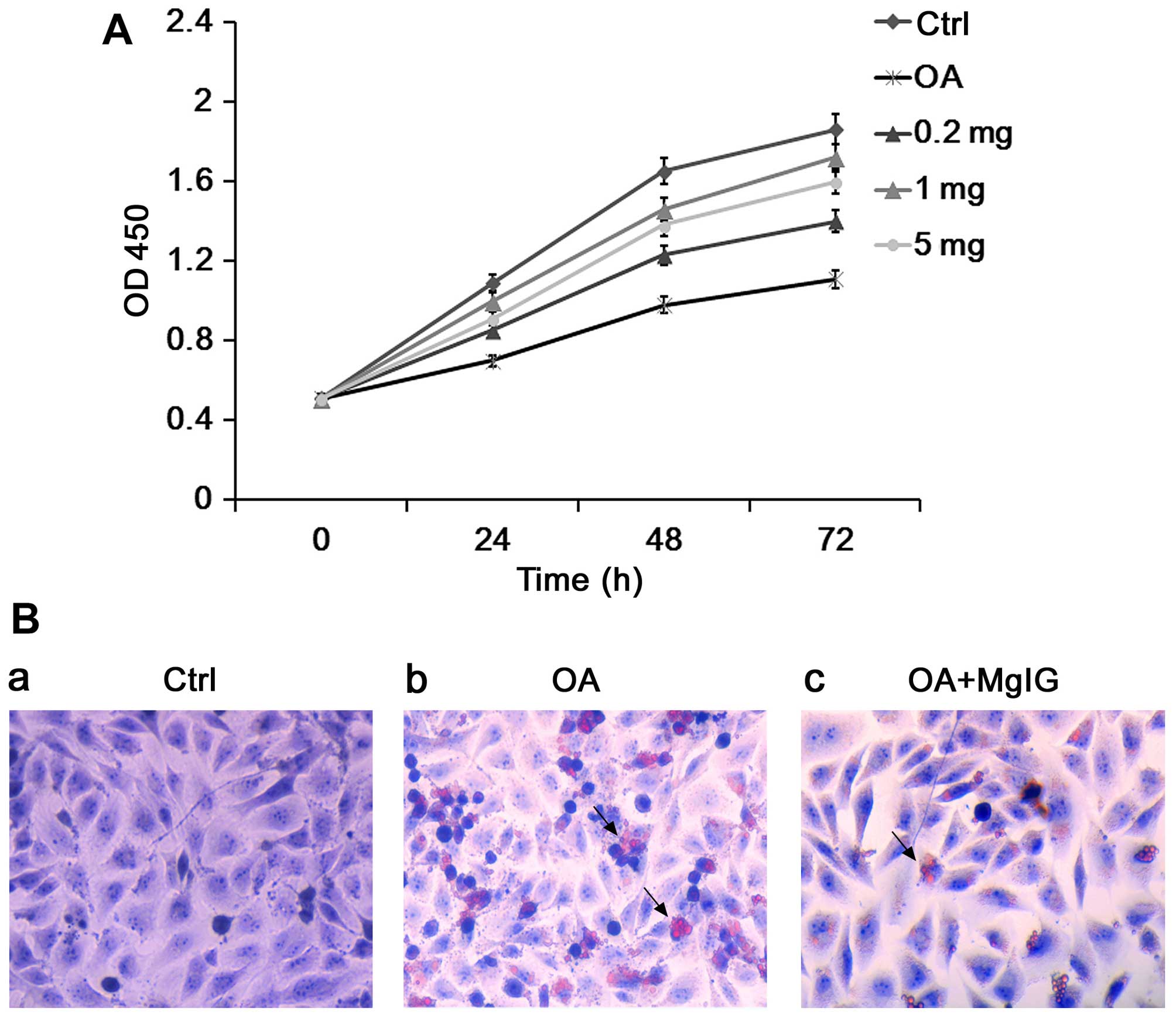
MgIG increases the viability of L02 cells
following exposure to OA
Lipotoxicity in hepatocytes induced by OA has been
implicated in the development of NAFLD. OA and hepatic L02 cells
have been previously used to establish a model of NAFLD in
vitro (21).
In the present study, we first treated the L02 cells
with OA and different doses of MgIG (0.2, 1 and 5 mg/ml) for 24 h.
Compared with the untreated control group, OA significantly reduced
the viability of the L02 cells (Fig.
1A). Furthermore, MgIG increased cell viability. The optimal
concentration of MgIG for protecting L02 cell viability was
identified as 1 mg/ml therefore this dose was selected for
subsequent experiments (Fig.
1A).
MgIG decreases the accumulation of lipid
droplets in L02 cells exposed to OA
The Oil red O staining assay was performed in order
to detect the accumulation of intracellular lipid droplets in L02
cells. After incubation with OA, the number of Oil Red O-positive
droplets was significantly increased in the L02 cells (Fig. 1B). MgIG treatment significantly
reduced the accumulation of Oil Red O-positive droplets in the
OA-treated L02 cells (Fig.
1B).
MgIG protects against the OA-induced
apoptosis of L02 cells
We performed Hoechst 33258 staining to evaluate the
effect of MgIG on the apoptosis of lipid-overloaded L02 cells. The
results indicated that L02 cells exposed to OA for 24 h exhibited
membrane dissolution as well as the formation of debris and nuclear
fragmentation, which was indicated by bright blue fluorescence
(Fig. 2A). The number of typical
apoptotic cells was decreased following treatment with MgIG
compared with that in the the cells incubated with OA alone
(Fig. 2A). The results of Annexin
V-PI staining showed that the portion of early- and late-stage
apoptotic cells increased markedly following exposure to OA
(Fig. 2B). However, MgIG
significantly suppressed the percentage of apoptotic cells
(Fig. 2B).
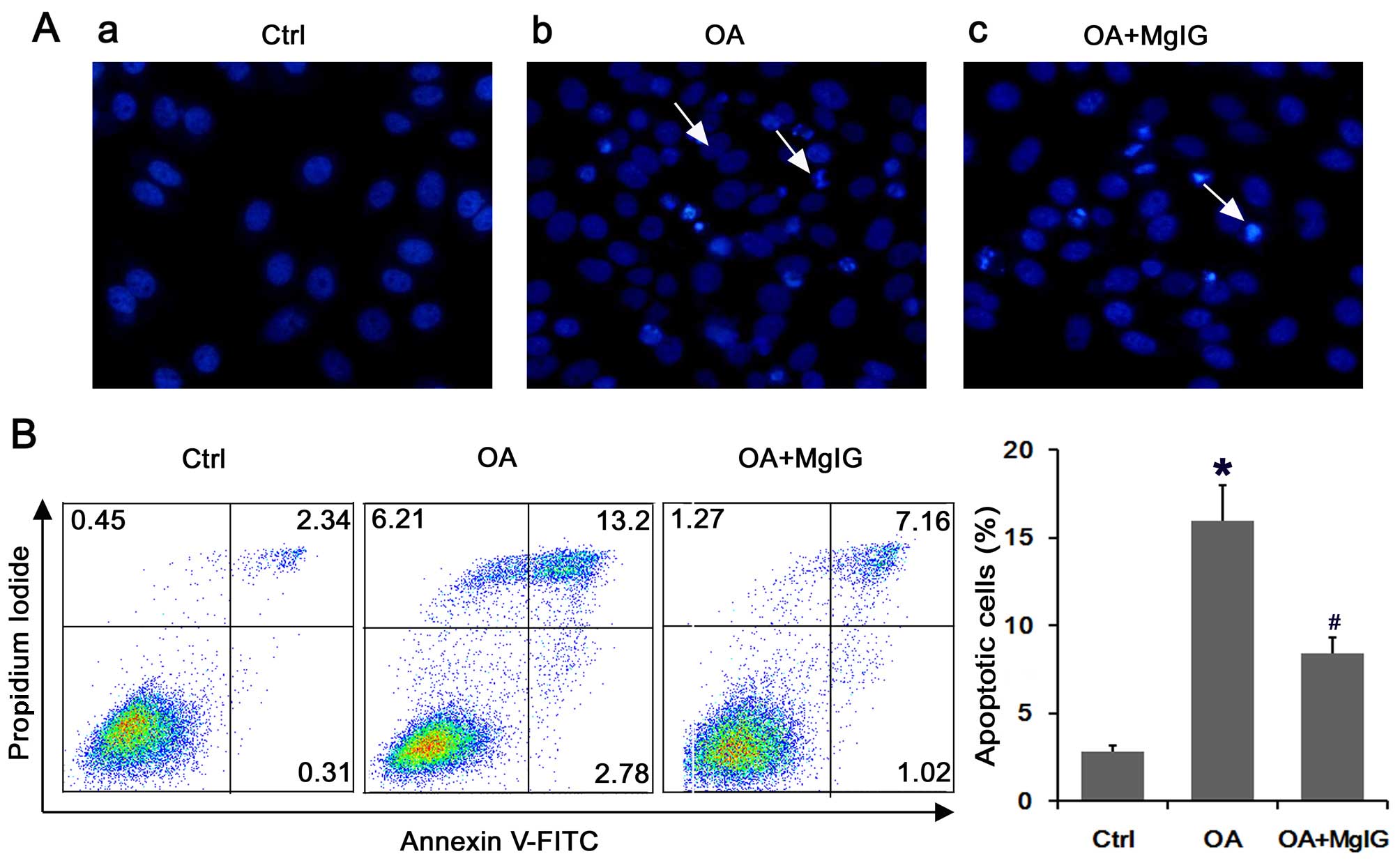 | Figure 2Magnesium isoglycyrrhizinate (MgIG)
protects against the oleic acid (OA)-induced apoptosis of L02
cells. (A) Fluorescence images of L02 cells treated individually
for 24 h and stained with Hoechst 33258, then imaged under a
fluorescence microscope (panel a, control (Ctrl); panel b, OA; and
panel c, OA with MgIG). The staining of apoptotic cells revealed
highly condensed, brightly stained nuclei, whereas the normal cells
were stained only slightly blue. Apoptotic cells are indicated by
white arrows. Magnification, ×400. (B) Annexin V-FITC/propidium
iodide (PI) double staining. After exposure to OA or OA with MgIG,
the cells were harvested and apoptosis was examined by performing
flow cytometry after Annexin V/PI staining. The horizontal and
vertical axes represent labeling with Annexin V-FITC and PI,
respectively. Cells are classified as healthy cells (Annexin
V−, PI−), early apoptotic cells (Annexin
V+, PI−), late apoptotic cells (Annexin
V+, PI+), and damaged cells (Annexin
V−, PI+). The bar chart shows the ratio of
apoptosis among the different experimental groups. The apoptotic
ratio was calculated from the percentage of early apoptotic cells
plus the percentage of late apoptotic cells. Data are presented as
the means ± SD of percentage of controls. *P<0.05 vs.
control group, #P<0.05 vs. OA group. |
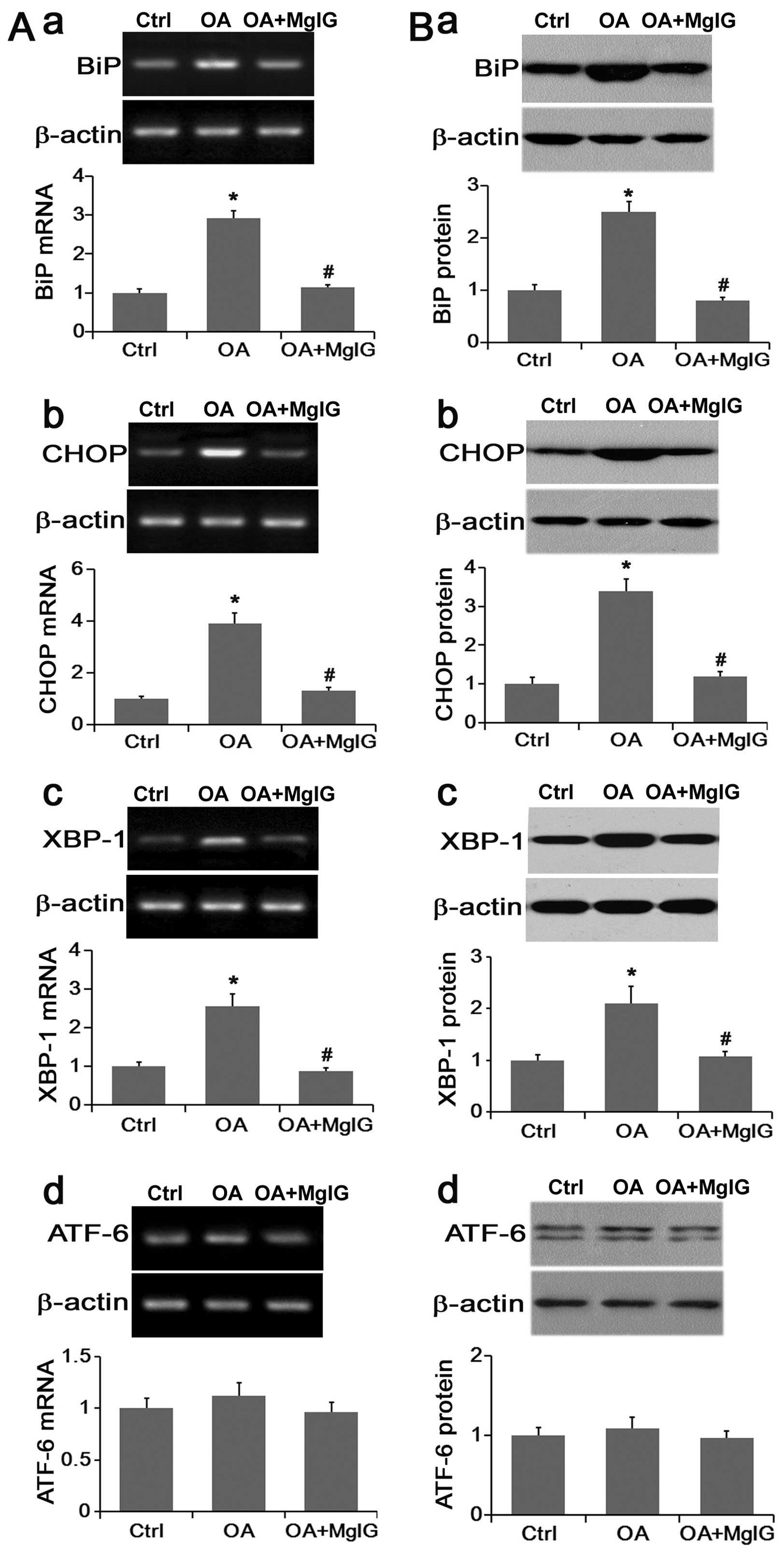
MgIG suppresses the lipotoxic effects of
OA through UPR signaling activation
BiP is an ER stress marker that reflects the
activation of UPR. The UPR signaling pathway consists of three
branches, namely PERK, IRE1 and ATF-6. The activation of PERK and
IRE1 promoted the activation of CHOP and XBP-1, respectively
(9,10). RT-qPCR and western blot analysis
were performed to evaluate the mRNA and protein expression of BiP,
CHOP, XBP-1 and ATF-6, respectively.
Compared with the control group, the expression of
Bip, CHOP and XBP-1 was significantly increased in lipid-overloaded
L02 cells, and MgIG effectively reversed their expression back to
normal both at the mRNA and the protein level (Fig. 3A and B). Compared with the other
endoplasmic reticulum stress-related genes, ATF-6 expression was
not significantly changed by either OA or OA with MgIG treatment
(Fig. 3A and B). These data
suggested that OA may induce the activation of UPR signaling mainly
through downstream PERK and IRE1 pathways whereas MgIG may greatly
suppress OA-induced UPR activation.
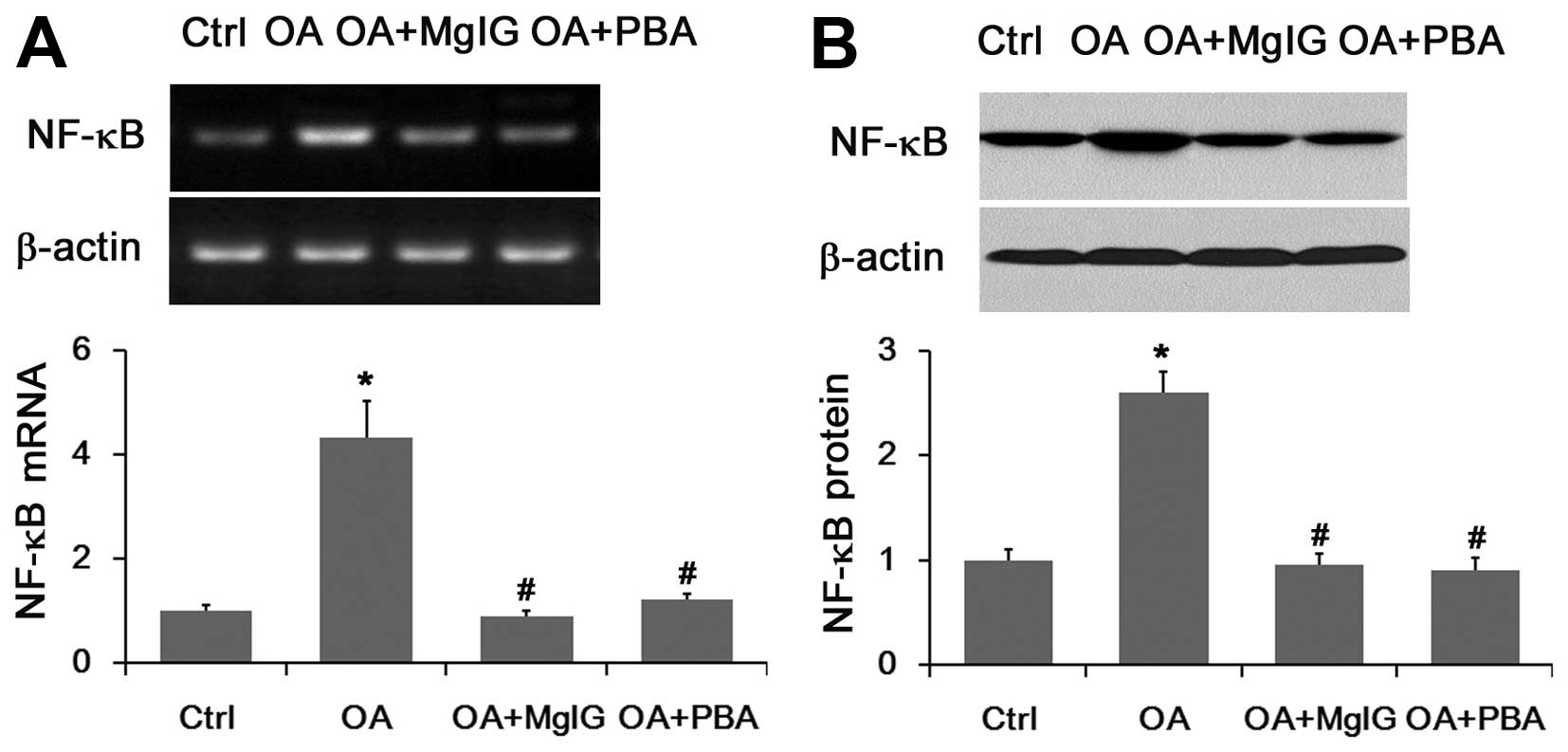
mRNA and protein expression of NF-κB is
altered in lipid-overloaded L02 cells in the presence or absence of
MgIG
Accumulating evidence indicates that the NF-κB
pathway in the liver is activated and NF-κB targets were
accordingly elevated by lipid accumulation (11). Our results showed that OA markedly
raised the mRNA and protein expression of NF-κB (Fig. 4). However, these levels was
significantly reduced when the cells were co-treated with MgIG
(Fig. 4). The UPR inhibitor 4-PBA
decreased the mRNA and protein expression levels of NF-κB as the
positive control (Fig. 4).
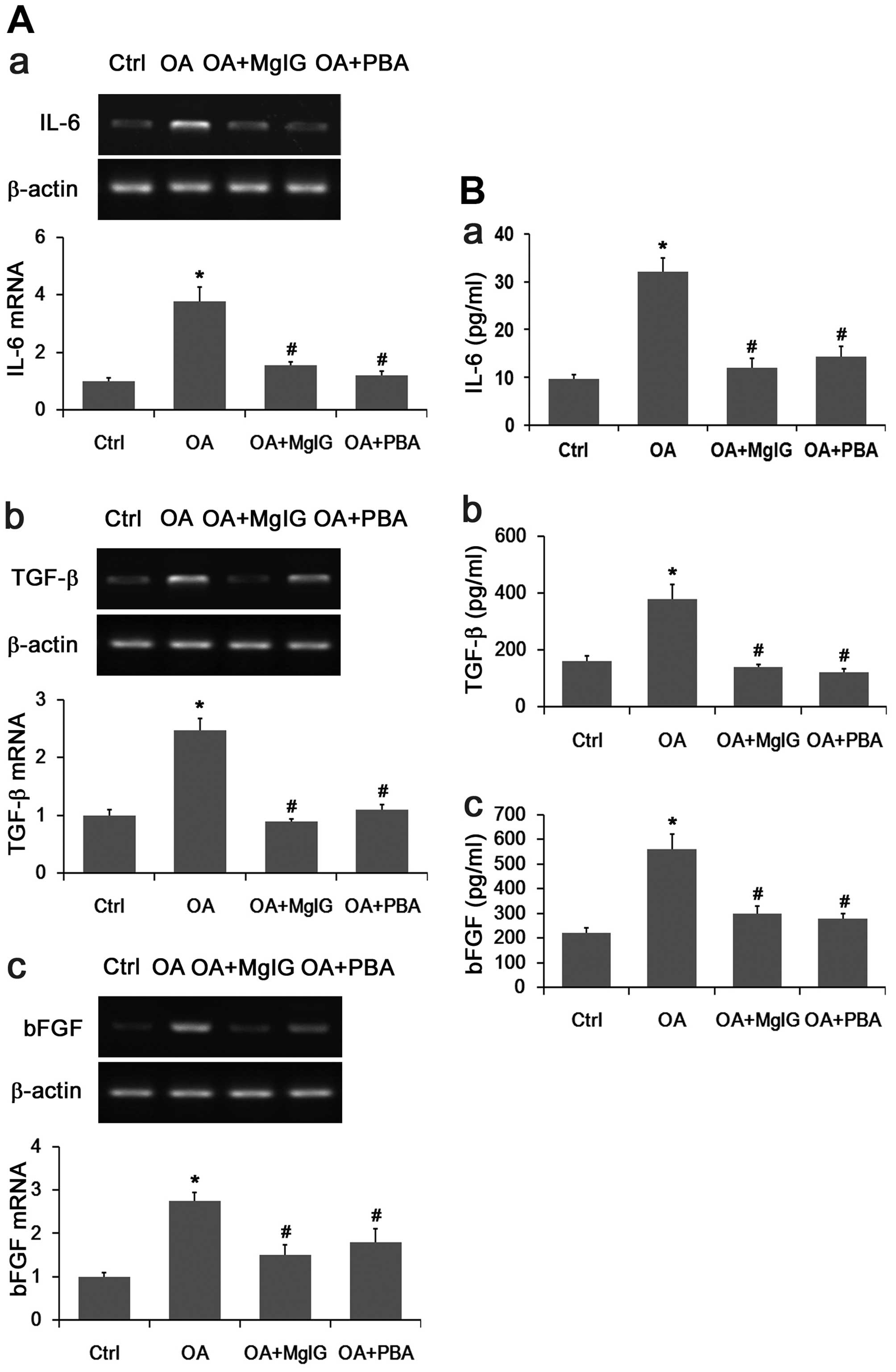
Expression of IL-6, TGF-β and bFGF is
partly regulated in lipid-overloaded L02 cells in the presence or
absence of MgIG
The mRNA and protein expression of IL-6, TGF-β and
bFGF were significantly increased in the lipid-overloaded L02 cells
compared with the control groups (Fig. 5A and B). A marked decrease in the
mRNA and protein expression of IL-6, TGF-β and bFGF was observed
following treatment with OA and MgIG or 4-PBA.
Discussion
In the present study, we established a model of
NAFLD by incubating hepatic L02 cells with OA in vitro.
Using this cell model, we examined the role of OA and MgIG in liver
cells and the associated molecular signaling events.
A previous study indicated that OA was capable of
decreasing cell viability and increasing cell apoptosis markedly
(22). It has also been
demonstrated that OA may induce the accumulation of lipid droplets
in lipid-overloaded L02 cells (23). Our current data are consistent
with these findings. In addition, our study suggested that MgIG
reduced cell death and decreased the apoptosis rate in
lipid-overloaded L02 cells significantly. Moreover, MgIG treatment
inhibited OA-induced lipid accumulation in L02 cells.
The UPR is regulated by three ER stress transducer
proteins PERK, IRE1, and ATF-6. BiP is an ER stress marker and a
central regulator of ER homeostasis as it plays multiple roles in
protein folding and the activation of transmembrane ER stress
sensors. The accumulation of unfolded proteins sequesters BiP so it
dissociates from transmembrane transducers thereby leading to the
activation of these three proteins. Thus, the expression of BiP is
thought to be an ER stress marker bound to these three pathways
(24). The activation of PERK
promotes the activation of CHOP after dissociating from BiP
(25). Activated IRE1α induces
the splicing of the mRNA encoding XBP-1 after dissociating from
BiP, which is a transcriptional activator that has many targets
including ER chaperones and genes involved in ER-associated
degradation (ERAD) (26). After
dissociating from BiP, ATF-6 is activated and translocates to the
nucleus where it transcriptionally activates ER chaperones and
genes associated with ERAD (27).
In the present study, the exposure of L02 cells to
OA, markedly upregulated the mRNA and protein expression of BiP,
CHOP and XBP-1, which was similar to the findings of a previous
study using sodium palmitate (28). However, we were surprised to
observe that the mRNA and protein expression of these endoplasmic
reticulum stress-related genes were significantly downregulated
when treated with MgIG whereas there was no significant change in
ATF-6 levels. The expression of ATF-6 was upregulated slightly in
the lipid-overloaded L02 cells, and suppressed slightly after MgIG
treatment. These results suggested that the lipid accumulation
caused by OA may activate the UPR and be involved in the activation
of the IRE1-XBP-1 and PERK-CHOP pathways. Furthermore, MgIG
suppresses the expression of these two pathways.
Previous findings have showed that the activation of
NF-κB pathway may be induced by lipid accumulation (11). A close examination of ER stress
and UPR pathways has demonstrated many links to major inflammatory
and stress signaling networks, including the activation of the
NF-κB pathways. These findings also indicated that lipid
accumulation in the liver leads to the increased hepatic production
of inflammatory cytokines, including IL-6, TGF-β and bFGF through
NF-κB activation and downstream cytokine production (12,29–31).
Changes to inflammatory factors accompany hepatocyte
injury and regulate lipid metabolism. IL-6 promotes inflammatory
signaling in the liver and increases insulin resistance in
hepatocytes (32). However, the
underlying mechanism remains unclear. TGF-β is a pleiotropic
cytokine involved in cell survival, proliferation, differentiation,
angiogenesis and wound healing responses; TGF-β and bFGF signaling
both participate in the fibrogenic response through hepatic
stellate cell (HSC) activation and play roles in the progression of
fibrosis in advanced NAFLD. TGF-β and bFGF signaling in hepatocytes
were also involved in insulin resistance (33–35). Insulin resistance represents the
most reproducible factor for the development of NAFLD (36).
The present study suggested that OA induces the
expression of NF-κB and the downstream inflammatory cytokines
markedly. 4-PBA, the UPR inhibitor, is used to suppress the
activation of the UPR thereby reducing the expression of NF-κB and
the downstream inflammatory cytokines (10). Our results also clearly showed
that the mRNA and protein expression of NF-κB, IL-6, TGF-β and bFGF
were significantly downregulated when the lipid-overloaded hepatic
cells were treated with MgIG which was consistent with the effects
of 4-PBA.
In conclusion, the data presented in this study
demonstrated that IRE1 and PERK signaling branches induced the
overexpression of inflammatory cytokines such as IL-6, TGF-β and
bFGF, through the activation of NF-κB in lipid-overloaded hepatic
cells. We suggest that MgIG significantly suppresses the activation
of the UPR thereby reducing the downstream inflammatory cytokines
and protecting hepatic cells from NAFLD-induced injury.
Acknowledgments
The present study was supported by grants awarded to
H. Zhu from the National Natural Science Foundation of China (no.
81572345) and the Tianqing Liver Disease Research Foundation (no.
20120024).
References
|
1
|
Angulo P and Lindor KD: Non-alcoholic
fatty liver disease. J Gastroenterol Hepatol. 17(Suppl): S186–S190.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Başaranoğlu M and Örmeci N: Nonalcoholic
fatty liver disease: diagnosis, pathogenesis, and management. Turk
J Gastroenterol. 25:127–132. 2014. View Article : Google Scholar
|
|
3
|
Du J, Ma YY, Yu CH and Li YM: Effects of
pentoxifylline on nonalcoholic fatty liver disease: a
meta-analysis. World J Gastroenterol. 20:569–577. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Duvnjak M, Lerotić I, Barsić N, Tomasić V,
Virović Jukić L and Velagić V: Pathogenesis and management issues
for non-alcoholic fatty liver disease. World J Gastroenterol.
13:4539–4550. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Takahashi Y, Sugimoto K, Inui H and
Fukusato T: Current pharmacological therapies for nonalcoholic
fatty liver disease/nonalcoholic steatohepatitis. World J
Gastroenterol. 21:3777–3785. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Papandreou D and Andreou E: Role of diet
on non-alcoholic fatty liver disease: an updated narrative review.
World J Hepatol. 7:575–582. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Puri P, Mirshahi F, Cheung O, Natarajan R,
Maher JW, Kellum JM and Sanyal AJ: Activation and dysregulation of
the unfolded protein response in nonalcoholic fatty liver disease.
Gastroenterology. 134:568–576. 2008. View Article : Google Scholar
|
|
8
|
Zhang K and Kaufman RJ: The unfolded
protein response: a stress signaling pathway critical for health
and disease. Neurology. 66(Suppl 1): S102–S109. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kim SR, Kim DI, Kang MR, Lee KS, Park SY,
Jeong JS and Lee YC: Endoplasmic reticulum stress influences
bronchial asthma pathogenesis by modulating nuclear factor κB
activation. J Allergy Clin Immunol. 132:1397–1408. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ayala P, Montenegro J, Vivar R, Letelier
A, Urroz PA, Copaja M, Pivet D, Humeres C, Troncoso R, Vicencio JM,
et al: Attenuation of endoplasmic reticulum stress using the
chemical chaperone 4-phenylbutyric acid prevents cardiac fibrosis
induced by isoproterenol. Exp Mol Pathol. 92:97–104. 2012.
View Article : Google Scholar
|
|
11
|
Hotamisligil GS: Endoplasmic reticulum
stress and the inflammatory basis of metabolic disease. Cell.
140:900–917. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cai D, Yuan M, Frantz DF, Melendez PA,
Hansen L, Lee J and Shoelson SE: Local and systemic insulin
resistance resulting from hepatic activation of IKK-beta and
NF-kappaB. Nat Med. 11:183–190. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li N and Karin M: Signaling pathways
leading to nuclear factor-kappa B activation. Methods Enzymol.
319:273–279. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bao QD, Yang LL and Wang L: Protective
effects of magnesium isoglycyrrhizinate against carbon
tetrachloride-induced acute liver injury in mice. World Chin J
Digestology. 16:1004–1007. 2008.
|
|
15
|
Dong LP, Yu F, Liu J and Mu XM: Protective
effect of magnesium isoglycyrrhizinate on acute hepatic injury in
mice. China Pharmacy. 17:902–904. 2006.
|
|
16
|
Mao YM, Zeng MD, Chen Y, Chen CW, Fu QC,
Cai X, Wu SM, Chen YG, Sun Y, Li J, et al: Magnesium
isoglycyrrhizinate in the treatment of chronic liver diseases: a
randomized, double-blind, multi-doses, active drug controlled,
multi-center study. Zhonghua Gan Zang Bing Za Zhi. 17:847–851.
2009.In Chinese. PubMed/NCBI
|
|
17
|
Cheng Y, Zhang J, Shang J and Zhang L:
Prevention of free fatty acid-induced hepatic lipotoxicity in HepG2
cells by magnesium isoglycyrrhizinate in vitro. Pharmacology.
84:183–190. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Xiao ZW, Zhang W, Ma L and Qiu ZW:
Therapeutic effect of magnesium isoglycyrrhizinate in rats on lung
injury induced by paraquat poisoning. Eur Rev Med Pharmacol Sci.
18:311–320. 2014.PubMed/NCBI
|
|
19
|
Chen KJ, Chen WY, Chen X, Jia YM, Peng GQ
and Chen L: Increased elimination of paclitaxel by magnesium
isoglycyrrhizinate in epithelial ovarian cancer patients treated
with paclitaxel plus cisplatin: a pilot clinical study. Eur J Drug
Metab Pharmacokinet. 39:25–31. 2014. View Article : Google Scholar
|
|
20
|
Abe K, Ikeda T, Wake K, Sato T, Sato T and
Inoue H: Glycyrrhizin prevents of
lipopolysaccharide/D-galactosamine-induced liver injury through
down-regulation of matrix metalloproteinase-9 in mice. J Pharm
Pharmacol. 60:91–97. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ziamajidi N, Khaghani S, Hassanzadeh G,
Vardasbi S, Ahmadian S, Nowrouzi A, Ghaffari SM and Abdirad A:
Amelioration by chicory seed extract of diabetes- and oleic
acid-induced non-alcoholic fatty liver disease
(NAFLD)/non-alcoholic steatohepatitis (NASH) via modulation of
PPARα and SREBP-1. Food Chem Toxicol. 58:198–209. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Alkhatatbeh MJ, Lincz LF and Thorne RF:
Low simvastatin concentrations reduce oleic acid-induced steatosis
in HepG2 cells: An in vitro model of non-alcoholic fatty liver
disease. Exp Ther Med. 11:1487–1492. 2016.PubMed/NCBI
|
|
23
|
Malhi H, Bronk SF, Werneburg NW and Gores
GJ: Free fatty acids induce JNK-dependent hepatocyte lipoapoptosis.
J Biol Chem. 281:12093–12101. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Henkel A and Green RM: The unfolded
protein response in fatty liver disease. Semin Liver Dis.
33:321–329. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Marciniak SJ, Yun CY, Oyadomari S, Novoa
I, Zhang Y, Jungreis R, Nagata K, Harding HP and Ron D: CHOP
induces death by promoting protein synthesis and oxidation in the
stressed endoplasmic reticulum. Genes Dev. 18:3066–3077. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yoshida H, Matsui T, Yamamoto A, Okada T
and Mori K: XBP1 mRNA is induced by ATF6 and spliced by IRE1 in
response to ER stress to produce a highly active transcription
factor. Cell. 107:881–891. 2001. View Article : Google Scholar
|
|
27
|
Adachi Y, Yamamoto K, Okada T, Yoshida H,
Harada A and Mori K: ATF6 is a transcription factor specializing in
the regulation of quality control proteins in the endoplasmic
reticulum. Cell Struct Funct. 33:75–89. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cao J, Dai DL, Yao L, Yu HH, Ning B, Zhang
Q, Chen J, Cheng WH, Shen W and Yang ZX: Saturated fatty acid
induction of endoplasmic reticulum stress and apoptosis in human
liver cells via the PERK/ATF4/CHOP signaling pathway. Mol Cell
Biochem. 364:115–129. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Deng J, Lu PD, Zhang Y, Scheuner D,
Kaufman RJ, Sonenberg N, Harding HP and Ron D: Translational
repression mediates activation of nuclear factor kappa B by
phosphorylated translation initiation factor 2. Mol Cell Biol.
24:10161–10168. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hu P, Han Z, Couvillon AD, Kaufman RJ and
Exton JH: Autocrine tumor necrosis factor alpha links endoplasmic
reticulum stress to the membrane death receptor pathway through
IRE1alpha-mediated NF-kappaB activation and down-regulation of
TRAF2 expression. Mol Cell Biol. 26:3071–3084. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Urano F, Wang X, Bertolotti A, Zhang Y,
Chung P, Harding HP and Ron D: Coupling of stress in the ER to
activation of JNK protein kinases by transmembrane protein kinase
IRE1. Science. 287:664–666. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Nerstedt A, Cansby E, Amrutkar M, Smith U
and Mahlapuu M: Pharmacological activation of AMPK suppresses
inflammatory response evoked by IL-6 signalling in mouse liver and
in human hepatocytes. Mol Cell Endocrinol. 375:68–78. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Dooley S and ten Dijke P: TGF-β in
progression of liver disease. Cell Tissue Res. 347:245–256. 2012.
View Article : Google Scholar
|
|
34
|
Yang L, Roh YS, Song J, Zhang B, Liu C,
Loomba R and Seki E: TGF-β signaling in hepatocytes participates in
steatohepatitis through regulation of cell death and lipid
metabolism. Hepatology. 59:483–495. 2014. View Article : Google Scholar
|
|
35
|
Henderson NC and Iredale JP: Liver
fibrosis: cellular mechanisms of progression and resolution. Clin
Sci (Lond). 112:265–280. 2007. View Article : Google Scholar
|
|
36
|
Marchesini G, Brizi M, Morselli-Labate AM,
Bianchi G, Bugianesi E, McCullough AJ, Forlani G and Melchionda N:
Association of nonalcoholic fatty liver disease with insulin
resistance. Am J Med. 107:450–455. 1999. View Article : Google Scholar : PubMed/NCBI
|