Introduction
Ischemic stroke is a major cause of death and is
associated with high rates of morbidity and disability in adults
worldwide (1). As an acute
cerebrovascular disease, it is associated with cerebral ischemia
and brain tissue damage due to significant deprivation of oxygen
and glucose caused by a reduction in or complete blockade of
arterial blood supplies to the brain (2). Transient cerebral ischemia induced
by the deprivation of blood to the brain may cause delayed neuronal
death in some specific vulnerable regions such as the hippocampus
(3). Early restoration of
cerebral blood flow is crucial for sustaining neuronal viability.
Nevertheless, reperfusion is believed to promote delayed secondary
brain injury as the freshly arriving oxygen will serve as a
substrate for the production of excessive reactive oxygen species
(ROS). Thus, antioxidant defenses including free radical scavengers
and antioxidant enzymes are considered a promising approach to
reduce the extent of damage caused by ischemia/reperfusion
injury.
Cumulative evidence suggests that oxidative stress
is a fundamental mechanism of cerebral ischemia/reperfusion injury
(2). Oxidative stress and
mitochondrial dysfunction are frequently implicated in the
pathology of secondary neuronal damage following cerebral
ischemia/reperfusion through the generation of ROS (4,5).
Mitochondria are abundant in cerebral tissues, and are a major
source of cerebral intracellular ROS, and the imbalance between the
generation and degradation of ROS leads to oxidative stress
(6). Moreover, the overall
process of ischemia/reperfusion injury is extremely complex. Cell
injury induced by ischemia/reperfusion is a multifactor,
multi-mechanism, malignant cascade reaction, which includes many
events, such as the increased release of excitatory amino acids,
calcium overload, mitochondrial membrane depolarization, free
radical production, apoptosis gene activation and so forth. These
factors reinforce each other and are interrelated, forming a
vicious cycle, which eventually results in cell apoptosis or
necrosis (7). For example, the
enhancement of concentrations of intracellular free calcium
[Ca2+]i and reduction in mitochondrial
membrane potential (MMP or Δψm) has been proven to
contribute to the secondary mitochondrial dysfunction induced by
cerebral ischemia/reperfusion injury in neonatal rat primary
cultured hippocampal neurons (7).
Thus, inhibition of oxidative stress and mitochondrial dysfunction
is beneficial in the treatment of cerebral ischemia/reperfusion
injury.
Polyphenols are found in many plants and possess
immunomodulatory and anti-inflammatory effects, as well as
antioxidant effects as they are capable of removing ROS formed by
lipid peroxidation, cellular damage and oxidative stress (8). In particular, syringic acid (SA;
4-hydroxy-3,5-di-methoxybenzoic acid), a polyphenolic derivative of
benzoic acid (9), has been shown
to exert antioxidant, chemoprotective and antimicrobial effects
(9,10,11). Recent studies in a rat model of
ischemia/reperfusion suggest that SA reduces oxidative stress
(9,12). However, the regulatory role of SA
in a model of hippocampal neurons subjected to oxygen-glucose
deprivation followed by reperfusion (OGD/R)-induced cell injury and
furthermore, the potential mechanisms involved, have not yet been
explored, to the best of our knowledge.
In the present study, we principally focused on the
protective effects of SA in OGD/R-treated primary hippocampal
neuronal cells by evaluating cell viability, oxidative stress
markers, [Ca2+]i, MMP and cell apoptosis, and
by elucidating the associated molecular mechanisms. Nimodipine is a
calcium antagonist, derived from the 1,4-dihydropyridine ring with
a preferential cerebrovascular action (13). This drug is light-sensitive and
also lipophilic, which enables it to cross the blood-brain barrier
and enter the brain (14). It has
been reported that nimodipine exerts potent cerebrovascular effects
in vitro (15). Thus, in
the present study, nimodipine was used as a positive control
drug.
Materials and methods
Animals, reagents and antibodies
Male Sprague-Dawley (SD) rats (n=20), 220–240 g,
were purchased from Vital River Laboratory Animal Technology Co.,
Ltd. (Beijing, China). All reagent-grade chemicals, SA (purity
>95%), dimethyl sulfoxide (DMSO), and
3-(4,5-dimethylthiazol-2-yl)-2,5-diphe-nyltetrazolium bromide (MTT)
were purchased from Sigma (St. Louis, MO, USA). The neurobasal
medium containing B27 supplements, fetal bovine serum (FBS), horse
serum and other culture products was purchased from Gibco
(Rockville, MD, USA). Nimodipine injection (5 mg/ml) was purchased
from Bayer AG (Leverkusen, Germany). The lactate dehydrogenase
(LDH) activity assay kit and the commercial kits for the detection
of superoxide dismutase (SOD) and ROS were purchased from
Jiangcheng Bioengineering (Nanjing, China). The commercial kits for
the detection of MMP and malondialdehyde (MDA) content were
purchased from Beyotime (Nanjing, China). Fluo-3-acetoxymethyl
ester (Fluo-3 AM) was purchased from Biotium, Inc. (Hayward, CA,
USA). Antibodies against phospho-p38 (#4631) and phospho-JNK
(#4668) antibodies were purchased from Cell Signaling Technology
(Beverly, MA, USA); JNK (sc-572), p38 (sc-535), caspase-3 (sc-1225)
and Bax (sc-493) were obtained from Santa Cruz Biotechnology (Santa
Cruz, CA, USA); Bcl-2 (#D038-3) was purchased from MBL (Nagoya,
Japan) and β-actin (A1978) was obtained from Sigma. Horseradish
peroxidase-conjugated anti-rabbit (A0208) or anti-mouse (A0216)
immunoglobulin G, and enhanced chemiluminescence (ECL) reagents
were purchased from Beyotime (Shanghai, China). The Pierce BCA
protein assay kit was purchased from Thermo Fisher Scientific
(Rockford, IL, USA). Polyvinylidene fluoride (PVDF) membranes were
purchased from Millipore Corp. (Billerica, MA, USA).
Cell isolation and cell culture
Primary hippocampal neuronal cells were prepared
from the brains of neonatal SD rats, as previously described
(16) with some modifications.
Briefly, the rats were sacrificed by decapitation and the
hippocampal tissues were dissected on ice and then dissociated in
0.25% trypsin-EDTA. The primary hippocampal neurons were maintained
in neuro-basal medium supplemented with GlutaMAX and B27 plus
glucose (4.5 g/l) for 7 days. Then, the cells were cultured in a
medium containing 5% horse serum and 5% FBS supplemented with 15 mM
glucose for 14 days. All cells were cultured in an incubator with a
humidified atmosphere of 5% CO2 at 37°C. All
experimental procedures were performed in accordance with the
regulations of the Ethics Committee of Jilin University for the
Care and Use of Laboratory Animals and were approved by the
Institutional Animal Care and Use Committee (IACUC) of Jilin
University (Changchun, China).
OGD/R procedure
We established an in vitro model of cerebral
ischemia by subjecting cells to OGD. The neurons were rinsed twice
and incubated in DMEM without glucose. The culture media were then
introduced into a specialized, humidified chamber filled with 1%
O2/94% N2/5% CO2 at 37°C for 3 h
in order to establish conditions of OGD. Thereafter, the culture
media were replaced with normal DMEM containing glucose under
normoxic conditions for an additional 24 h as OGD/R. SA (0.1, 1,
10, and 20 µM) or nimodipine (final concentration 5 mg/l)
was applied to the cell cultures 24 h prior to OGD/R. The cultures
in the blank control group were maintained in normal DMEM medium
under normoxic conditions at 37°C without OGD/R exposure and SA
pre-treatment. The cultures in the control group were subjected to
OGD/R in the absence of SA pre-treatment.
Cell viability assay
Neuronal cell viability was measured using the MTT
assay. Briefly, the cultured cells were seeded into 96-well plates
(Corning Inc., Corning, NY, USA) at a density of 5×103
cells/well in DMEM with 10% FBS for 24 h at 37°C in 5%
CO2. After exposure to OGD/R, 20 µl modified
tetrazolium salt MTT (5 mg/ml) was added to each well and the
samples were incubated at 37°C for 4 h. The supernatant was then
carefully removed and 100 µl DMSO was added to lyse the
cells. Once the dark-blue MTT crystals had dissolved, absorbance
was measured at 490 nm using a Benchmark microplate reader
(Bio-Rad, Hercules, CA, USA).
Cytotoxicity assay
Neuronal injury in the cells was also quantitatively
assessed by measuring the activity of LDH released from damaged or
dead cells. Previous findings have shown that the activity of LDH
from either necrotic or apoptotic cells is proportional to the
number of neurons damaged or destroyed (17). Briefly, the cells were seeded in
96-well plates (Corning Inc.) at a density of 5×103
cells/well and incubated for 24 h. After exposure to OGD/R, a 50
µl volume of medium was removed, and the amount of LDH
leakage from the cells was determined using the LDH activity assay
kit according to the manufacturer's instructions. The absorbance of
the samples was read spectrophotometrically at 490 nm. The results
are expressed as the percentage of LDH release relative to the
control cells.
Measurement of cellular SOD and MDA
levels
After exposure to OGD/R, the cells were harvested
and resuspended in 0.1 M ice cold PBS. The cell suspensions were
sonicated for 25 sec on ice and centrifuged at 12,000 rpm at 4°C
for 10 min, and the supernatants were collected. SOD levels were
assayed by a modification of the xanthine/xanthine oxidase method
(18). Cellular SOD levels were
determined by spectrophotometry. The levels of MDA, a compound
produced during lipid peroxidation, were measured using the
commercial kit based on a reaction with thiobarbituric acid
(19). The optical density at 532
nm was determined using a microplate reader (Spectra Max 190;
Molecular Devices, Sunnyvale, CA, USA).
Measurement of ROS content
ROS formation was determined using the fluorescent
probe 2′,7′-dichlorfluorescein-diacetate (DCFH-DA). Cell-permeable
non-fluorescent DCFH-DA has been shown to be oxidized to the highly
fluorescent 2,7-dichlo-rofluorescein in the presence of ROS. The
neurons were washed twice with PBS, and then incubated with 10 mM
DCFH-DA for 30 min at 37°C in the dark. The cells were then
harvested and suspended in PBS. The fluorescence intensity was
measured by a fluorospectrophotometer (Hitachi, Tokyo, Japan) at
excitation/emission maxima of 485/525 nm (20).
Measurement of
[Ca2+]i
After exposure to OGD/R, the cells were harvested,
washed with PBS, centrifuged at 1,000 rpm for 5 min, and then
resuspended in PBS. Thereafter, the cell suspensions were incubated
with 5 µM Fluo-3 AM in the dark at 37°C for 40 min. After
centrifugation and one wash, the cells were resuspended in PBS. The
fluorescence of 1×104 cells was analyzed by flow
cytometry at 488 nm and the relative fluorescence intensity of
Fluo-3 was used as an indicator of the quantity of
[Ca2+]i.
Measurement of MMP
The cationic dye, JC-1 was used to evaluate the loss
of MMP in hippocampal cultures subjected to OGD/R under a
fluorescence microscope (Olympus, Tokyo, Japan). JC-1 is a
convenient voltage sensitive probe used to monitor MMP (21). The cells containing J-aggregates
have high Δψm, and show fluorescence. The cells with low
Δψm are those in which JC-1 maintains (or reacquires)
its monomeric form, and show only green fluorescence (22). The relative proportions of red and
green fluorescence were used to measure the ratio of mitochondrial
depolarization.
Western blot analysis
The cell lysates were collected and protein
concentrations were determined using the Pierce BCA Protein assay
kit. Equal amounts of protein were separated by SDS-PAGE and
transferred onto PVDF membranes. The membranes were blocked in 5%
nonfat milk in TBST buffer (5 mM Tris-HCl, pH 7.4, 136 mM NaCl,
0.1% Tween-20) for 1 h at room temperature prior to hybridization
with a primary antibody overnight at 4°C, followed by three washes
for 5 min with TBST. Following incubation with HRP-conjugated
secondary antibodies for 1 h at room temperature and three washes
with TBST, the resultant protein bands were visualized by ECL
reagents according to the manufacturer's instructions. The
absorbance values of the target proteins were obtained through
Gel-Pro Analyzer version 4.0 software (Media Cybernetics, Silver
Spring, MD, USA).
Statistical analysis
Data are expressed as the means ± SD of results
derived from three independent experiments performed in triplicate.
Statistical analysis was performed using the Student's t-test and
ANOVA. A P-value <0.05 was considered to indicate a
statistically significant difference.
Results
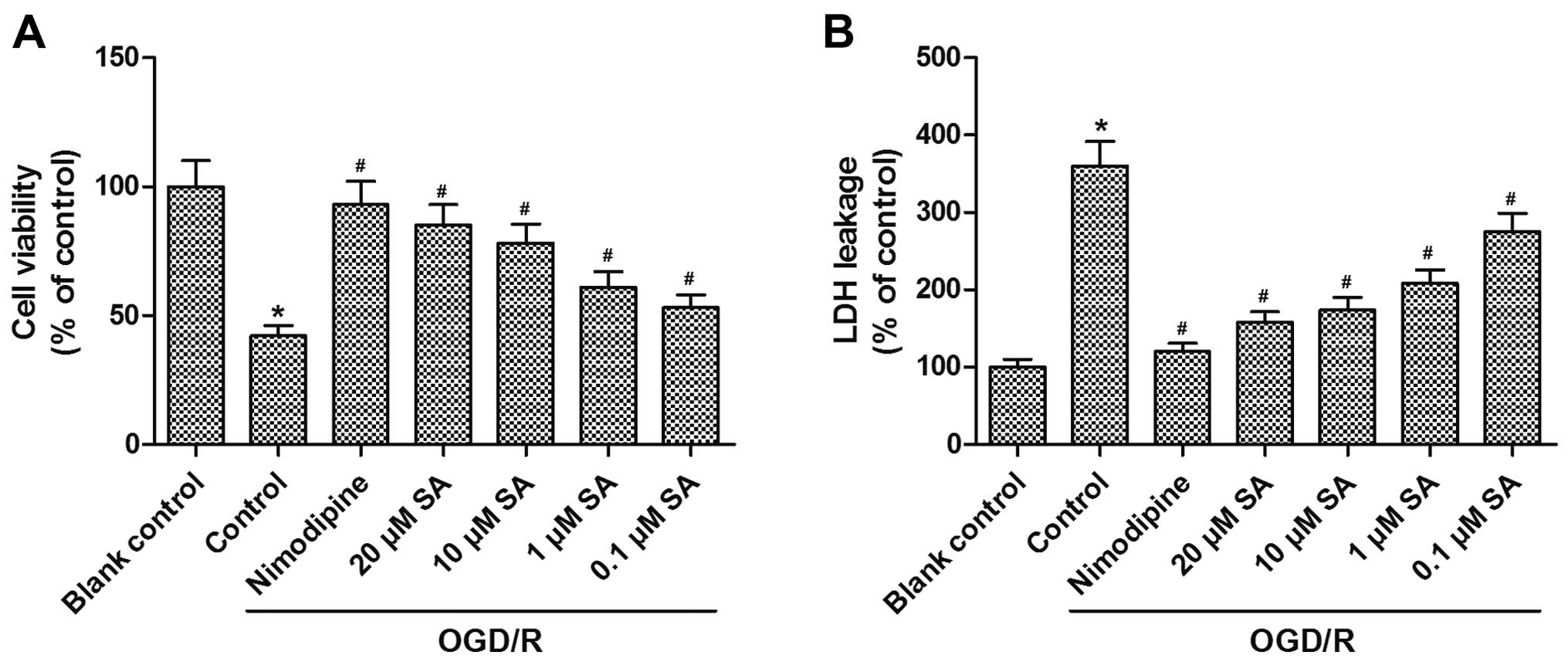
SA pre-treatment ameliorates
OGD/R-induced loss of viable hippocampal neuronal cells
In order to examine the protective effects of SA
against cell cytotoxicity caused by OGD/R, MTT and LDH assays were
performed to assess the viability of hippocampal neurons. Compared
with the blank control group, viability was significantly decreased
after subjecting the cells to OGD/R, and this effect was reversed
by pre-treatment with SA at concentrations of 0.1, 1, 10, and 20
µM, in a dose-dependent manner (Fig. 1A). In addition, LDH leakage was
increased after OGD/R, and incubation with SA attenuated
OGD/R-induced LDH leakage at concentrations of 0.1, 1, 10, and 20
µM in a dose-dependent manner (Fig. 1B). These results demonstrated that
pre-treatment with SA attenuated the OGD/R-induced loss of viable
hippocampal neuronal cells.
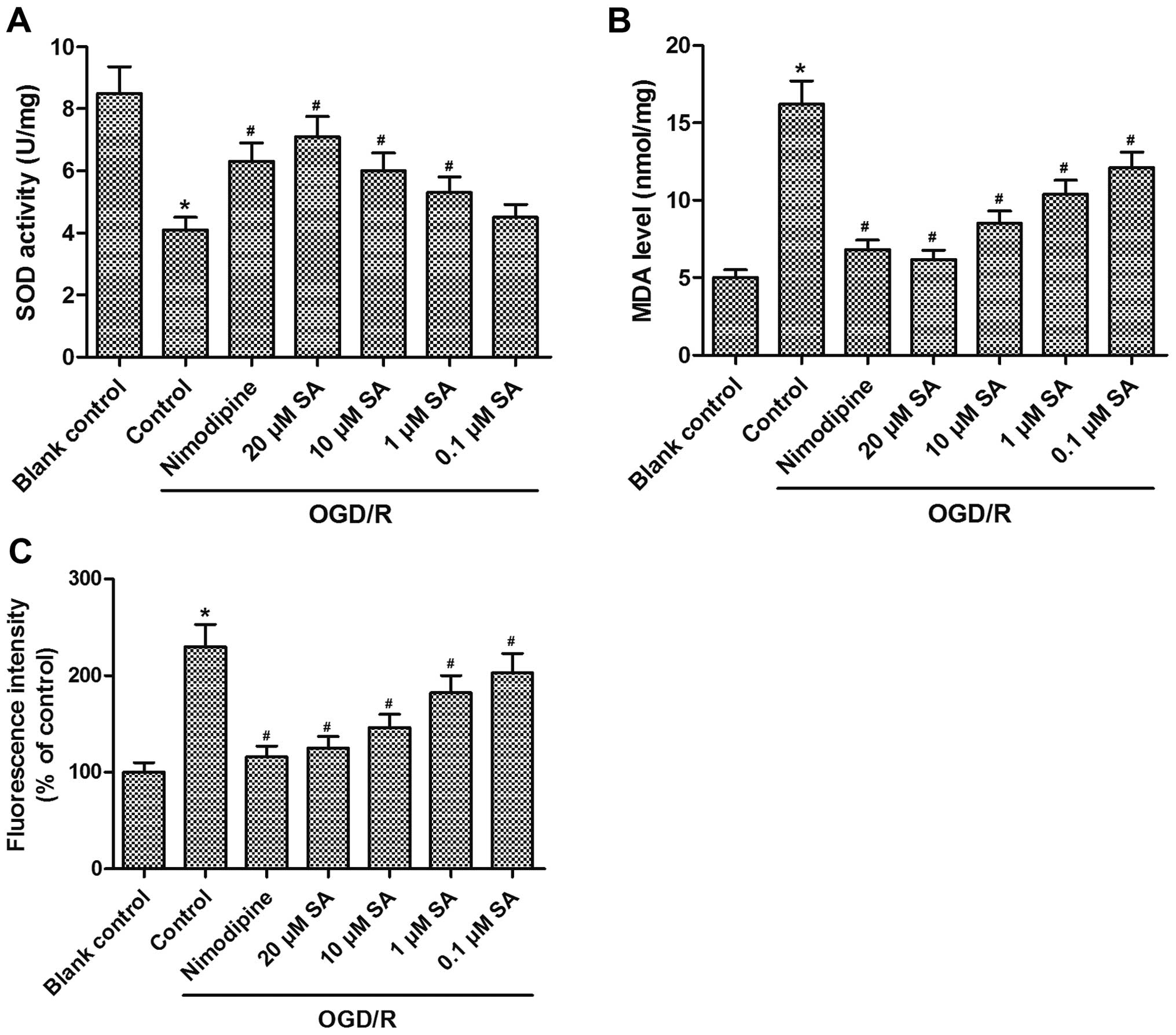
SA pre-treatment ameliorates
OGD/R-induced oxidative stress in hippocampal neuronal cells
We next determined the effects of SA on
OGD/R-induced oxidative stress in hippocampal neurons. OGD exposure
markedly reduced the antioxidant SOD activity in the neuronal
cultures, whereas SA (0.1, 1, 10 and 20 µM) pre-treatment
resulted in a noticeable enhancement of SOD activity in a
dose-dependent manner (Fig. 2A).
Additionally, a 2.3-fold increase in intracellular ROS generation
and a 3.2-fold elevation in MDA levels (a cellular lipid
peroxidation product) were found after subjecting the hippocampal
neuronal cells to OGD/R. Conversely, pre-treatment with SA (0.1, 1,
10, and 20 µM) decreased MDA content and ROS production in a
dose-dependent manner (Fig. 2B and
C). Taken together, these findings demonstrate that SA
pre-treatment distinctly ameliorated OGD/R-induced oxidative stress
in hippocampal neuronal cells.
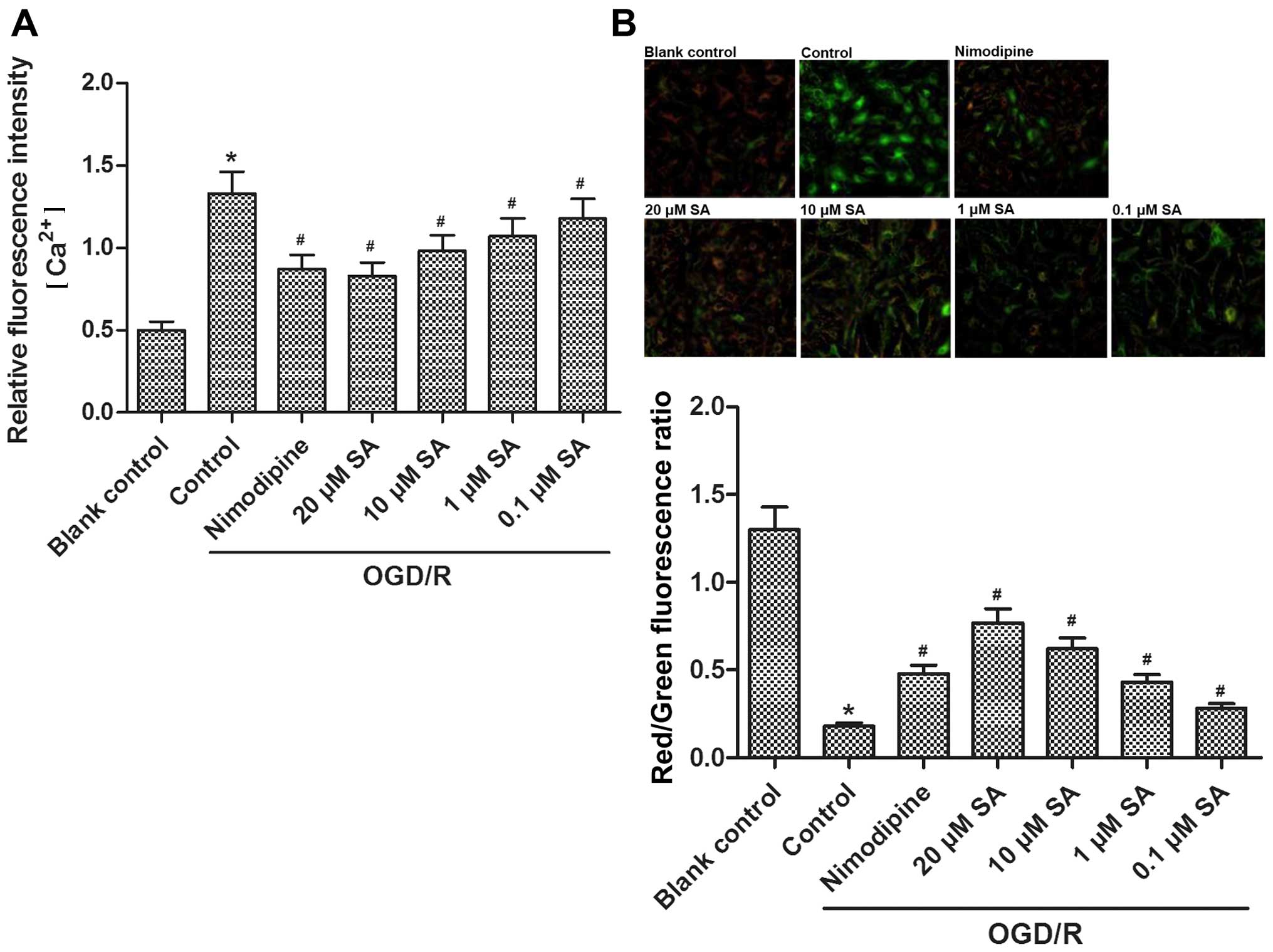
SA pre-treatment prevents OGD/R-induced
elevations in [Ca2+]i and MMP dissipation in
hippocampal neuronal cells
Compared with the blank control group,
[Ca2+]i in the hippocampal neurons was
significantly enhanced after exposure to OGD/R. However, nimodipine
and SA (0.1, 1, 10, and 20 µM) pre-treatment decreased the
enhanced [Ca2+]i in a dose-dependent manner
(Fig. 3A). Moreover, levels in
the 20 µM SA-treated group decreased to a similar level as
those in the nimodipine-treated group.
JC-1 was used to assess the extent of mitochondrial
depolarization in the hippocampal cultures exposed to OGD/R. After
OGD/R, the red/green fluorescence intensity of JC-1 in the
hippocampal neurons was reduced, suggesting a dissipation of the
MMP whereas pre-treatment with SA (0.1, 1, 10, and 20 µM)
significantly stabilized the MMP in a dose-dependent manner
(Fig. 3B). Moreover, a greater
stabilizing effect was achieved in the 20 µM SA-treated
group than in the other groups.
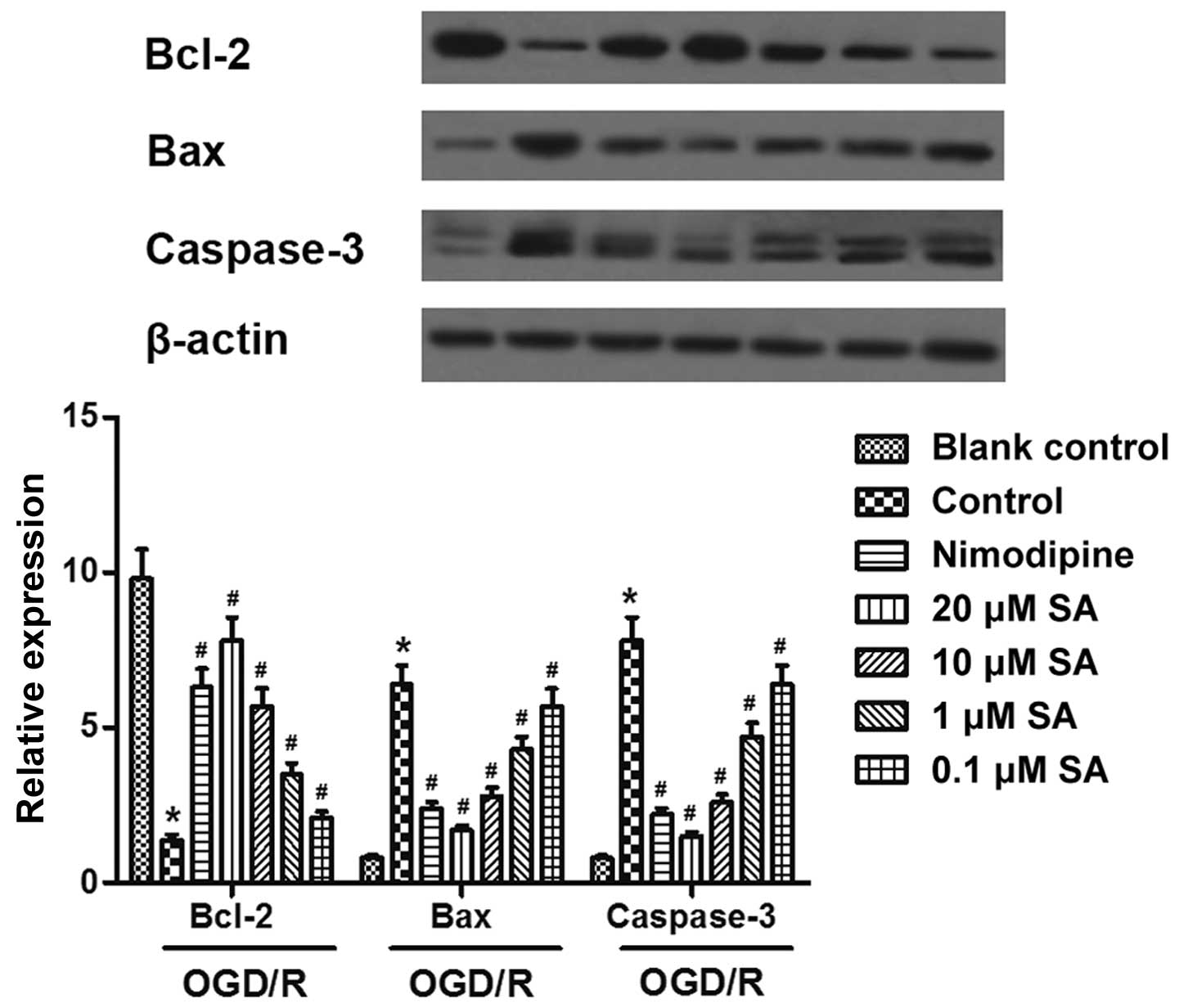
SA pre-treatment attenuates OGD/R-induced
apoptosis of hippocampal neuronal cells
Western blot analysis revealed that exposure to OGD
resulted in the apoptosis of hippocampal neurons, accompanied by a
marked increase in the expression of Bax and caspase-3 proteins,
and a significant reduction in Bcl-2 protein. However, in the SA
(0.1, 1, 10, and 20 µM)-pre-treated groups, the expression
of Bax and caspase-3 was downregulated, and the expression of Bcl-2
was upregulated in a dose-dependent manner (Fig. 4). Thus, these data suggested that
SA pre-treatment attenuated the OGD/R-induced apoptosis of
hippocampal neuronal cells.
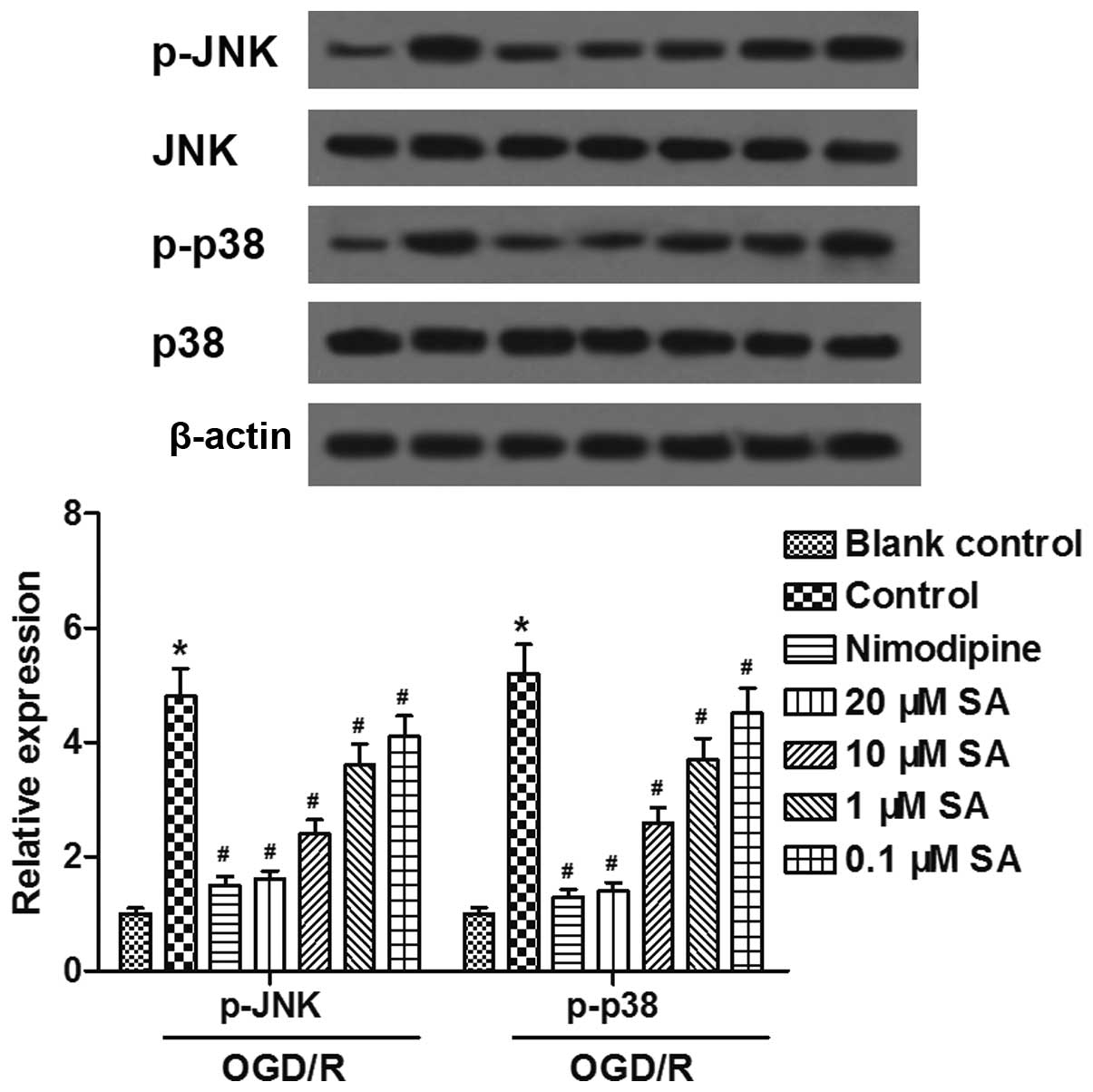
SA pre-treatment inhibits OGD/R-induced
activation of JNK and p38 signaling pathways in hippocampal
neuronal cells
Furthermore, we examined whether the JNK and p38
pathways are associated with the neuroprotective effects of SA
against OGD/R-induced injury in neuronal cells. Western blot
analysis revealed that the levels of phosphorylated (p-)JNK and
p-p38 expression were enhanced by exposure to OGD/R, and inhibited
by pre-treatment with SA in a dose-dependent manner (Fig. 5). The results indicated that SA
pre-treatment inhibited the OGD/R-induced activation of the JNK and
p38 signaling pathways in hippocampal neuronal cells.
Discussion
Cerebral ischemia is one of the most common causes
of death worldwide after cardiovascular diseases and cancer, with a
higher incidence in aged individuals (23). The therapeutic aim in treating
cerebral ischemia is to reduce the extent of brain injury and thus,
minimize neurological impairment. In order to develop effective
strategies for the prevention, treatment and prognosis of the
disease, it is necessary to identify safe and effective drugs or
natural substances and furthermore, to have a sufficient
understanding of the pathological mechanisms involved in cerebral
ischemia/reperfusion injury. In the present study, we examined the
protective effects of SA in primary cultures of hippocampal neurons
injured by OGD/R.
Studies have shown that antioxidants reduce ischemic
injury in the brain. Indeed, the efficacy of plant extracts,
including gallic acid, (S)-ZJM-289, aloperine and SA, in the
prevention of stroke and cerebral ischemia has been evaluated in
some cell and animal models of ischemic insults (12,24–26). In particular, SA is an active
compound isolated from Isatis indigotica or Radix isatidis,
which has been demonstrated to exert strong antioxidant and
hepatoprotective effects (11,27). On the other hand, it is well known
that ROS have been associated with high levels of MDA and suggested
to cause lipid peroxidation thereby initiating the cascade of cell
membrane damage. The synthesis of SOD is the principal factor
involved in controlling ROS levels. Previous findings have
demonstrated that SA is a strong inhibitor of low-density
lipoprotein oxidation, contributing to the scavenging of free
radicals, reducing the production of MDA, and thus, slowing the
development of atherosclerosis (28). SA reduces the levels of oxidative
stress markers and exerts antioxidant effects, augmenting the
antioxidant capacity in L-arginine-induced acute pancreatitis in
rats (11). In the present study,
we found that pre-treatment with SA ameliorated OGD/R-induced
oxidative stress in hippocampal neuronal cells in a dose-dependent
manner.
Caspase-3 is a major cell death effector protease in
the adult and neonatal nervous system (29), which is crucial among multiple
distinct caspases during neuronal development and under
pathological conditions including cerebral ischemia (30). As a neuroprotective agent, SA
reduced oxidative stress and neuronal degeneration, with increased
SOD generation and decreased levels of MDA and caspase-3, in rats
with spinal cord ischemia/reperfusion (12) or in rats with cerebral ischemia
(9). Moreover, phenolic acids,
including SA, exerted distinct inhibitory effects on the viability,
cytotoxicity and apoptosis of Neuro-2A cells induced by
methylglyoxal (31). Similarly,
our data revealed that pre-treatment with SA dose-dependently
attenuated the OGD/R-induced loss of viability and apoptosis of
hippocampal neuronal cells, suggesting that SA may exert
cytoprotective effects against OGD/R-induced neuronal injury.
To further explore the mechanisms through which SA
protects against OGD/R injury, we performed additional experiments
measuring [Ca2+]i and MMP. Calcium is an
important second messenger involved in neurotransmitter release and
signal transduction. Numerous studies have definitely indicated
that alterations in intracellular Ca2+ homeostasis play
a central role in initiating the apoptotic response (32). In addition, it has been suggested
that mitochondrial membrane depolarization is a determining factor
in the final step to apoptosis (7). Previously, both elevations of
[Ca2+]i and MMP loss lead to destabilization
of the neuronal cell structure and caused cell damage, eventually
leading to cell death (7,26). Polyphenols from the plant
Zanthoxylum heitzii, including SA as a major component,
exerted an inhibitory effect on HL-60 cells through ROS generation,
MMP loss and cell cycle destabilization (33). Similarly, in our experiments, the
results demonstrated that SA significantly reduced
[Ca2+]i elevation, whereas it increased the
MMP after OGD/R exposure in hippocampal neuronal cells, inferring
that the antioxidant effects of SA in cultured hippocampal neurons
injured by OGD/R may be associated with the inhibition of
[Ca2+]i overload and the stabilization of the
MMP.
As suggested by previous findings, OGD/R induced the
activation of the apoptotic signaling pathways of JNK and p38, and
increased the activity of caspase-3 in primary cultures of rat
cortical neuronal cells (34). In
addition, the activation of JNK and p38 signaling pathways
participated in the methylglyoxal-induced apoptosis of Neuro-2A
cells (31), which was inhibited
by pre-treatment with SA. Our data demonstrated that SA inhibited
the OGD/R-induced activation of JNK and p38 signaling pathways in
hippocampal neuronal cells.
In conclusion, the data revealed that SA exerted
strong neuroprotective effects against OGD/R-induced neuronal
injury in vitro. Our results demonstrated that pre-treatment
with SA may inhibit the OGD/R-induced loss of viability, oxidative
stress, [Ca2+]i overload, MMP dissipation and
apoptosis of cultured hippocampal neuronal cells through the JNK
and p38 signaling pathways.
References
|
1
|
Donnan GA, Fisher M, Macleod M and Davis
SM: Stroke. Lancet. 371:1612–1623. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chen H, Yoshioka H, Kim GS, Jung JE, Okami
N, Sakata H, Maier CM, Narasimhan P, Goeders CE and Chan PH:
Oxidative stress in ischemic brain damage: mechanisms of cell death
and potential molecular targets for neuroprotection. Antioxid Redox
Signal. 14:1505–1517. 2011. View Article : Google Scholar :
|
|
3
|
Schmidt-Kastner R and Freund TF: Selective
vulnerability of the hippocampus in brain ischemia. Neuroscience.
40:599–636. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Christophe M and Nicolas S: Mitochondria:
a target for neuro-protective interventions in cerebral
ischemia-reperfusion. Curr Pharm Des. 12:739–757. 2006. View Article : Google Scholar
|
|
5
|
Turrens JF: Mitochondrial formation of
reactive oxygen species. J Physiol. 552:335–344. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Radermacher KA, Wingler K, Langhauser F,
Altenhöfer S, Kleikers P, Hermans JJ, Hrabě de Angelis M,
Kleinschnitz C and Schmidt HH: Neuroprotection after stroke by
targeting NOX4 as a source of oxidative stress. Antioxid Redox
Signal. 18:1418–1427. 2013. View Article : Google Scholar :
|
|
7
|
Rui C, Yuxiang L, Yinju H, Qingluan Z,
Yang W, Qipeng Z, Hao W, Lin M, Juan L, Chengjun Z, et al:
Protective effects of Lycium barbarum polysaccharide on neonatal
rat primary cultured hippocampal neurons injured by oxygen-glucose
deprivation and reperfusion. J Mol Histol. 43:535–542. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Scalbert A and Williamson G: Dietary
intake and bioavailability of polyphenols. J Nutr. 130(8S Suppl):
2073S–2085S. 2000.PubMed/NCBI
|
|
9
|
Güven M, Aras AB, Topaloğlu N, Özkan A,
Şen HM, Kalkan Y, Okuyucu A, Akbal A, Gökmen F and Coşar M: The
protective effect of syringic acid on ischemia injury in rat brain.
Turk J Med Sci. 45:233–240. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kumar S, Prahalathan P and Raja B:
Syringic acid ameliorates (L)-NAME-induced hypertension by reducing
oxidative stress. Naunyn Schmiedebergs Arch Pharmacol.
385:1175–1184. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cikman O, Soylemez O, Ozkan OF, Kiraz HA,
Sayar I, Ademoglu S, Taysi S and Karaayvaz M: Antioxidant activity
of syringic acid prevents oxidative stress in L-arginine-induced
acute pancreatitis: an experimental study on rats. Int Surg.
100:891–896. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tokmak M, Yuksel Y, Sehitoglu MH, Guven M,
Akman T, Aras AB, Cosar M and Abbed KM: The neuroprotective effect
of syringic acid on spinal cord ischemia/reperfusion injury in
rats. Inflammation. 38:1969–1978. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kazda S and Towart R: Nimodipine: a new
calcium antagonistic drug with a preferential cerebrovascular
action. Acta Neurochir (Wien). 63:259–265. 1982. View Article : Google Scholar
|
|
14
|
Scriabine A and van den Kerckhoff W:
Pharmacology of nimodipine. A review. Ann N Y Acad Sci.
522:698–706. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wadworth AN and McTavish D: Nimodipine. A
review of its pharmacological properties, and therapeutic efficacy
in cerebral disorders. Drugs Aging. 2:262–286. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Krohn AJ, Preis E and Prehn JH:
Staurosporine-induced apoptosis of cultured rat hippocampal neurons
involves caspase-1-like proteases as upstream initiators and
increased production of superoxide as a main downstream effector. J
Neurosci. 18:8186–8197. 1998.PubMed/NCBI
|
|
17
|
McCord JM and Fridovich I: Superoxide
dismutase. An enzymic function for erythrocuprein (hemocuprein). J
Biol Chem. 244:6049–6055. 1969.PubMed/NCBI
|
|
18
|
Gwag BJ, Lobner D, Koh JY, Wie MB and Choi
DW: Blockade of glutamate receptors unmasks neuronal apoptosis
after oxygen-glucose deprivation in vitro. Neuroscience.
68:615–619. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ohkawa H, Ohishi N and Yagi K: Assay for
lipid peroxides in animal tissues by thiobarbituric acid reaction.
Anal Biochem. 95:351–358. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wu W, Xia Q, Luo RJ, Lin ZQ and Xue P: In
vitro study of the antagonistic effect of low-dose liquiritigenin
on gemcitabine-induced capillary leak syndrome in pancreatic
adenocarcinoma via inhibiting ROS-mediated signalling pathways.
Asian Pac J Cancer Prev. 16:4369–4376. 2015. View Article : Google Scholar
|
|
21
|
Reers M, Smith TW and Chen LB: J-aggregate
formation of a carbocyanine as a quantitative fluorescent indicator
of membrane potential. Biochemistry. 30:4480–4486. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhu XJ, Shi Y, Peng J, Guo CS, Shan NN,
Qin P, Ji XB and Hou M: The effects of BAFF and BAFF-R-Fc fusion
protein in immune thrombocytopenia. Blood. 114:5362–5367. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Margaill I, Plotkine M and Lerouet D:
Antioxidant strategies in the treatment of stroke. Free Radic Biol
Med. 39:429–443. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sun J, Li YZ, Ding YH, Wang J, Geng J,
Yang H, Ren J, Tang JY and Gao J: Neuroprotective effects of gallic
acid against hypoxia/reoxygenation-induced mitochondrial
dysfunctions in vitro and cerebral ischemia/reperfusion injury in
vivo. Brain Res. 1589:126–139. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang C, Zhang Z, Zhao Q, Wang X, Ji H and
Zhang Y: (S)-ZJM-289 preconditioning induces a late phase
protection against nervous injury induced by transient cerebral
ischemia and oxygen-glucose deprivation. Neurotox Res. 26:16–31.
2014. View Article : Google Scholar
|
|
26
|
Ma NT, Zhou R, Chang RY, Hao YJ, Ma L, Jin
SJ, Du J, Zheng J, Zhao CJ, Niu Y, et al: Protective effects of
aloperine on neonatal rat primary cultured hippocampal neurons
injured by oxygen-glucose deprivation and reperfusion. J Nat Med.
69:575–583. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Itoh A, Isoda K, Kondoh M, Kawase M,
Watari A, Kobayashi M, Tamesada M and Yagi K: Hepatoprotective
effect of syringic acid and vanillic acid on CCl4-induced liver
injury. Biol Pharm Bull. 33:983–987. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Morton LW, Croft KD, Puddey IB and Byrne
L: Phenolic acids protect low density lipoproteins from
peroxynitrite-mediated modification in vitro. Redox Rep. 5:124–125.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Le DA, Wu Y, Huang Z, Matsushita K,
Plesnila N, Augustinack JC, Hyman BT, Yuan J, Kuida K, Flavell RA
and Moskowitz MA: Caspase activation and neuroprotection in
caspase-3-deficient mice after in vivo cerebral ischemia and in
vitro oxygen glucose deprivation. Proc Natl Acad Sci USA.
99:15188–15193. 2002. View Article : Google Scholar
|
|
30
|
Nicholson DW: Caspase structure,
proteolytic substrates, and function during apoptotic cell death.
Cell Death Differ. 6:1028–1042. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Huang SM, Chuang HC, Wu CH and Yen GC:
Cytoprotective effects of phenolic acids on methylglyoxal-induced
apoptosis in Neuro-2A cells. Mol Nutr Food Res. 52:940–949. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Orrenius S, Zhivotovsky B and Nicotera P:
Regulation of cell death: the calcium-apoptosis link. Nat Rev Mol
Cell Biol. 4:552–565. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Pieme CA, Santosh GK, Tekwu EM, Askun T,
Aydeniz H, Ngogang JY, Bhushan S and Saxena AK: Fruits and barks
extracts of Zanthoxylum heitzii a spice from Cameroon induce
mitochondrial dependent apoptosis and Go/G1 phase arrest in human
leukemia HL-60 cells. Biol Res. 47:542014. View Article : Google Scholar
|
|
34
|
Wang CP, Li JL, Zhang LZ, Zhang XC, Yu S,
Liang XM, Ding F and Wang ZW: Isoquercetin protects cortical
neurons from oxygen-glucose deprivation-reperfusion induced injury
via suppression of TLR4-NF-κB signal pathway. Neurochem Int.
63:741–749. 2013. View Article : Google Scholar : PubMed/NCBI
|