Introduction
Pterygium is a common benign fibrovascular lesion of
the ocular surface in which the wedge-shaped ingrowth of
conjunctival tissue invades the peripheral cornea (1). It becomes a significant
sight-threatening complication once the ingrowth covers the pupil.
Surgical excision can be a useful therapy for pterygia; however,
recurrences are common. Thus, there is a significant need to gain
more insight into pterygium formation and recurrence, in order to
enable the design of novel therapeutic strategies, either for
inhibiting pterygium growth, regressing pterygia or preventing
recurrence (2). Recent studies
provide evidence that pterygium is a stem cell disorder with
prema-lignant characteristics (3,4),
and that epithelial-mesenchymal transition (EMT) may play a key
role in the pathogenesis of this disease (5,6).
Although considerable progress has been made towards understanding
the etiology of the disease, the pathogenesis of pterygium is not
yet completely understood (7,8).
Long non-coding RNAs (lncRNAs) are recognized as
being longer than 200 nucleotides in length without protein coding
properties (9). They can be
classified into 6 categories according to their genomic proximity
to protein-coding genes: exon sense-overlapping, intron
sense-overlapping, intronic antisense, natural antisense,
bidirectional and intergenic lncRNAs (10). Recently, lncRNAs have been
implicated in regulating a number of biological processes, such as
gene imprinting, dosage compensation, cell cycle regulation, cell
pluripotency, meiotic entry and telomere length (10,11). lncRNAs regulate these processes at
the transcriptional, post-transcriptional and epigenetic levels
(12,13). Moreover, dysregulated lncRNAs have
been linked to the states of different diseases, such as cancer
development and neurological disorders. Pterygium is a complex
pathological process mediated by the dysfunction of the gene
regulatory network; however, there is little data available on
lncRNAs in pterygium. The features of lncRNAs expression and their
potential biological function in pterygium require further
investigation.
We hypothesized that lncRNAs may be involved in the
molecular pathogenesis of pterygium. To examine this hypothesis, in
this study, the global expression profiles of lncRNAs were analyzed
by microarray analysis and quantitative polymerase chain reaction
(qPCR) in human pterygium samples compared to adjacent normal
conjunctival samples. Our results are likely to provide important
insight into the pathogenesis of pterygium.
Patients and methods
Patient sample preparation
A total of 12 pterygium specimens were obtained
during pterygium surgery at the Second Affiliated Hospital of
Nanjing Medical University. The patients were in good health and
their age ranged from 49 to 79 years, with 5 males and 7 females
(Table I). All patients underwent
conventional excision of pterygium with auto-transplantation of the
conjunctiva. In the same patient, 1.5×1.5 mm conjunctival tissues
were obtained from the inferior temporal quadrant of the bulbar
conjunctiva during surgery. Clinical details pertaining to the
study patients are described in Table
I. The study protocol was approved by the Ethics Review
Committee of the Second Affiliated Hospital of Nanjing Medical
University, Nanjing, China and all participants gave informed
consent to the treating surgeon. All tissues were immediately
frozen in liquid nitrogen and stored in a freezer at −80°C until
use.
 | Table IClinical and demographic information
for patients. |
Table I
Clinical and demographic information
for patients.
| Case no. | Age | Gender | Case details |
|---|
| 1 | 58 | M | Primary |
| 2 | 63 | F | Primary |
| 3 | 49 | M | Primary |
| 4 | 72 | M | Recurrent |
| 5 | 50 | F | Primary |
| 6 | 66 | F | Primary |
| 7 | 79 | M | Primary |
| 8 | 68 | F | Primary |
| 9 | 61 | F | Primary |
| 10 | 70 | M | Primary |
| 11 | 54 | F | Primary |
| 12 | 72 | F | Primary |
RNA extraction and quality control
To extract RNA from tissues, we resuspended the
frozen tissues in TRIzol reagent (Life Technologies, Carlsbad, CA,
USA) and finally eluted into 50 µl of elution solution
according to the manufacturer's instructions. RNA quantity and
quality were measured using a NanoDrop ND-1000 spectrophotometer.
RNA integrity was assessed by standard denaturing agarose gel
electrophoresis. We stored all the RNA samples at −80°C until
further processing. Complementary DNA (cDNA) was synthesized from 1
µg of total RNA using a PrimesScript™ RT Master Mix kit
(Takara, Dalian, China) with random hexamer primers in a final
volume of 20 µl. The reverse transcription reactions were
incubated at 37°C for 15 min, 85°C for 5 sec and 4°C for 10
min.
Microarray analysis
Arraystar Human LncRNA Microarray V3.0 is designed
for the global profiling of human lncRNAs and protein-coding
transcripts, which is updated from the previous Microarray V2.0.
Approximately 30,586 lncRNAs and 26,109 coding transcripts can be
detected by the third-generation lncRNA microarray. The lncRNAs
were obtained from authoritative databases (RefSeq, Ensembl and
UCSC_knowngenes, and others). The mRNAs were collected from RefSeq
and GENCODE.
RNA labeling and array hybridization
RNA labeling and array hybridization were performed
according to the Agilent One-Color Microarray-Based Gene Expression
Analysis protocol (Agilent Technologies, Santa Clara, CA, USA) with
minor modifications. Briefly, mRNA was purified from total RNA
following the removal of rRNA (mRNA-ONLY™ Eukaryotic mRNA Isolation
kit; Epicentre, Madison, WI, USA). Each sample was then amplified
and transcribed into fluorescent cRNA along the entire length of
the transcripts without 3′ bias utilizing a mixture of oligo(dT)
and random primers (Arraystar Flash RNA Labeling kit; Arraystar,
Rockville, MD, USA). The labeled cRNAs were purified using an
RNeasy Mini kit (Qiagen, Hilden, Germany). The concentration and
specific activity of the labeled cRNAs (pmol Cy3/µg cRNA)
were measured using a NanoDrop ND-1000 spectrophotometer. A total
of 1 µg of each labeled cRNA was fragmented by the addition
of 5 µl 10X blocking agent and 1 µl of 25X
fragmentation buffer, then the mixture was heated at 60°C for 30
min. Finally, 25 µl 2X GE hybridization buffer were added to
dilute the labeled cRNA. Subsequently, 50 µl of
hybridization solution were dispensed into the gasket slide and
assembled to the lncRNA expression microarray slide. The slides
were incubated for 17 h at 65°C in an Agilent Hybridization Oven.
The hybridized arrays were washed, fixed and scanned with using the
Agilent DNA Microarray Scanner (part no. G2505C; Agilent
Technologies).
qPCR
The selected lncRNAs and the primers (Table II) used for qPCR were designed
and synthesized by Generay Biotech (Shanghai, China). β-actin was
used as an internal control for the tissue samples. qPCR was
performed using the ViiA 7 Real-time PCR system (Applied
Biosystems, Foster City, CA, USA) with the SYBR expression assay
system (Takara). The PCR reaction conditions were as follows: an
initial denaturation at 95°C for 10 min, followed by
40*PCR cycles at 95°C for 10 sec and 60°C for 60 sec.
Finally annealing and extension at 95°C for 10 sec, 60°C for 60 sec
and 95°C for 15 sec. Each sample was assayed in triplicate. We used
the standard curve method to determine the fold change in gene
expression in the pterygium samples relative to the adjacent normal
conjunctival samples.
| Table IIPrimers used for PCR in this
study. |
Table II
Primers used for PCR in this
study.
| lncRNAs | Primers | Length (bp) |
|---|
| PISRT1 | F:
5′-GATCCAGGCTCTTGACCACTC-3′ | |
| R:
5′-CGGCTTCTCTCTGGAACTCAG-3′ | 88 |
| LOC283761 | F:
5′-AAGACCCTCCCTTGAACCGT-3′ | |
| R:
5′-TCAAACCCTGTGGGGCTTTC-3′ | 153 |
| FOXD2-AS1 | F:
5′-AGTGGGGAATGAGGATGGGT-3′ | |
| R:
5′-CGGCGTGTAATTGGTAGGAG-3′ | 142 |
| LPAL2 | F:
5′-ACGCCACACCAGCACAGTAG-3′ | |
| R:
5′-GCACGAACACAGTCCCTTCAT-3′ | 178 |
| SNHG1 | F:
5′-ATGAAACAGCAGTTGAGGGT-3′ | |
| R:
5′-AAAAACAAAAGGGCAGGTAG-3′ | 102 |
| β-actin | F:
5′-GTGGCCGAGGACTTTGATTG-3′ | |
| R:
5′-CCTGTAACAACGCATCTCATATT-3′ | 73 |
Gene Ontology (GO) enrichment and pathway
analysis
The GO project (http://www.geneontology.org) provides a controlled
vocabulary to describe gene and gene product attributes (14). GO categories were considered as
significantly enriched only if the Fisher's exact probability
(P-value) was <0.05. Pathway analysis is a functional analysis
mapping genes to KEGG pathways (15). The P-value (EASE-score, Fisher
P-value or hypergeometric P-value) denotes the significance of the
pathway correlated to the conditions. The lower the P-value, the
more significant the pathway.
Statistical analysis
Agilent Feature Extraction software (version
11.0.1.1; Agilent Technologies) was used to analyze acquired array
images. Quantile normalization and subsequent data processing were
performed with using the GeneSpring GX v12.1 software package
(Agilent Technologies). Following quantile normalization of the raw
data, lncRNAs and mRNAs that at least 2 out of 4 samples have flags
in Present or Marginal ('All Targets Value') were selected for
further data analysis. Differentially expressed lncRNAs and mRNAs
with statistical significance between the 2 groups were identified
through P-value/FDR filtering and fold change filtering.
Hierarchical clustering and combined analysis were performed using
homemade scripts.
The data were analyzed using the SPSS 20.0 software
package (SPSS Inc., Chicago, IL, USA). Differential expression
levels of lncRNAs were compared by an independent-samples t-test
between 2 groups. Fisher's exact test was used in GO and pathway
analyses. All values are expressed as the means + SEM. All
experiments were repeated at least 3 times. A value of P<0.05
was considered to indicate a statistically significant
difference.
Results
Profile of microarray data
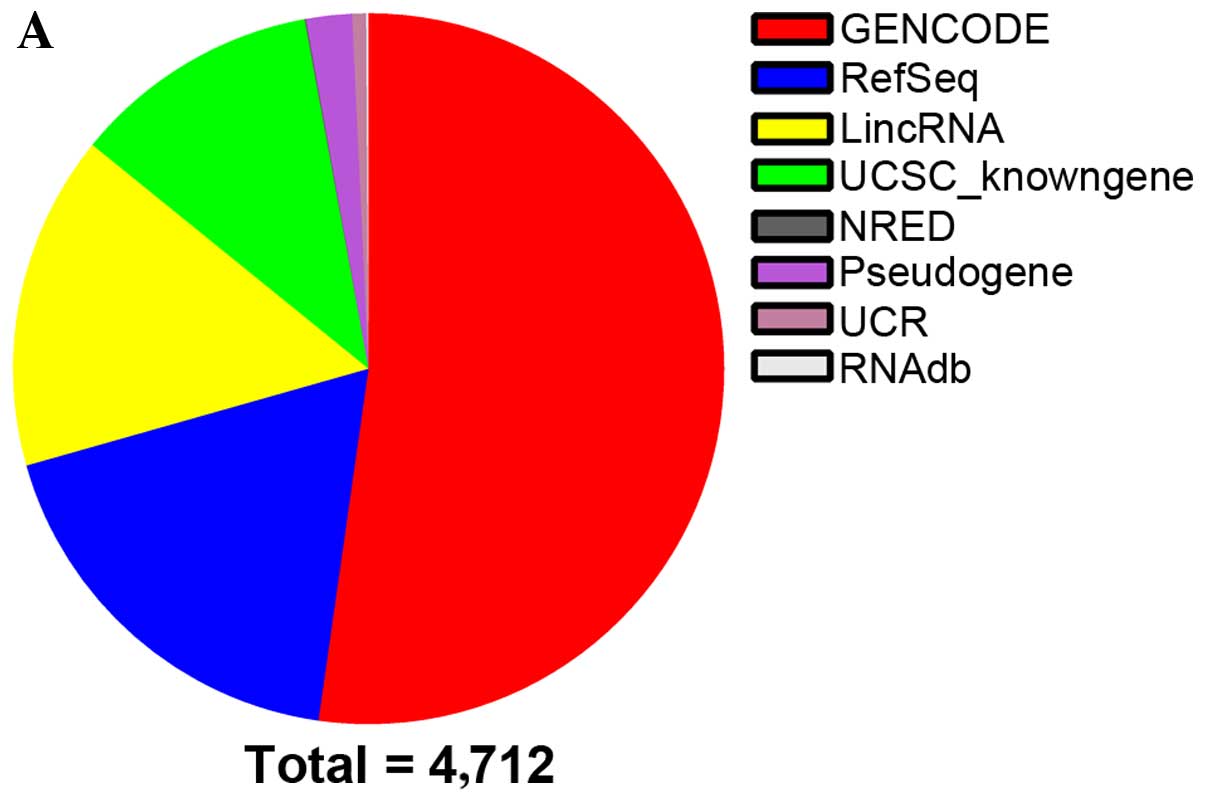
According to microarray expression profiling data,
25,770 lncRNAs were detected. In addition, 4,712 lncRNAs (log fold
change >2.0) were found to be differentially expressed between
the pterygium samples and the paired adjacent normal conjunctival
tissue samples (Table III and
IV). All these lncRNAs were
obtained from authoritative databases,such as RefSeq,
UCSC_Knowngenes, Ensembl and other related databases (Fig. 1A). Hierarchical Cluster analysis
revealed lncRNA expression patterns (Fig. 1B). A scatter plot was used for
assessing variations in lncRNA expression between the 2 groups of
pterygium samples and adjacent normal conjunctival samples
(Fig. 1C). A total of 3,066
upregulated and 1,646 downregulated lncRNAs were identified in the
pterygium tissues compared with the paired adjacent normal
conjunctival tissues. A volcano plot filtering between the 2 groups
initially revealed differentially expressed lncRNAs with
statistical significance (fold change >2.0, P<0.05) (Fig. 1D).
| Table IIINumbers of significantly
differentially expressed lncRNAs. |
Table III
Numbers of significantly
differentially expressed lncRNAs.
| | Fold change 2–5 | Fold change 5–10 | Fold change
>10 |
|---|
| lncRNA | Upregulation | 1308 | 943 | 815 |
| Downregulation | 827 | 554 | 265 |
| Table IVTop 10 significantly differently
expressed lncRNAs. |
Table IV
Top 10 significantly differently
expressed lncRNAs.
| Seqname | GeneSymbol | Regulation | Fold-change |
|---|
| NR_026878 | FOXD2-AS1 | Up | 92.676596 |
|
ENST00000580781 | RP11-78F17.1 | Up | 88.7604945 |
|
ENST00000530941 | RP11-702F3.4 | Up | 82.0072128 |
|
ENST00000423020 | RP5-963E22.4 | Up | 75.080159 |
|
ENST00000549419 | RP11-611E13.2 | Up | 70.6282429 |
|
ENST00000445586 | AF196972.9 | Up | 66.0127502 |
|
ENST00000563678 | KIAA0664L3 | Up | 63.4678339 |
| NR_027074 | LOC283761 | Up | 61.2789639 |
|
ENST00000441203 | RP3-416J7.2 | Up | 58.0589993 |
| NR_024564 | LOC100130264 | Up | 56.0539117 |
| NR_024396 | LINC00638 | Down | 113.4657051 |
|
ENST00000510017 | WI2-2373I1.2 | Down | 104.5215055 |
| NR_024526 | ARL6IP6 | Down | 89.9350702 |
| NR_024602 | CLCA4 | Down | 68.6207695 |
| NR_038103 | TECR | Down | 57.4518562 |
|
ENST00000554583 | RP11-398J10.2 | Down | 54.8515259 |
|
ENST00000493239 | RP11-61L23.2 | Down | 48.1050017 |
|
ENST00000505133 | RP11-420A23.1 | Down | 46.1669142 |
|
ENST00000562356 | RP11-217B1.2 | Down | 43.4260703 |
|
ENST00000430751 | RP3-434O14.8 | Down | 38.9352585 |
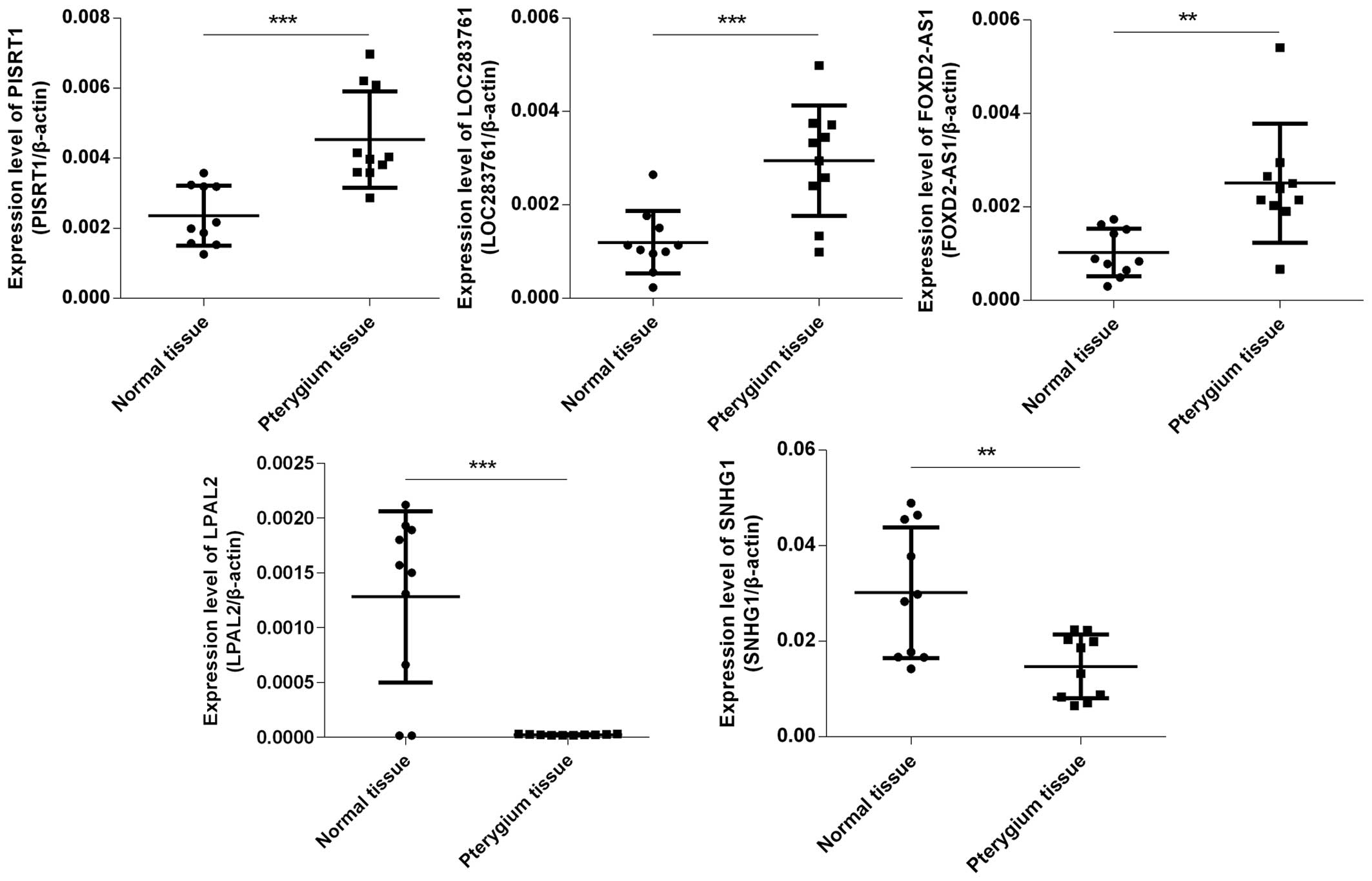
Confirmation by qPCR
By using qPCR, 3 upregulated lncRNAs [polled
intersex syndrome regulated transcript 1 (PISRT1), LOC283761 and
FOXD2 antisense RNA 1 (FOXD2-AS1)] and 2 downregulated lncRNAs
[lipoprotein, Lp(A)-like 2, pseudogene (LPAL2) and small nucleolar
RNA host gene 1 (SNHG1)] with log fold changes >10 were randomly
selected to test and verify the microarray data in 10 different
samples of pterygium tissues and paired adjacent normal
conjunctival tissues (Fig. 2).
The results of qPCR and microarray data analysis were consistent.
Thus, microarray data profiling indicated a series of lncRNAs which
were constantly differentially expressed between the pterygium
tissues and the paired adjacent normal conjunctival tissues.
Expression signatures of deregulated
lncRNAs between pterygium tissues and paired adjacent normal
conjunctival tissues
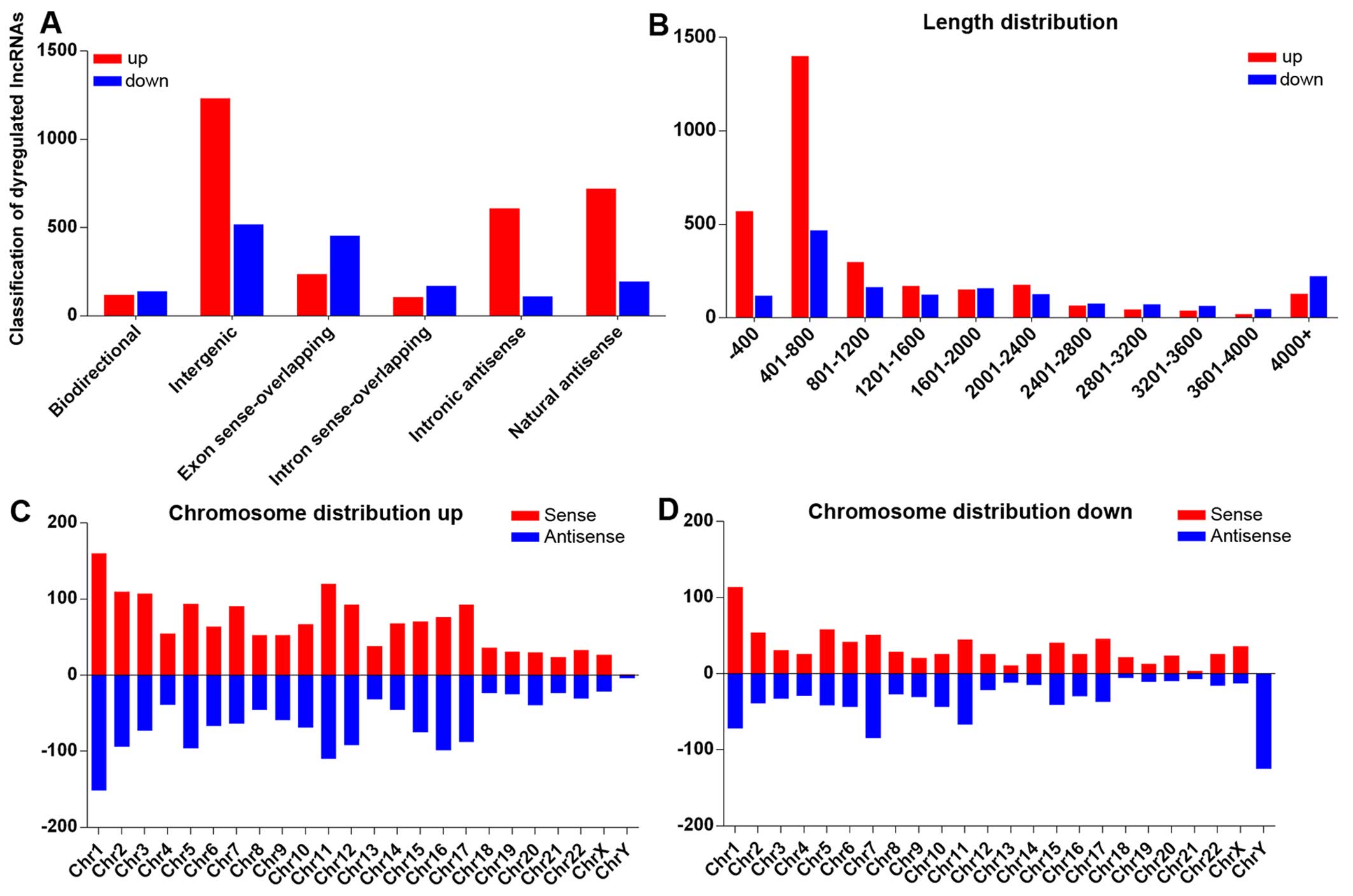
As lncRNA expression was tissue-specific, to further
study the lncRNA expression patterns in pterygium tissues, we first
investigated general signatures of deregulated lncRNAs which log
fold changes >2.0, including lncRNA classification, length
distribution and chromosome distribution. According to the lncRNA
position on the genome, we attributed lncRNA into 6 categories
(bidirectional, exon sense-overlapping, intergenic, intronic
antisense and natural antisense). Classification analysis revealed
that the majority of the lncRNAs in our microarray belong to
intergenic lncRNAs (Fig. 3A) and
are distributed 401–800 nt in length (Fig. 3B). Chromosome distribution shows
that upregulated and downregulated lncRNAs have different
chromosomal locations (Fig. 3C and
D). For the downregulated lncRNAs on the Y chromosome, all 125
transcripts were antisense, indicating that the mechanisms of the Y
chromosome involved in pterygium formation may differ from those of
other chromosomes.
GO analysis
GO analysis was performed to determine the gene and
gene product enrichment, which covered 3 domains: biological
processes, cellular components and molecular functions. Fisher's
exact test was used to determine whether the overlap between the
differentially expressed gene list and the GO annotation list was
greater than that expected by chance (a p-value <0.05 is
recommended). We found that the highest GO classification targeted
by over-regulated transcripts were single-organism process
(ontology:biological process), membrane (ontology:cellular
component) and protein binding (ontology:molecular function).
Furthermore, the highest GO classification targeted by the
downregulated transcripts were cellular process
(ontology:biological process), cell part (ontology:cellular
component) and binding (ontology:molecular function). To analyze
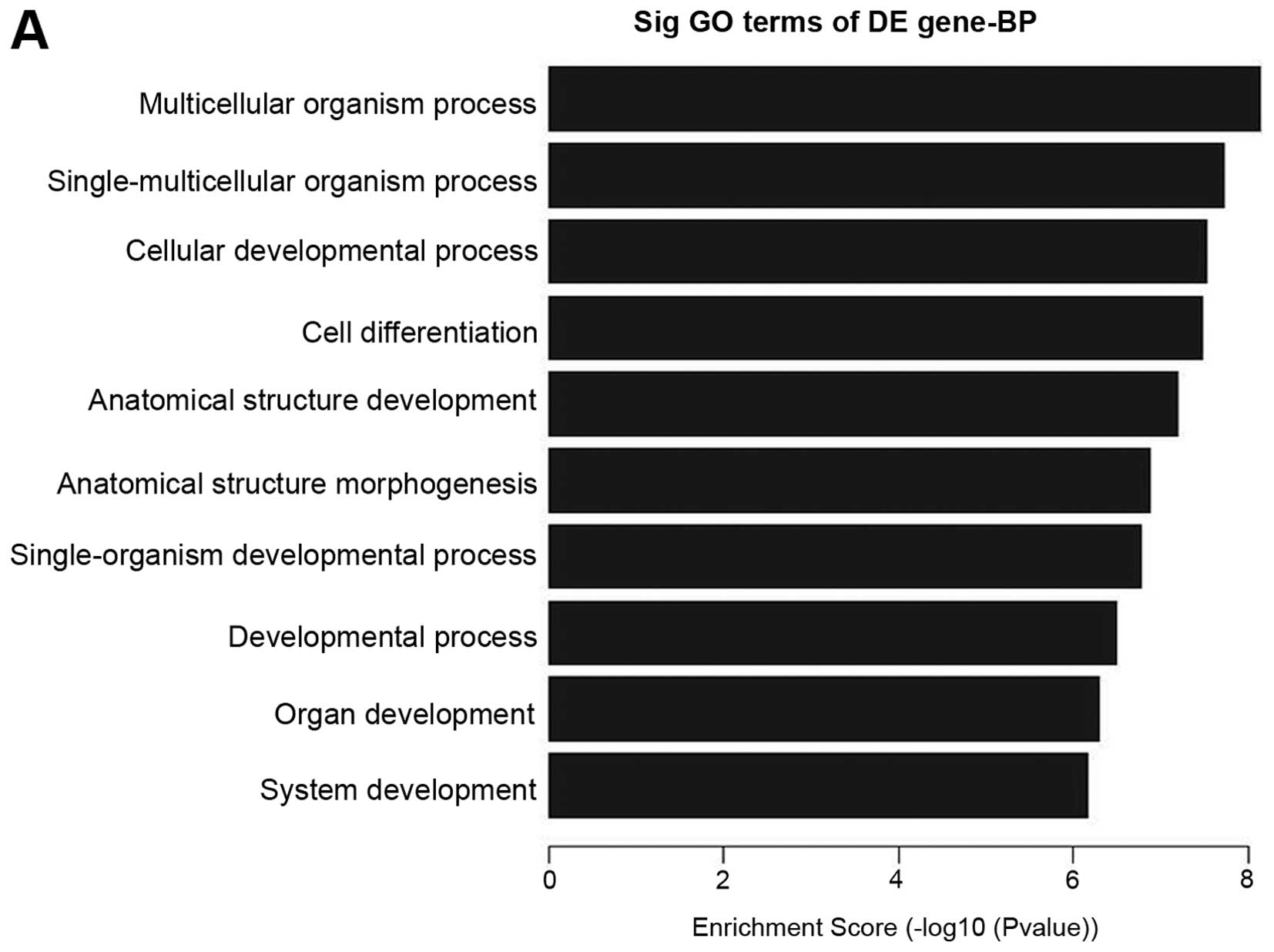
the candidate biological process of lncRNAs, we enriched the
biological process of both the up- and downregulated lncRNAs. It is
interesting that most biological processes of upregulated lncRNAs
are involved in proliferation and differentiation, such as
muticellular organismal process, single-multicellular organism
process, cellular developmental process, cell differentiation,
anatomical structure development, anatomical structure
morphogenesis, single-organism developmental process, developmental
process, organ development and system development, which shows that
upregulated lncRNAs in pterygium tissues have a tight association
with proliferation and differentiation (Fig. 4A). To the best of our knowlege, we
are the first to report that pterygium-related lncRNAs are
associated with proliferation and differentiation by microarray
analysis. On the other hand, we found that the biological process
of downregulated lncRNAs was enriched in cellular metabolic
process, cellular macromolecule metabolic process and other
metabolic processes (Fig.
4B).
Pathway analysis
We performed pathway analysis by mapping genes to
KEGG pathways. Pathway analysis indicated that 82 pathways
corresponded to downregulated transcripts and 15 pathways
corresponded to upregulated transcripts. The top 10 pathways that
were associated with upregulated and downregulated lncRNAs were
both analyzed (Fig. 4C and D).
The top pathways that were associated with the upregulated lncRNAs
were enriched in the category 'hypertrophic cardiomyopathy',
closely related with proliferation. Furthermore, the majority of
the pathways that were associated with the downregulated lncRNAs
were related to tumorigenesis, such as proteoglycans in cancer,
colorectal cancer and prostate cancer. This analysis revealed that
lncRNAs may play a key role in proliferation and may be involved in
the pterygium developmental process.
As we found that upregulated lncRNAs may be
associated with proliferation and the downregulated lncRNAs may
influence pterygium development, we selected upregulated lncRNAs
with a log fold change >2.0 in pterygium and the paired adjacent
normal conjunctival tissues and their associated coding genes with
a function of proliferation; we also selected downregulated lncRNAs
with a log fold change >2.0 and their associated coding genes
with a function of apoptosis.
Discussion
Pterygium has been considered a degenerative and
proliferative process of the corneal limbus and is characterized by
the invasion of a fleshy triangle of conjunctival tissue onto the
cornea (16). The condition is
associated with chronic ultraviolet radiation exposure and is
characterized by the induction of cell proliferation, squamous
metaplasia, goblet cell hyperplasia, inflammation, fibrosis,
angiogenesis and extracellular matrix breakdown (8). Indeed, pterygium tissue shares the
characteristics of many premalignant tissues, including epithelial
proliferation, goblet cell hyperplasia, angiogenesis, inflammation
and elastosis (3,17). However, the mechanisms responsible
for pterygium formation have not been fully clarified. This is
critical to determine the expression profiles of lncRNAs in
pterygium and to understand its pathogenesis.
Recent microarray analysis has demonstrated that
microRNAs (miRNAs) and their targeted mRNAs, are functionally
important in processes relevant to pterygium pathogenesis (1). The concerted upregulation of miR-221
is known to regulate downstream p27Kip1 gene expression (16), and evidence has indicated that the
miR-200 family is a potential regulator of EMT in pterygium
(1).
Mounting evidence has indicated that numerous
lncRNAs play roles in multiple biological processes and regulate
different diseases. Of all the functions of lncRNAs, the most
important is their involvement in tumorigenesis, which has been
shown in different cancer cells (18,19). The prolifera tive capacities of
pterygial cells have a mechanism similar to tumorigenesis. However,
the roles of pterygium pathogenesis remain unclear.
In the present study, we investigated the lncRNA
expression profiles of pterygium using microarray analysis and
found that the lncRNA expression levels were altered compared to
adjacent normal conjunctival tissues. From the microarray
expression profiles, we found that 3,066 upregulated and 1,646
downregulated lncRNAs were significantly differentially expressed
(>2.0-fold) in all pterygium samples. Five lncRNAs were
evaluated by qPCR to validate the consistency. The qPCR results and
microarray data were consistent. Furthermore, we utilized GO
analysis, pathway analysis and constructed a co-expression network
to preliminarily examine the biological functions of these lncRNAs
in the development of pterygium.
For pterygium, genetic mutations in development
genes are common, as observed in other proliferative diseases, such
as cancers. An over-regulated lncRNA, PISRT1, was found to be
located near mitochondrial ribosomal protein S22 (MRPS22), which
interacts with p53 and is considered a potential driver gene whose
targets were shown to be involved in DNA replication, cell cycle,
mismatch repair, p53 signaling pathway and other lung cancer
related signaling pathways, and many immunological pathways
(20). An intronic antisense
relationship between the over-regulated lncRNA, PISRT1, and MRPS22
may help us learn more about the association between pterygium and
lncRNAs at the transcriptional level.
To understand the functions of lncRNAs, we applied
pathway analysis to study the differentially expressed lncRNAs and
found that 82 pathways corresponded to the downregulated
transcripts and 15 pathways corresponded to the upregulated
transcripts. Chronic inflammation is a critical process involved in
the development and progression of pterygium, including the
promotion of angiogenesis (7,21,22). Pterygium research has uncovered
multiple pro-inflammatory genes that are activated in pterygium
tissue, including the pro-inflammatory transcription factor,
nuclear factor-κB (NF-κB) (22),
and several cytokines, including tumor necrosis factor-α (TNF-α)
(23). Some of those pathways
include the NF-κB signaling pathway and TNF signaling pathway,
which is consistent with previous research. Moreover, one of these
pathways, the 'Focal adhesion' signaling pathway has shown to
modulate tumor signatures (24).
These pathways were associated with outcomes for pterygium. In this
study, 97 pathways corresponding to differentially expressed
transcripts were found, and some or all may be involved in the
pathogenesis of pterygium.
In conclusion, to the best of our knowledge, in this
study, we report for the first time that lncRNAs are differentially
expressed in pterygium compared with paired adjacent normal
conjunctival tissues. Some of the changes identified in this study
suggest new directions in the formation of pterygium. In the
future, we will expand the number of enrolled patients, and further
studies are warranted applying immunofluorescence, proteomics or
protein level quantification of the lncRNA targets in patients with
pterygium staged according to the severity level. Through further
investigation, these aberrant lncRNAs may be biomarkers, molecular
targets and prognostic markers in the medical treatment of
pterygium.
Acknowledgments
The authors wish to thank Dr Chun Lu for his
technical assistance and critical review of this manuscript.
References
|
1
|
Engelsvold DH, Utheim TP, Olstad OK,
Gonzalez P, Eidet JR, Lyberg T, Trøseid AM, Dartt DA and Raeder S:
miRNA and mRNA expression profiling identifies members of the
miR-200 family as potential regulators of epithelial-mesenchymal
transition in pterygium. Exp Eye Res. 115:189–198. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Al-Swailem S, Xu Z, Wu L, Hartsock MJ, Yiu
SC and Duh EJ: Induction of endothelial RAGE expression in
pterygium. Mol Vis. 20:1740–1748. 2014.
|
|
3
|
Chui J, Coroneo MT, Tat LT, Crouch R,
Wakefield D and Di Girolamo N: Ophthalmic pterygium: A stem cell
disorder with premalignant features. Am J Pathol. 178:817–827.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hirst LW, Axelsen RA and Schwab I:
Pterygium and associated ocular surface squamous neoplasia. Arch
Ophthalmol. 127:31–32. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ando R, Kase S, Ohashi T, Dong Z, Fukuhara
J, Kanda A, Murata M, Noda K, Kitaichi N and Ishida S: Tissue
factor expression in human pterygium. Mol Vis. 17:63–69.
2011.PubMed/NCBI
|
|
6
|
Kato N, Shimmura S, Kawakita T, Miyashita
H, Ogawa Y, Yoshida S, Higa K, Okano H and Tsubota K: Beta-catenin
activation and epithelial-mesenchymal transition in the
pathogenesis of pterygium. Invest Ophthalmol Vis Sci. 48:1511–1517.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bradley JC, Yang W, Bradley RH, Reid TW
and Schwab IR: The science of pterygia. Br J Ophthalmol.
94:815–820. 2010. View Article : Google Scholar
|
|
8
|
Chui J, Di Girolamo N, Wakefield D and
Coroneo MT: The pathogenesis of pterygium: Current concepts and
their therapeutic implications. Ocul Surf. 6:24–43. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rinn JL and Chang HY: Genome regulation by
long noncoding RNAs. Annu Rev Biochem. 81:145–166. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ponting CP, Oliver PL and Reik W:
Evolution and functions of long noncoding RNAs. Cell. 136:629–641.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yan B, Wang ZH, Liu JY, Tao ZF, Li XM and
Qin J: Long noncoding RNAs: Versatile players in biologcial
processes and human disorders. Epigenomics. 6:375–379. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hung T and Chang HY: Long noncoding RNA in
genome regulation: Prospects and mechanisms. RNA Biol. 7:582–585.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang KC and Chang HY: Molecular mechanisms
of long noncoding RNAs. Mol Cell. 43:904–914. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Huntley RP, Harris MA, Alam-Faruque Y,
Blake JA, Carbon S, Dietze H, Dimmer EC, Foulger RE, Hill DP,
Khodiyar VK, et al: A method for increasing expressivity of Gene
Ontology annotations using a compositional approach. BMC
Bioinformatics. 15:1552014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Khatri P, Sirota M and Butte AJ: Ten years
of pathway analysis: Current approaches and outstanding challenges.
PLoS Comput Biol. 8:e10023752012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wu CW, Cheng YW, Hsu NY, Yeh KT, Tsai YY,
Chiang CC, Wang WR and Tung JN: MiRNA-221 negatively regulated
downstream p27Kip1 gene expression involvement in pterygium
pathogenesis. Mol Vis. 20:1048–1056. 2014.PubMed/NCBI
|
|
17
|
Tung JN, Chiang CC, Tsai YY, Chou YY, Yeh
KT, Lee H and Cheng YW: CyclinD1 protein expressed in pterygia is
associated with β-catenin protein localization. Mol Vis.
16:2733–2738. 2010.PubMed/NCBI
|
|
18
|
Wang J, Liu X, Wu H, Ni P, Gu Z, Qiao Y,
Chen N, Sun F and Fan Q: CREB upregulates long non-coding RNA, HULC
expression through interaction with microRNA-372 in liver cancer.
Nucleic Acids Res. 38:5366–5383. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Huang JL, Ren TY, Cao SW, Zheng SH, Hu XM,
Hu YW, Lin L, Chen J, Zheng L and Wang Q: HBx-related long
non-coding RNA DBH-AS1 promotes cell proliferation and survival by
activating MAPK signaling in hepatocellular carcinoma. Oncotarget.
6:33791–33804. 2015.PubMed/NCBI
|
|
20
|
Lazar V, Suo C, Orear C, van den Oord J,
Balogh Z, Guegan J, Job B, Meurice G, Ripoche H, Calza S, et al:
Integrated molecular portrait of non-small cell lung cancers. BMC
Med Genomics. 6:532013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Detorakis ET and Spandidos DA:
Pathogenetic mechanisms and treatment options for ophthalmic
pterygium: Trends and perspectives (Review). Int J Mol Med.
23:439–447. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mauro J and Foster CS: Pterygia:
Pathogenesis and the role of subconjunctival bevacizumab in
treatment. Semin Ophthalmol. 24:130–134. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hong S, Choi JY, Lee HK, Seong GJ, Seo KY,
Kim EK and Byeon SH: Expression of neurotrophic factors in human
primary pterygeal tissue and selective TNF-alpha-induced
stimulation of ciliary neurotrophic factor in pterygeal
fibroblasts. Exp Toxicol Pathol. 60:513–520. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
O'Donoghue LE, Ptitsyn AA, Kamstock DA,
Siebert J, Thomas RS and Duval DL: Expression profiling in canine
osteosarcoma: Identification of biomarkers and pathways associated
with outcome. BMC Cancer. 10:5062010. View Article : Google Scholar : PubMed/NCBI
|