Introduction
Ischemic brain injury (IBI) is a complex pathology
that leads to inflammation (1),
apoptosis (2) and excitotoxicity
(3) and causes neuronal damage.
Excessive extracellular glutamate is released in brain ischemia. As
one of the neurotransmitters, high-level glutamate causes
excitotoxicity and cell death (4). Glutamate-mediated excitotoxicity is
rescued by glutamate transporters (5), such as glial glutamate transporter-1
(GLT-1).
GLT-1 is a transporter that can regulate glutamate
homeostasis in the extracellular fluid, which lacks the enzyme to
catabolize glutamate (6). GLT-1
is so named due to its main distribution on glial cells. GLT-1 can
remove excess glutamate and reduce excitotoxicity (7). Over 90% of glutamate uptake is
carried out by GLT-1 in the adult forebrain (6). GLT-1 knockdown in the hippocampal
CA1 region has been shown to lead to high levels of extracellular
glutamate (8). A study showed
that ischemia-reperfusion led to a significant decrease in GLT-1 in
the hippocampal CA1 region (9).
The excitotoxicity caused by the decrease in GLT-1 levels can be
reversed by cerebral ischemic preconditioning and transient
sublethal cerebral ischemia, which protects neurons against brain
ischemic damage (10). There is
evidence to suggest that the upregulation of GLT-1 results in
neuroprotection against excitotoxicity and ischemic injury
(11–13). In addition, a high level of GLT-1
reduces the activity of N-methyl-D-aspartate (NMDA) receptors in
morphine-induced analgesia in rats (14). The excessive activation of NMDA
leading to apoptosis and excitotoxicity underlies normal synaptic
plasticity (15). These findings
indicate that NMDA-induced excitotoxicity plays a critical role in
neuronal damage, such as ischemia-reperfusion insults (16). The effect of NMDA-induced
excitotoxicity can be attenuated by the upregulation of GLT-1
(17).
Propofol, as one of the common anesthetics, is
widely used in clinical settings. The neuroprotective effects of
propofol have previously been demonstrated (18,19). Hypoxia-induced hippocampal neuron
injury was also attenuated by propofol (20). A recent study demonstrated that
propofol upregulates the expression of GLT-1, which leads to a
decrease in the concentration of glutamate in the hippocampus of
depressed rats (21). In a study
on arterioles, propofol reduced both the dilatation and superoxide
production caused by NMDA (22).
In another study, NMDA-induced excitotoxicity was attenuated by
treatment with propofol in cultured rat cortical neurons (23).
However, it remains unclear as to whether propofol
protects hippocampal neurons by upregulating GLT-1 expression in
IBI. In the present study, we explored the underlying mechanisms
and the effect of propofol on hippocampal neurons in IBI.
Materials and methods
Primary hippocampal neuronal culture
The use of all animals in the experiments conformed
to the guidelines of the National Council on Animal Care. All
experiments were conducted in accordance with the rules of the
Committee on the Ethics of Animal Experimentation of the First
Affiliated Hospital of Xinxiang Medical University, Weihui, China.
Pregnant Wistar rats (n=20; the Laboratory Animal Center of the
First Affiliated Hospital of Xinxiang Medical University, Henan,
China) were sacrificed by cervical dislocation, and 15–17 day
embryos (E15–E17) were removed and the brains were rapidly
collected, as previously described (24,25). These hippocampal tissues were
dissociated in 0.125% trypsin-EDTA solution for 20 min at 37°C in
an incubator. The cells were collected by centrifugation. The cells
seeded on 6-well dishes pre-coated with poly-L-lysine and chamber
slides were cultured in a 5% CO2 incubator at 37°C at a
density of 1×106/ml, and supplied with 1% glutamine, 10%
heat-inactivated fetal bovine serum, 10% horse serum, 1% penicillin
and streptomycin. After 24 h, 100 mM cytosine arabinoside was added
to the culture medium to inhibit neuroglial cells. The culture
medium supplied with 2% B27 and 1% N2, was changed every 3 days.
All reagents were purchased from Sigma (St. Louis, MO, USA).
Culture cells were used after 7 days in vitro.
Establishment of model of IBI by exposure
to hypoxia
After 7 days of normoxic cultivation (20% oxygen),
the culture conditions of the hippocampal neurons were changed to
5% CO2 and 95% N2 at 37°C for 3 h (26) to establish the model of IBI. After
2 h of incubation under hypoxic conditions, the hippocampal
neuronal cells were treated with propofol (Sigma; 15 µM) and
either pre-incubated (or not) with inhibitors of PI3-kinase
(LY294002; Sigma; 5 µM) or anisomycin (Sigma; 5 µM)
followed by culture for 1 h.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
The hippocampal neuronal cells cultured for 7 days
were used for the detection of the mRNA levels of GLT-1. The RNeasy
mini kit (Qiagen, Hilden, Germany) and PrimeScript™ RT reagent kit
(Takara Biotechnology, Dalian, China) were used for total RNA
extraction, reverse transcription and PCR amplification.
Gene-specific primers for GLT-1 (forward,
5′-AGTATGTGGCGGGCTGCTTC-3′ and reverse,
5′-GGAAATGATGAGAGGGAGGATGAG-3′); NR1 (forward,
5′-GCACGCCTTTATCTGGGACTC-3′ and reverse,
5′-GTCGGGCTCTGCTCTACCACT-3′); NR2A (forward,
5′-GCTACACACTCTGCACCAATT-3′ and reverse, 5′-CACCTGATAGCCTTCCTCAG
TGA-3′); NR2B (forward, 5′-TCCGTCTTTCTTATGTGGATATGC-3′ and reverse,
5′-CCTCTAGGCGGACAGATTAAGG-3′) and the internal control primers for
glyceraldehyde 3-phosphate dehydro-genase (GAPDH) (forward,
5′-GCCAAAAGGGTCATCATCT CTG-3′ and reverse,
5′-CATGCCAGTGAGCTTCCCGT-3′) were synthesized by Sangon Biotech
(Sangon, Shanghai, China). The mRNA expression levels were
normalized to those of GAPDH and were calculated using the
2−ΔΔCt method, as previously described (27).
GLT-1 overexpression and silencing
cDNA for rat GLT-1 was amplified by PCR. The
obtained GLT-1 cDNA was subcloned into the pIRES vector (Invitrogen
Life Technologies, Carlsbad, CA, USA) to generate the
overexpression vector of GLT-1, named GLT-1-pIRES. The GLT-1-pIRES
vector was transfected into the hippocampal neurons pre-incubated
(or not) with NMDA (Sigma; 5 µM) using Lipofectamine 2000
(Invitrogen) following the manufacturer's instructions. The empty
pIRES vector was used as a control. Small interfering RNA (siRNA)
against GLT-1 and a negative control siRNA were purchased from
Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). The siRNA
targeting GLT-1 were transfected into the hippocampal neurons using
Lipofectamine 2000 according to the manufacturer's instructions.
Six hours after transfection, the cells were used for the
subsequent experiments.
Cell viability assay
Hippocampal neuronal cell viability was assessed by
AlamarBlue (Sigma). The cells were plated onto 96-well plates at a
density of 1×106 cells/well and 10 µl AlamarBlue
reagent were added to each well followed by incubatiohn at 5%
CO2 and 37°C. After 4 h, the percentage reduction in
light absorption at 570 and 600 nm was detected in each well for
oxidized and reduced AlamarBlue using a microplate reader (Thermo
Fisher Scientific, Waltham, MA, USA). The experiment was performed
at least 3 times. Results were calculated as previously described
(28).
Cell apoptosis assay
The Annexin V-FITC/propidium iodide (PI) apoptosis
detection kit (Sigma) was used to detect cell apoptosis using a
flow cytometer (Gibco, Rockville, MD, USA). Briefly, the
hippocampal neuron cell medium plated in 6-well dishes was removed
and trypsin was added. The cells were then washed twice with
phosphate-buffered saline (PBS). Annexin V-FITC and PI were added
and the cells were incubated for 30 min at 37°C. The stained
neurons were analyzed by flow cytometry and the rate of cell
apoptosis was determined.
Western blot analysis
Total proteins were extracted from the cultured
hippocampal neuronal cells in RIPA lysis buffer (Sigma). The BCA
protein assay kit (Beyotime Institute of Biotechnology, Haimen,
China) was used to measure the protein concentration using a
microplate reader (Thermo Fisher Scientific, Uppsala, Sweden).
Protein (20 µg) from each sample was separated by 12% sodium
dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gel
and transferred to a nitrocellulose membrane. Primary antibodies
(Abcam, Cambridge, UK) against GLT-1 (Cat. no. ab178401), Jun
N-terminal kinase (JNK; Cat. no. ab179461), p-JNK (Cat. no.
ab76572), p38 (Cat. no. ab31828), p-p38 (Cat. no. ab4822),
extracellular signal-regulated kinase 1/2 (ERK1/2; Cat. no.
ab54230), p-ERK1/2 (Cat. no. ab47339), Akt (Cat. no. ab8805), p-Akt
(Cat. no. ab38449), NR1 (Cat. no. ab17345), NR2A (Cat. no.
ab14596), NR2B (Cat. no. ab28373) and β-actin (Cat. no. ab8226)
were used to detect the protein levels. The horseradish peroxidase
(HRP)-labeled secondary antibodies were purchased from Abcam.
β-actin was utilized as an internal control. The gray value was
analyzed using ImageJ software.
Statistical analysis
All results are reported as the means ± SD.
Two-group comparisons were analyzed using the Student's t-test. A
value of P<0.05 was considered to indicate a statistically
significant difference.
Results
Neuroprotective effects of propofol on
hippocampal neurons
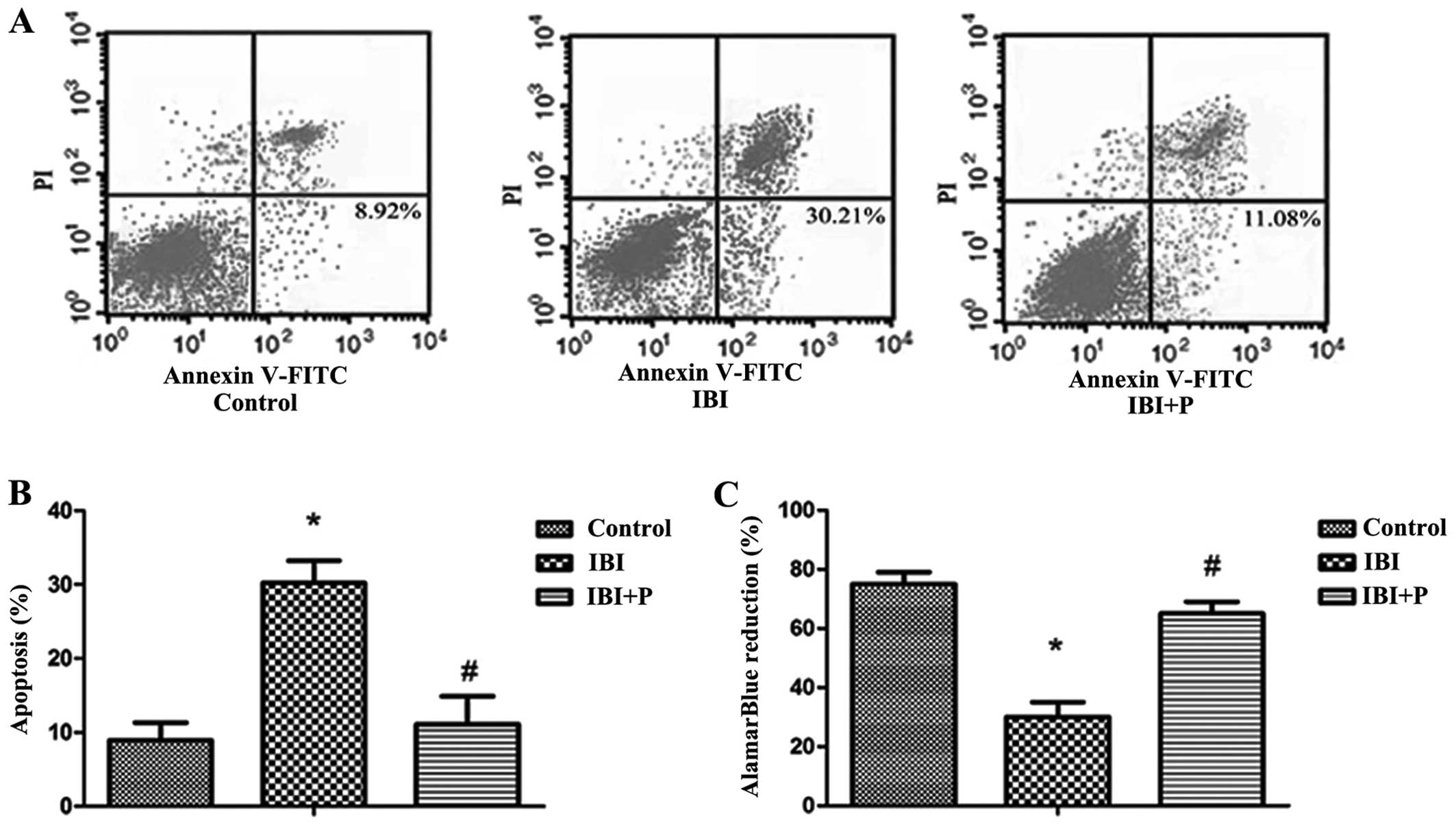
To investigate the effects of propofol on IBI, a
model of IBI was established using primary hippocampal neurons. We
found that exposure to hypoxia induced a significant decrease in
cell viability and an increase in cell apoptosis in the IBI group
compared with the control group (P<0.05; Fig. 1). The increase in cell apoptosis
and the decrease in cell viability induced by IBI were
significantly attenuated by treatment with propofol (P<0.05;
Fig. 1B and C).
Propofol protects hippocampal neurons in
IBI via the upregulation of GLT-1
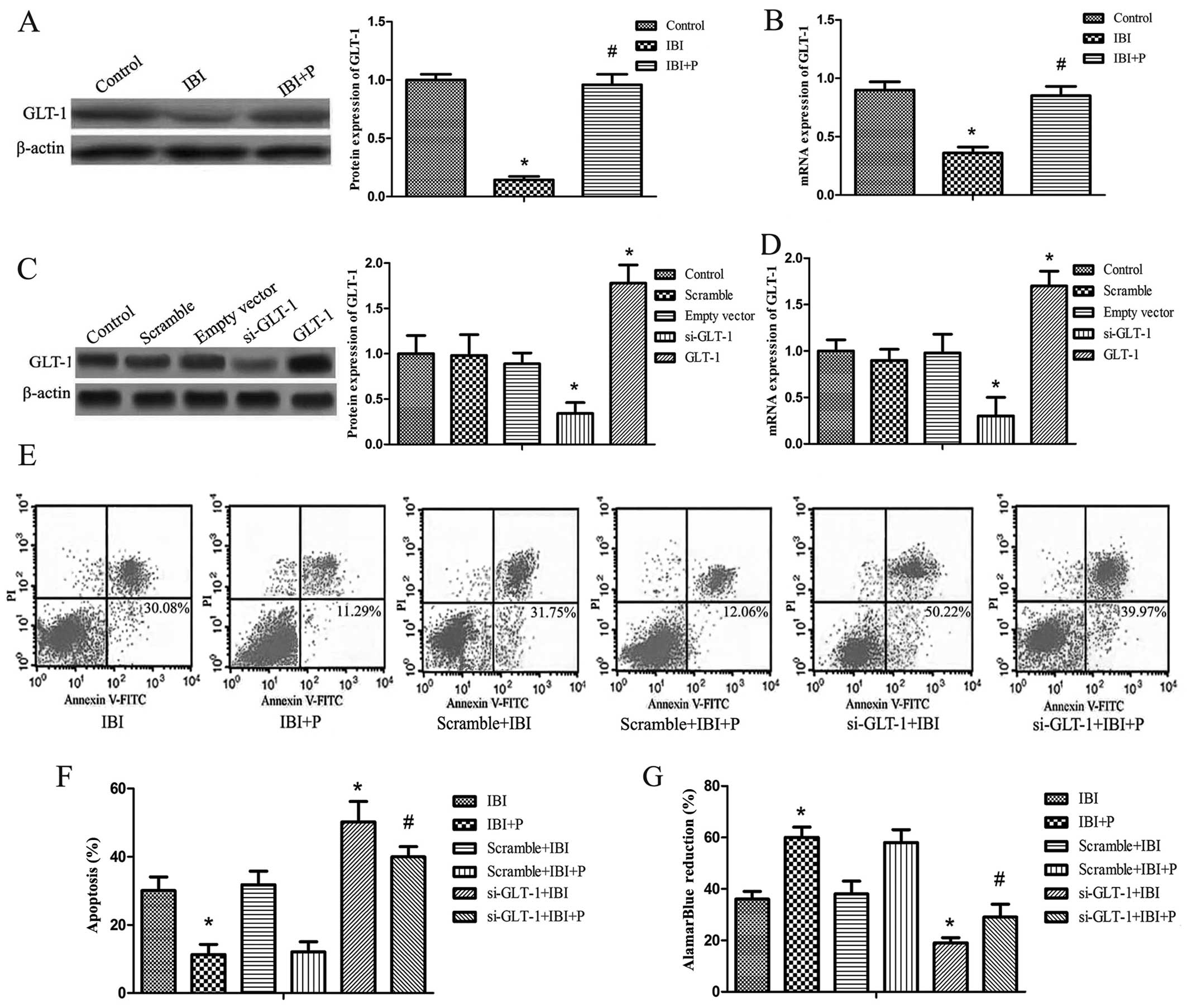
To determine whether propofol protects hippocampal
neurons in IBI via the upregulation of GLT-1, western blot analysis
and RT-qPCR were performed. It was found that the protein and mRNA
expression levels of GLT-1 were significantly decreased in the IBI
group compared with the control group (P<0.05; Fig. 2A and B). The decrease in GLT-1
expression caused by IBI was significantly attenuated by treatment
with propofol (IBI + P group) compared with the IBI group
(P<0.05; Fig. 2A and B). GLT-1
silencing by siRNA and the overexpression of GLT-1 was established
by transfection with GLT-1-pIRES vector or siRNA against GLT-1. The
efficiency of GLT-1 overexpression and silencing was detected by
western blot analysis and RT-qPCR. The results revealed that the
protein expression of GLT-1 correlated with its mRNA expression
(Fig. 2C and D). The protein and
mRNA expression of GLT-1 did not differ significantly among the
control, scramble and empty vector groups (Fig. 2C and D). The expression of GLT-1
was significantly decreased in the si-GLT-1 group compared with the
control group (P<0.05) and it was increased in the GLT-1 group
compared with the control group (P<0.05; Fig. 2C and D). Cell apoptosis and
viability were examined in the hippocampal neurons following GLT-1
silencing under hypoxic conditions. As shown in the dual-parameter
fluorescent dot plots and bar graph, cell apoptosis was not
affected by transfection with control siRNA (Scramble + IBI group
or the Scramble + IBI + P group compared with the IBI and IBI + P
groups, respectively). The knockdown of GLT-1 markedly increased
the apoptotic rate (si-GLT-1 + IBI and si-GLT-1 + IBI + P groups
compared with the IBI and IBI + P groups, respectively) (P<0.05;
Fig. 2E and F). Propofol did not
attenuate the increase in the apoptotic rate when GLT-1 was knocked
down (Fig. 2E and F). In the cell
viability assay, treatment with propofol yielded similar results to
those of the apoptosis assay (Fig.
2G). The si-GLT-1 + IBI and si-GLT-1 + IBI + P groups exhibited
markedly reduced cell survival rates compared with the IBI and IBI
+ P groups, respectively (P<0.05; Fig. 2G). The effect of GLT-1 knockdown
was slightly reversed by propofol (Fig. 2G).
JNK/Akt signaling pathway is involved in
propofol-mediated neuroprotection
Our findings revealed that propofol protected the
nerve cells against ischemic injury via the upregulation of the
expression of GLT-1. To explore the potential signaling pathways
through which propofol upregulated GLT-1 expression in our model of
IBI in hippocampal neuronal cells, the expression of JNK, p-JNK,
p38, p-p38, ERK1/2, p-ERK1/2, Akt and p-Akt was detected by western
blot analysis (Fig. 3A). It was
found that the expression of p38, ERK1/2, JNK and Akt exhibited no
obvious change between the groups, whereas the phosphorylation
levels of p38, ERK1/2, JNK and Akt exhibited marked differences in
some groups (Fig. 3B–E). The
levels of p-p38, p-ERK1/2 and p-JNK in the IBI + P group were
significantly decreased when compared to the IBI group (P<0.05;
Fig. 3B–D), while the levels of
p-Akt were significantly increased in the IBI + P group compared
with IBI group (P<0.05; Fig.
3E). Following the knockdown of GLT-1, no significant
differences in the levels of p-p38 and p-ERK1/2 were observed
between the si-GLT-1 + IBI and IBI groups, and between the si-GLT-1
+ IBI + P and IBI + P groups (Fig. 3B
and C). However, GLT-1 knockdown significantly increased the
levels of p-JNK in the si-GLT-1 + IBI and si-GLT-1 + IBI + P groups
compared with IBI and IBI + P groups, respectively (P<0.05;
Fig. 3D). GLT-1 knockdown led to
a significant decrease in the levels of p-Akt in the si-GLT-1 + IBI
and si-GLT-1 + IBI + P groups compared with the IBI and IBI + P
groups, respectively (P<0.05; Fig.
3E). No significant differences in the levels of p-Akt were
observed between the si-GLT-1 + IBI and si-GLT-1 + IBI + P groups
(Fig. 3E).
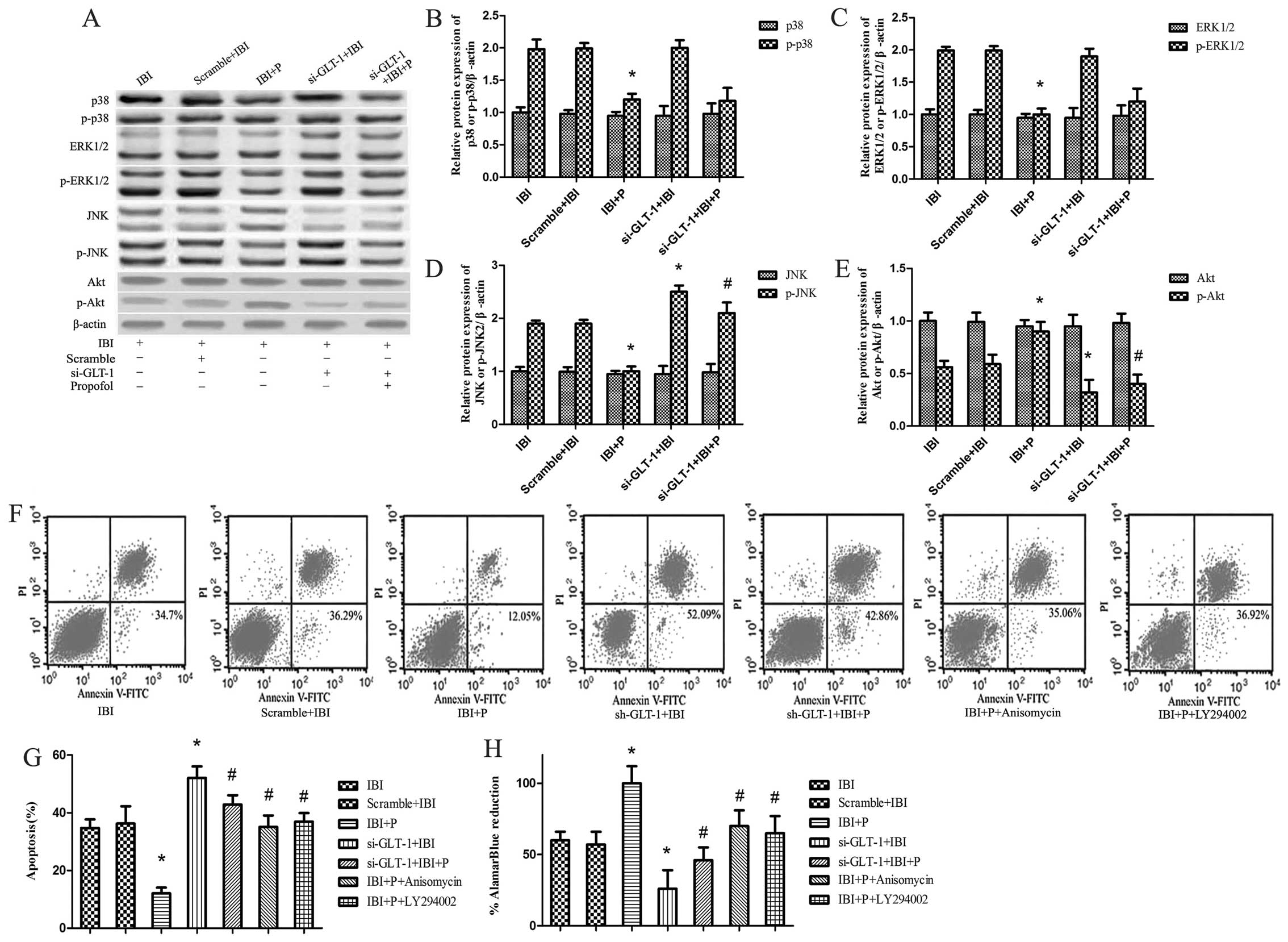 | Figure 3Signaling pathways of
propofol-mediated neuroprotection under glial glutamate
transporter-1 (GLT-1) knockdown conditions. Ischemic brain injury
(IBI) + P+ anisomycin group cells were treated with propofol and
the Jun N-terminal kinases (JNK) agonist, anisomycin, under hypoxic
conditions. IBI + P + LY294002 group cells were treated with
propofol and the Akt inhibitor, LY294002, under hypoxic conditions.
(A) The protein expression of ERK, p-ERK, p38, p-p38, JNK, p-JNK,
Akt and p-Akt was determined by western blot analysis with
corresponding antibodies. β-actin was utilized as an internal
control. (B-E) These data were calculated using ImageJ software.
(F) Cell apoptosis was detected in the presence of anisomycin and
LY294002 by Annexin V-FITC/propidium iodide (PI) in dual-parameter
fluorescent dot plots. (G) Quantitative graphs of (F). (H) Cell
viability was detected using AlamarBlue. *P<0.05 vs.
IBI group; #P<0.05 vs. IBI + P group. |
In order to further investigate the functional role
of JNK and Akt in propofol-mediated neuroprotection, we examined
the effects of the JNK agonist, anisomycin, and the Akt inhibitor,
LY294002, on cell viability and cell apoptosis. It was found that
the changes in cell viability and cell apoptosis did not differ
significantly between the IBI and Scramble + IBI groups (Fig. 3F–H). The increase in cell
apoptosis and the decrease in cell viability were significantly
attenuated by propofol in the IBI + P group compared with the IBI
group (P<0.05; Fig. 3G and H).
The decrease in cell apoptosis and the increase in cell viability
caused by propofol were attenuated by anisomycin and LY294002,
respectively (P<0.05; Fig. 3G and
H).
NMDAR plays a key role in
propofol-mediated neuroprotection
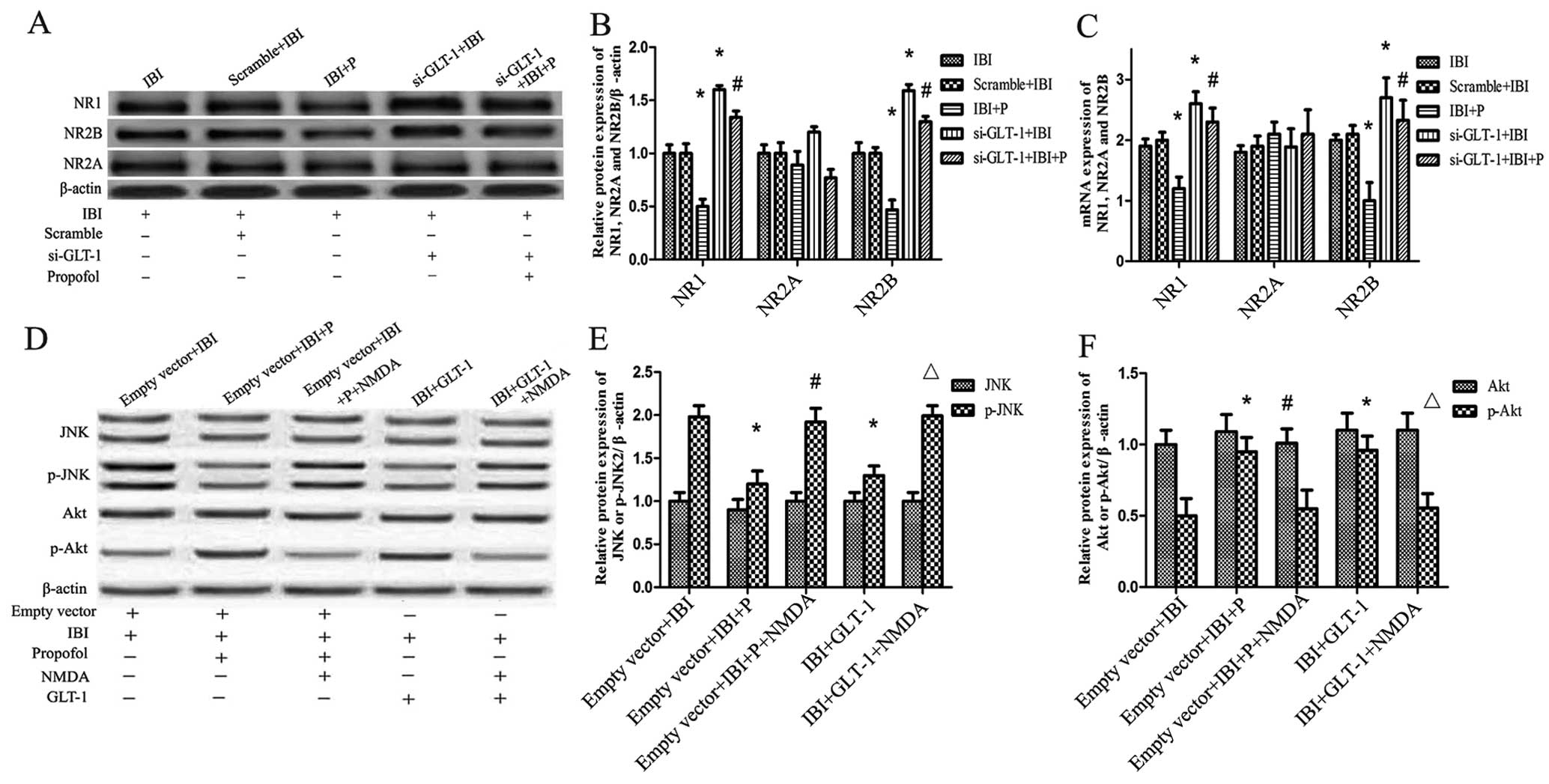
To explore the role of NMDAR in propofol-mediated
neuroprotection, the protein and mRNA expression levels of the
NMDAR subunits NR1, NR2A and NR2B were detected in each group by
western blot analysis and RT-qPCR. It was found that the protein
expression level of NMDAR correlated with its mRNA expression
level. There was a slight difference in the expression of NR2A
between the groups (Fig. 4A–C).
The protein and mRNA expression were coincident between the IBI and
Scramble+IBI groups (Fig. 4A–C).
Propofol significantly attenuated the increase in NR1 and NR2B
expression caused by IBI in the IBI + P group compared with the IBI
group (P<0.05; Fig. 4B and C).
This attenuation caused by propofol was significantly reversed by
GLT-1 knockdown (si-GLT-1 + IBI + P group compared with IBI + P
group) (P<0.05; Fig. 4B and
C). GLT-1 knockdown led to a significant increase in the levels
of NR1 and NR2B in the si-GLT-1 + IBI group compared with the IBI
group (P<0.05; Fig. 4A–C).
Propofol only slightly attenuated the increase in the levels of NR1
and NR2B caused by GLT-1 knockdown (si-GLT-1 + IBI + P group
compared with the si-GLT-1 + IBI group) (Fig. 4B and C). As regards the levels of
NR2A, no significant differences were observed between the
groups.
The activation of JNK and Akt was also detected in
the presence of NMDA. As shown in Fig. 4D–F, the high level p-JNK and the
low level p-Akt caused by IBI were significantly reversed by
treatment with propofol (empty vector + IBI + P group compared with
the empty vector + IBI group) (P<0.05). Treatment with NMDA
significantly increased the levels of p-JNK and decreased the
levels of p-Akt (empty vector + IBI + P + NMDA group compared with
the empty vector + IBI + P group) (P<0.05; Fig. 4E and F). The effects of GLT-1
overexpression on the levels of p-JNK and p-Akt were similar to
those observed with propofol treatment. GLT-1 overexpression led to
a decrease in p-JNK levels and an increase in p-Akt levels (IBI +
GLT-1 group compared with the IBI group) (P<0.05; Fig. 4E and F). NMDA significantly
increased the levels of p-JNK and decreased the levels of p-Akt
(IBI + GLT-1 + NMDA group compared with IBI + GLT-1 group)
(P<0.05; Fig. 4E and F).
Discussion
IBI is a complex pathology that causes disability
and dementia. Increasing evidence has indicated that IBI induces
excessive extracellular glutamate release, which causes
neurotoxicity and neuronal injury (4,29).
GLT-1 is one of the major glutamate transporters and GLT-1 can take
up 90% of glutamate, protecting against IBI (30,31). Propofol is a commonly used
clinical anesthetic. The neuroprotective effects of propofol have
been previously demonstrated (32). However, it remains unclear as to
whether propofol can regulate the expression of GLT-1 to protect
neuronal cells in IBI.
In this study, the effects of propofol on neuronal
cells in IBI were explored. The results revealed that hippocampal
neuronal injury induced by hypoxia was attenuated by propofol.
Consistent with our findings, the post-conditioning of rats with
transient middle cerebral artery occlusion by propofol was shown to
inhibit neuronal apoptosis in ischemia/reperfusion (33). Peroxynitrite-induced apoptosis in
astroglial cells has also been shown to be attenuated by propofol
treatment (34). An in
vitro study demonstrated that propofol exerts a neuroprotective
effect on hippocampal neurons against hypoxic damage (20). By contrast, a high dose propofol
(200 µM) has been shown to be cytotoxic in the neuroblastoma
SH-SY5Y cell line (35).
The expression of GLT-1 was reduced in hippocampal
neurons under hypoxic conditions. The decrease in GLT-1 expression
caused by hypoxia was attenuated by propofol treatment in
vitro. Similarly, propofol has been shown to attenuate the
decrease in GLT-1 expression in depressed rats (21). The reduced cell viability and
increased cell apoptosis caused by the downregulation of GLT-1 were
markedly reversed by propofol treatment. It has also been shown
that the knockdown of GLT-1 causes hippocampal excitotoxicity and
exacerbates hippocampal neuronal cell damage and mortality, which
are attenuated by propofol treatment (21,36). However, when ischemia is elongated
to a duration of 20 min, GLT-1 leads to glutamate release and
triggers neuronal death (8).
Potential signaling pathways were explored by
western blot nalysis. Previous studies have revealed that MAPK
signaling pathways, particularly JNK, are involved in
neuroprotection (37,38). The functional relatedness of
classical JNK-c-Jun signaling was restricted to the
hypoxic-ischemic and excitotoxic sets of effects (37). The MAPK family includes JNK, the
ERKs and p38 MAPK. Thus, the signaling pathway of p38, ERK1/2 and
JNK was explored in the present study. In addition, Akt as a
general signaling pathway in cell proliferation (39) and apoptosis (40) also was explored. The results of
western blot analysis revealed that the p38, ERK1/2, JNK and Akt
signaling pathways were involved in the propofol-mediated
neuroprotection, whereas the increase in p-JNK and the decrease in
p-Akt levels caused by GLT-1 knockdown were not attenuated by
propofol. Similarly, the strong expression of the c-Jun gene and
protein are known to precede or coincide with periods of intense
cell death during embryonic development (41). JNK inhibition attenuated neuronal
apoptosis in the developing rat brain after hypoxia-ischemia
(42) and the activation of Akt
decreased cell apoptosis (43).
These findings suggested that the increase in the p-Akt level and
the decrease in the p-JNK level were involved in neuroprotection
(44). In this study, the JNK
agonist, anisomycin, and the Akt, inhibitor LY294002, reversed the
increase in p-Akt and the decrease in p-JNK levels caused by
propofol treatment, suggesting that propofol protected hippocampal
neurons by upregulating the expression of GLT-1 partly via the
JNK/Akt signaling pathway.
A previous study showed that the excitotoxicity
caused by the activation of NMDAR led to neuronal death following
ischemia in some chronic neurodegenerative diseases (45). In our study, the excessive
activation of NMDAR (NR1 and NR2B) caused by IBI was attenuated by
propofol treatment. After GLT-1 knockdown, the effect of IBI on
NMDAR was not reversed by propofol and GLT-1 knockdown led to an
increase in NMDAR activation. Consistent with this study, it has
been previously demonstrated that NMDAR, as the downstream molecule
of GLT-1 (46), is possibly
involved in propofol-mediated neuroprotection. In this study, NMDA
reversed the decrease in p-JNK and the increase in p-Akt levels
caused by propofol and GLT-1 overexpression. This result suggested
that NMDA can induce JNK activation (15) and attenuate Akt activation
(47) in IBI. It is implied that
the NMDA receptor (NR1 and NR2B) could be the upstream molecule of
JNK signaling and Akt signaling and, at least in part, may be
involved in propofol-mediated neuroprotection.
In conclusion, the findings of our study suggest
that propofol upregulates GLT-1 and inhibits NMDAR to protect
hippocampal neuronal cells against IBI through the JNK/Akt
signaling pathway. All experiments were performed in vitro,
but the resulting details were not clear. We aim to perform in
vivo experiments in future studies.
References
|
1
|
Bayat M, Azami Tameh A, Hossein Ghahremani
M, Akbari M, Mehr SE, Khanavi M and Hassanzadeh G: Neuroprotective
properties of Melissa officinalis after hypoxic-ischemic injury
both in vitro and in vivo. Daru. 20:422012. View Article : Google Scholar
|
|
2
|
Gong G, Yuan L, Cai L, Ran M, Zhang Y,
Gong H, Dai X, Wu W and Dong H: Tetramethylpyrazine suppresses
transient oxygen-glucose deprivation-induced connexin32 expression
and cell apoptosis via the ERK1/2 and p38 MAPK pathway in cultured
hippocampal neurons. PLoS One. 9:e1059442014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chen H, Qu Y, Tang B, Xiong T and Mu D:
Role of mammalian target of rapamycin in hypoxic or ischemic brain
injury: Potential neuroprotection and limitations. Rev Neurosci.
23:279–287. 2012.PubMed/NCBI
|
|
4
|
Choi DW and Rothman SM: The role of
glutamate neurotoxicity in hypoxic-ischemic neuronal death. Annu
Rev Neurosci. 13:171–182. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gegelashvili G, Robinson MB, Trotti D and
Rauen T: Regulation of glutamate transporters in health and
disease. Prog Brain Res. 132:267–286. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Danbolt NC: Glutamate uptake. Prog
Neurobiol. 65:1–105. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ouyang YB, Xu L, Liu S and Giffard RG:
Role of astrocytes in delayed neuronal death: GLT-1 and its novel
regulation by MicroRNAs. Adv Neurobiol. 11:171–188. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mitani A and Tanaka K: Functional changes
of glial glutamate transporter GLT-1 during ischemia: An in vivo
study in the hippocampal CA1 of normal mice and mutant mice lacking
GLT-1. J Neurosci. 23:7176–7182. 2003.PubMed/NCBI
|
|
9
|
Bruhn T, Levy LM, Nielsen M, Christensen
T, Johansen FF and Diemer NH: Ischemia induced changes in
expression of the astrocyte glutamate transporter GLT1 in
hippocampus of the rat. Neurochem Int. 37:277–285. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gong J, Gong S, Zhang M, Zhang L, Hu Y,
Liu Y and Li W: Cerebral ischemic preconditioning reduces glutamate
excitotoxicity by up-regulating the uptake activity of GLT-1 in
rats. Amino Acids. 46:1537–1545. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Fang Q, Hu WW, Wang XF, Yang Y, Lou GD,
Jin MM, Yan HJ, Zeng WZ, Shen Y, Zhang SH, et al: Histamine
up-regulates astrocytic glutamate transporter 1 and protects
neurons against ischemic injury. Neuropharmacology. 77:156–166.
2014. View Article : Google Scholar
|
|
12
|
Sun P, Zhang S, Li Y and Wang L: Harmine
mediated neuroprotection via evaluation of glutamate transporter 1
in a rat model of global cerebral ischemia. Neurosci Lett.
583:32–36. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hu YY, Xu J, Zhang M, Wang D, Li L and Li
WB: Ceftriaxone modulates uptake activity of glial glutamate
transporter-1 against global brain ischemia in rats. J Neurochem.
132:194–205. 2015. View Article : Google Scholar
|
|
14
|
Shen N, Mo LQ, Hu F, Chen PX, Guo RX and
Feng JQ: A novel role of spinal astrocytic connexin 43: Mediating
morphine antinociceptive tolerance by activation of NMDA receptors
and inhibition of glutamate transporter-1 in rats. CNS Neurosci
Ther. 20:728–736. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nisticò R, Florenzano F, Mango D, Ferraina
C, Grilli M, Di Prisco S, Nobili A, Saccucci S, D'Amelio M, Morbin
M, et al: Presynaptic c-Jun N-terminal Kinase 2 regulates NMDA
receptor-dependent glutamate release. Sci Rep. 5:90352015.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Barone FC and Feuerstein GZ: Inflammatory
mediators and stroke: New opportunities for novel therapeutics. J
Cereb Blood Flow Metab. 19:819–834. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Furuya T, Pan Z and Kashiwagi K: Role of
retinal glial cell glutamate transporters in retinal ganglion cell
survival following stimulation of NMDA receptor. Curr Eye Res.
37:170–178. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Harman F, Hasturk AE, Yaman M, Arca T,
Kilinc K, Sargon MF and Kaptanoglu E: Neuroprotective effects of
propofol, thiopental, etomidate, and midazolam in fetal rat brain
in ischemia-reperfusion model. Childs Nerv Syst. 28:1055–1062.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhou R, Yang Z, Tang X, Tan Y, Wu X and
Liu F: Propofol protects against focal cerebral ischemia via
inhibition of microglia-mediated proinflammatory cytokines in a rat
model of experimental stroke. PLoS One. 8:e827292013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang DX, Ding HZ, Jiang S, Zeng YM and
Tang QF: An in vitro study of the neuroprotective effect of
propofol on hypoxic hippocampal slice. Brain Inj. 28:1758–1765.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhu X, Hao X, Luo J, Min S, Xie F and
Zhang F: Propofol inhibits inflammatory cytokine-mediated glutamate
uptake dysfunction to alleviate learning/memory impairment in
depressed rats undergoing electroconvulsive shock. Brain Res.
1595:101–109. 2015. View Article : Google Scholar
|
|
22
|
Hama-Tomioka K, Kinoshita H, Nakahata K,
Kondo T, Azma T, Kawahito S, Hatakeyama N and Matsuda N: Roles of
neuronal nitric oxide synthase, oxidative stress, and propofol in
N-methyl-D-aspartate-induced dilatation of cerebral arterioles. Br
J Anaesth. 108:21–29. 2012. View Article : Google Scholar
|
|
23
|
Kingston S, Mao L, Yang L, Arora A, Fibuch
EE and Wang JQ: Propofol inhibits phosphorylation of
N-methyl-D-aspartate receptor NR1 subunits in neurons.
Anesthesiology. 104:763–769. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Brewer GJ: Serum-free B27/neurobasal
medium supports differentiated growth of neurons from the striatum,
substantia nigra, septum, cerebral cortex, cerebellum, and dentate
gyrus. J Neurosci Res. 42:674–683. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Junghans U and Kappler J: Rat neocortex.
The neuron in tissue culture, IBRO Handbook series: Methods in the
neurosciences. Haynes LW: John Wiley and Sons; 18. Chichester: pp.
545–553. 1999
|
|
26
|
Cui D, Wang L, Qi A, Zhou Q, Zhang X and
Jiang W: Propofol prevents autophagic cell death following oxygen
and glucose deprivation in PC12 cells and cerebral
ischemia-reperfusion injury in rats. PLoS One. 7:e353242012.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
28
|
Suchanek W and Yoshimura M: Processing and
properties of hydroxyapatite-based biomaterials for use as hard
tissue replacement implants. J Mater Res. 13:94–117. 1998.
View Article : Google Scholar
|
|
29
|
Hinzman JM, Thomas TC, Quintero JE,
Gerhardt GA and Lifshitz J: Disruptions in the regulation of
extracellular glutamate by neurons and glia in the rat striatum two
days after diffuse brain injury. J Neurotrauma. 29:1197–1208. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tanaka K, Watase K, Manabe T, Yamada K,
Watanabe M, Takahashi K, Iwama H, Nishikawa T, Ichihara N, Kikuchi
T, et al: Epilepsy and exacerbation of brain injury in mice lacking
the glutamate transporter GLT-1. Science. 276:1699–1702. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kim K, Lee SG, Kegelman TP, Su ZZ, Das SK,
Dash R, Dasgupta S, Barral PM, Hedvat M, Diaz P, et al: Role of
excitatory amino acid transporter-2 (EAAT2) and glutamate in
neurodegeneration: Opportunities for developing novel therapeutics.
J Cell Physiol. 226:2484–2493. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Fan W, Zhu X, Wu L, Wu Z, Li D, Huang F
and He H: Propofol: An anesthetic possessing neuroprotective
effects. Eur Rev Med Pharmacol Sci. 19:1520–1529. 2015.PubMed/NCBI
|
|
33
|
Wang HY, Wang GL, Yu YH and Wang Y: The
role of phosphoinositide-3-kinase/Akt pathway in propofol-induced
postconditioning against focal cerebral ischemia-reperfusion injury
in rats. Brain Res. 1297:177–184. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Acquaviva R, Campisi A, Raciti G, Avola R,
Barcellona ML, Vanella L and Li Volti G: Propofol inhibits
caspase-3 in astroglial cells: Role of heme oxygenase-1. Curr
Neurovasc Res. 2:141–148. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wu GJ, Chen WF, Hung HC, Jean YH, Sung CS,
Chakraborty C, Lee HP, Chen NF and Wen ZH: Effects of propofol on
proliferation and anti-apoptosis of neuroblastoma SH-SY5Y cell
line: New insights into neuroprotection. Brain Res. 1384:42–50.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Rao VL, Dogan A, Todd KG, Bowen KK, Kim
BT, Rothstein JD and Dempsey RJ: Antisense knockdown of the glial
glutamate transporter GLT-1, but not the neuronal glutamate
transporter EAAC1, exacerbates transient focal cerebral
ischemia-induced neuronal damage in rat brain. J Neurosci.
21:1876–1883. 2001.PubMed/NCBI
|
|
37
|
Raivich G: c-Jun expression, activation
and function in neural cell death, inflammation and repair. J
Neurochem. 107:898–906. 2008.PubMed/NCBI
|
|
38
|
Zhu Z, Li R, Stricker R and Reiser G:
Extracellular α-crystallin protects astrocytes from cell death
through activation of MAPK, PI3K/Akt signaling pathway and blockade
of ROS release from mitochondria. Brain Res. 1620:17–28. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Liu K, Zhang Q, Lan H, Wang L, Mou P, Shao
W, Liu D, Yang W, Lin Z, Lin Q, et al: GCN5 Potentiates Glioma
Proliferation and Invasion via STAT3 and AKT Signaling Pathways.
Int J Mol Sci. 16:21897–21910. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Gu Q, Zhai L, Feng X, Chen J, Miao Z, Ren
L, Qian X, Yu J, Li Y, Xu X and Liu CF: Apelin-36, a potent
peptide, protects against ischemic brain injury by activating the
PI3K/Akt pathway. Neurochem Int. 63:535–540. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Sun W, Gould TW, Newbern J, Milligan C,
Choi SY, Kim H and Oppenheim RW: Phosphorylation of c-Jun in avian
and mammalian motoneurons in vivo during programmed cell death: An
early reversible event in the apoptotic cascade. J Neurosci.
25:5595–5603. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Li D, Li X, Wu J, Li J, Zhang L, Xiong T,
Tang J, Qu Y and Mu D: Involvement of the JNK/FOXO3a/Bim pathway in
neuronal apoptosis after hypoxic-ischemic brain damage in neonatal
rats. PLoS One. 10:e01329982015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Tovilovic G, Zogovic N, Soskic V,
Schrattenholz A, Kostic-Rajacic S, Misirkic-Marjanovic M,
Janjetovic K, Vucicevic L, Arsikin K, Harhaji-Trajkovic L and
Trajkovic V: Arylpiperazine-mediated activation of Akt protects
SH-SY5Y neuroblastoma cells from 6-hydroxydopamine-induced
apoptotic and autophagic death. Neuropharmacology. 72:224–235.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Xu L, Li Y, Fu Q and Ma S: Perillaldehyde
attenuates cerebral ischemia-reperfusion injury-triggered
overexpression of inflammatory cytokines via modulating Akt/JNK
pathway in the rat brain cortex. Biochem Biophys Res Commun.
454:65–70. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Zhou Q and Sheng M: NMDA receptors in
nervous system diseases. Neuropharmacology. 74:69–75. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Joe N, Scott V and Brown CH: Glial
regulation of extrasynaptic NMDA receptor-mediated excitation of
supraoptic nucleus neurones during dehydration. J Neuroendocrinol.
26:35–42. 2014. View Article : Google Scholar
|
|
47
|
Dong C, Rovnaghi CR and Anand KJ: Ketamine
affects the neurogenesis of rat fetal neural stem progenitor cells
via the PI3K/Akt-p27 signaling pathway. Birth Defects Res B Dev
Reprod Toxicol. 101:355–363. 2014. View Article : Google Scholar : PubMed/NCBI
|