Introduction
Acute myocardial infarction (AMI) is a leading cause
of clinical morbidity and mortality (1–3).
Currently, various reperfusion therapies, including pharmacological
interventions (e.g., thrombolytics) and/or invasive manipulations
(e.g., angioplasty) constitute the most effective strategies for
the treatment of AMI (4).
Paradoxically, the restoration of oxygen-rich blood to the ischemic
tissue leads to additional myocardial impairment, a situation
collectively referred to as myocardial ischemia/reperfusion (I/R)
injury (MIRI) (5,6). Finding ways to limit MIRI and
thereby achieve the maximal cardioprotective effects of clinical
reperfusion strategies has always been a problem. Recent studies
indicated that the apoptosis and autophagy of cardiomyocytes played
an important pathogenic role in MIRI (5,7,8).
Although the molecular mechanisms underlying MIRI have not been
completely clarified, the mechanistic exploration of apoptosis and
autophagy may unveil novel therapeutic approaches for the treatment
of MIRI.
Apoptosis, named as type I programmed cell death, is
genetically determined by the appropriate genes in response to MIRI
(9). Among these, the Bcl-2
family, mainly consisting of pro-apoptotic Bax and anti-apoptotic
Bcl-2, determine the possibility of cardiomyocytes undergoing
apoptosis or surviving under conditions of I/R (7,10,11). Autophagy, or type II programmed
cell death, plays a critical role in the pathogenesis of MIRI
(5,12,13). Beclin-1 and light chain 3 (LC3)
are two pivotal regulatory genes involved in autophagy, and are
both commonly recognized as indispensable markers of autophagy
(3,14,15). The transfer of LC3-I to LC3-II
leads to the formation of double-membrane vesicles named
autophagosomes (3). Furthermore,
close crosstalk occurs between these two forms of programmed cell
death, which is modulated by intracellular signaling pathways
(16,17). For instance, the cell surface
receptor-mediated signaling pathways including pathways involving
Toll-like receptor 4 (TLR4), simultaneously regulate apoptosis- and
autophagy-associated pathogenesis in I/R injury (18). Furthermore, blocking TLR4-induced
apoptosis and autophagy may confer a protective effect against MIRI
(5,7). Thus, a better understanding of the
mechanisms which initiate apoptotic and autophagic responses is
critical for the study of MIRI and for the development of novel
treatments.
TLR4, as an upstream common sensor of multiple
intracellular pathways including TLR4/nuclear factor-κB (NF-κB)
signaling, is commonly recognized as an important hallmark and
potent stimulus of the apoptotic cascade as well as autophagic
signaling (18–20). Previous findings have confirmed
that TLR4 activity stimulated the downstream transcription factor
NF-κB; it induced the expression of NF-κB target genes including
interleukin (IL)-6 and tumor necrosis factor α (TNF-α), and led to
the activation of the apoptotic cascade and autophagic responses
(18). It has been demonstrated
that IL-6 and TNF-α are the principal cytokines involved in the
initiation of the apoptotic process (18). The precise mechanisms detailing
how each of these signaling molecules participates in apoptosis and
autophagy during MIRI remain elusive.
The radioprotective 105 kDa protein (RP105) is a
TLR4 homologue which lacks the intracytoplasmic Toll-IL-1 receptor
(TIR) domain of TLR4 (21,22).
RP105 is widely involved in the pathophysiological process of
various cardiovascular diseases through directly inhibiting
TLR4-driven pathogenesis during myocardial infarction,
atherosclerosis and post-interventional vascular remodeling
(22–24). Previously, we showed that RP015
protected the myocardium against I/R injury, and the underlying
mechanism was ascribed to the inhibition of the apoptotic cascade
response (25). There exists
close crosstalk between apoptosis and autophagy, although the role
of RP105 in reducing myocardial apoptosis and autophagy
simultaneously during I/R injury is yet to be defined. Thus, we
aimed to examine the molecular mechanisms through which RP105
protects cardiomyocytes exposed to I/R injury against apoptosis and
autophagy. In this study, we demonstrated that the overexpression
of RP105 ameliorates MIRI by limiting apoptosis and autophagy.
RP105 may inhibit these two types of programmed cell death
synergistically through inhibiting the activation of the TLR4/NF-κB
signaling pathways. Moreover, we hypothesized that the
anti-autophagic potency of RP105 may be closely associated with the
increase in binding between Bcl-2 and Beclin1.
Materials and methods
Preparation of adenoviral vector
An adenoviral vector encoding EGFP-RP105
(Ad-EGFP-RP105) or EGFP (Ad-EGFP) was generated under the control
of AdMax adenovirus packing system (Microbix Biosystems Inc.,
Mississauga, ON, Canada) in accordance with the manufacturer's
instructions. The resulting recombinant adenoviruses were picked
and amplified in 293 cells (American Type Culture Collection,
Manassas, VA, USA). The virus was then purified using an Adeno-XTM
Virus Purification kit (BD Biosciences; Clontech, Mountain View,
CA, USA). The viral titer was determined by a plaque assay, and
finally concentrated to 1.5E+10 plaque-forming units (PFU).
Animals and experimental design
Adult male Sprague-Dawley (SD) rats weighing 220–250
g were used for all experiments in the present study. The
experimental procedures were approved by the Institutional Animal
Care and Use Committee of Wuhan University (Wuhan, China). Animal
care conformed to the Guide for the Care and Use of Laboratory
Animals published by the US National Institutes of Health. One week
prior to surgery, SD rats were randomly divided into four groups
(n=10), namely, the sham-operated group (SO group), the myocardial
I/R group (I/R group), the myocardial I/R with Ad-EGFP group (I/R +
Ad-E group), and the myocardial I/R with Ad-EGFP-RP105 group (I/R +
Ad-E-R group). Three days after adenovirus delivery or saline
injection, each rat underwent 30 min of left anterior descending
(LAD) coronary artery occlusion and subsequently, 6 h of
reperfusion.
Gene transfer in vivo and establishment
of a rat model of MIRI
Ad-EGFP or Ad-EGFP-RP105 was transferred or saline
was injected into the left ventricular wall of the rat heart. The
animals were then subjected to a MIRI surgical operation three days
post-transfection as reported previously (25). Briefly, the SD rats were
anesthetized by an intraperitoneal injection of sodium
pentobarbital (40 mg/kg), and were intubated orally with 100%
oxygen using a rodent ventilator. After a left thoracotomy between
the fourth and fifth intercostal space, Ad-EGFP
(1.5×1010 PFU), Ad-EGFP-RP105 (1.5×1010 PFU)
or saline in volume of 100 µl was injected intramyocardially
into five separate sites via a 30-gauge needle. All the five
injection sites were situated in the left ventricular anterior wall
around the LAD artery. The chest was closed and all rats received a
single intramuscular injection of penicillin sodium (0.8 mg/g;
North China Pharmaceutical Co., Ltd., Shijiazhuang, China).
Three days after virus delivery or saline injection,
the MIRI model was established as previously described (7). Briefly, all rats were
re-anesthetized and ventilated with 100% oxygen. A thoracotomy
through the original incision site was re-performed. A 6-0 silk
suture with a curved needle was placed under the origin of the LAD
artery and medical latex tubing was located over the LAD artery.
Myocardial ischemia was induced by tightening the suture for 30
min. Ischemia was confirmed by electrocardiogram changes (S-T
segment elevation) and the immediate regional cyanosis in the
anterior ventricular wall. The suture was then loosened to restore
coronary circulation. At 6 h post-reperfusion, the rats were
re-anesthetized and blood samples were collected through the
jugular vein. The rats were then sacrificed by air embolism methods
and the myocardial tissue near the cardiac apex was obtained for
further analysis. The SO group rats underwent the same operation
with the exception of LAD artery ligation.
Immunohistochemistry analysis and
fluorescence microscopy
We detected the efficiency of adenoviral
transfection by immunohistochemistry and fluorescence microscopy
[immunofluorescence (IF) microscopy] as previously described
(26). Briefly, the heart
specimens were quickly fixed in 4.0% paraformaldehyde, embedded in
paraffin, and then sectioned. After dewaxing and antigen retrieval
using microwave processing, the slices were incubated in 1% goat
serum albumin at room temperature (RT) for 30 min, and incubated
with anti-EGFP antibody (Santa Cruz Biotechnology, Inc., Santa
Cruz, CA, USA) overnight at 4°C. The sections were then incubated
for 60 min at 20–37°C in the dark with fluorescein isothiocyanate
(FITC)-labeled goat anti-rabbit IgG secondary antibody (Boster
Biotech, Wuhan, China). Finally, the slices underwent
4′,6′-diamidino-2-phenylindole (DAPI; Beyotime Biotech, Shanghai,
China) staining to redye the nuclei of the cardiomyocytes. Images
were captured using a fluorescence microscope (BX51; Olympus
America, Inc., Center Valley, PA, USA).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from myocardial tissue using
TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the
manufacturer's instructions. RNA (4.0 µg) was reverse
transcribed into cDNA according to the manufacturer's instructions
(Fermentas, Glen Burnie, MD, USA). RT-qPCR was performed using a
SYBR-Green/fluorescein qPCR Master Mix kit (Fermentas) with the ABI
Prism 7500 system (Foster City, CA, USA). Data were normalized to
β-actin gene expression, and calculated using the comparative
quantification method (2−ΔΔCt). The following
sequence-specific primers were used to amplify gene products: RP105
forward, 5′-TGAGGGCCTCTGTGAAATGT-3′ and reverse,
5′-GGAAGCACTGATTTGGCACA-3′; β-actin forward,
5′-CACGATGGAGGGGCCGGACTCATC-3′ and reverse,
5′-TAAAGACCTCTATGCCAACACAGT-3′.
Western blot analysis
Western blot analysis was performed to assess the
protein levels of RP105, TLR4, NF-κB/p65, phosphorylated
(p-)NF-κB/p65, Bax, Bcl-2, Beclin-1, LC3, IL-6 and TNF-α according
to the manufacturer's instructions. Briefly, the extracted proteins
were separated by electrophoresis with SDS-polyacrylamide gel and
then transferred onto nitrocellulose membranes. After blocking with
5% (w/v) nonfat dry milk for 2 h at RT, the membranes were
incubated with primary antibodies overnight at 4°C. After washing 5
times, the membranes were then incubated with peroxidase-conjugated
secondary antibodies for 2 h at RT. Bands were scanned and analyzed
using an enhanced chemiluminescence (ECL) detection kit (Thermo
Fisher Scientific, Waltham, MA, USA). GAPDH served as a loading
control.
Histological analysis of myocardial
injury
For histopathological observations, heart tissues
were obtained at 6h post-reperfusion and immediately fixed in 4.0%
paraformaldehyde solution overnight. After dehydration at RT by a
graded alcohol series, specimen slices were embedded in paraffin
and sectioned at 4 µm. The sections were then stained with
hematoxylin and eosin (H&E; Beijing Solarbio Science &
Technology Co., Ltd., Beijing, China) and examined under a light
microscope (Leica Microsystems, Wetzlar, Germany).
Determination of serum activity levels of
lactate dehydrogenase (LDH) and creatine kinase (CK)
Blood samples were collected 6 h after reperfusion.
LDH and CK assay commercial kits (Elabscience Biotechnology Co.,
Ltd., Wuhan, China) were used to analyze enzyme activity levels.
All procedures were performed in accordance with the manufacturer's
instructions. The results are expressed as international units per
liter.
Determination of myocardial
apoptosis
The terminal deoxynucleotidyl-transferase
(TdT)-mediated dUTP nick-end labeling (TUNEL) assay was performed
in order to detect myocardial apoptosis. The TUNEL detection kit
(Roche Applied Science, Basel, Switzerland) was used according to
the manufacturer's instructions. In addition, the sections were
co-stained with hematoxylin after TUNEL staining. Five microscopic
fields of each section (×400 magnification; Leica Microsystems)
were randomly chosen to count the number of TUNEL-positive cells
and total cells. An apoptotic index (AI) score represented the
ratio of apoptotic cardiomyocytes/total myocytes.
Statistical analysis
Statistical analysis was performed using SPSS
software (version 14.0). Data are expressed as the means ± standard
deviation (SD). Differences between groups were assessed using
one-way analysis of variance (ANOVA) and the Student-Newman-Keuls
(SNK)-q test. A p-value <0.05 was considered to indicate a
statistically significant difference.
Results
Efficiency of adenoviral transfection
into the rat myocardium
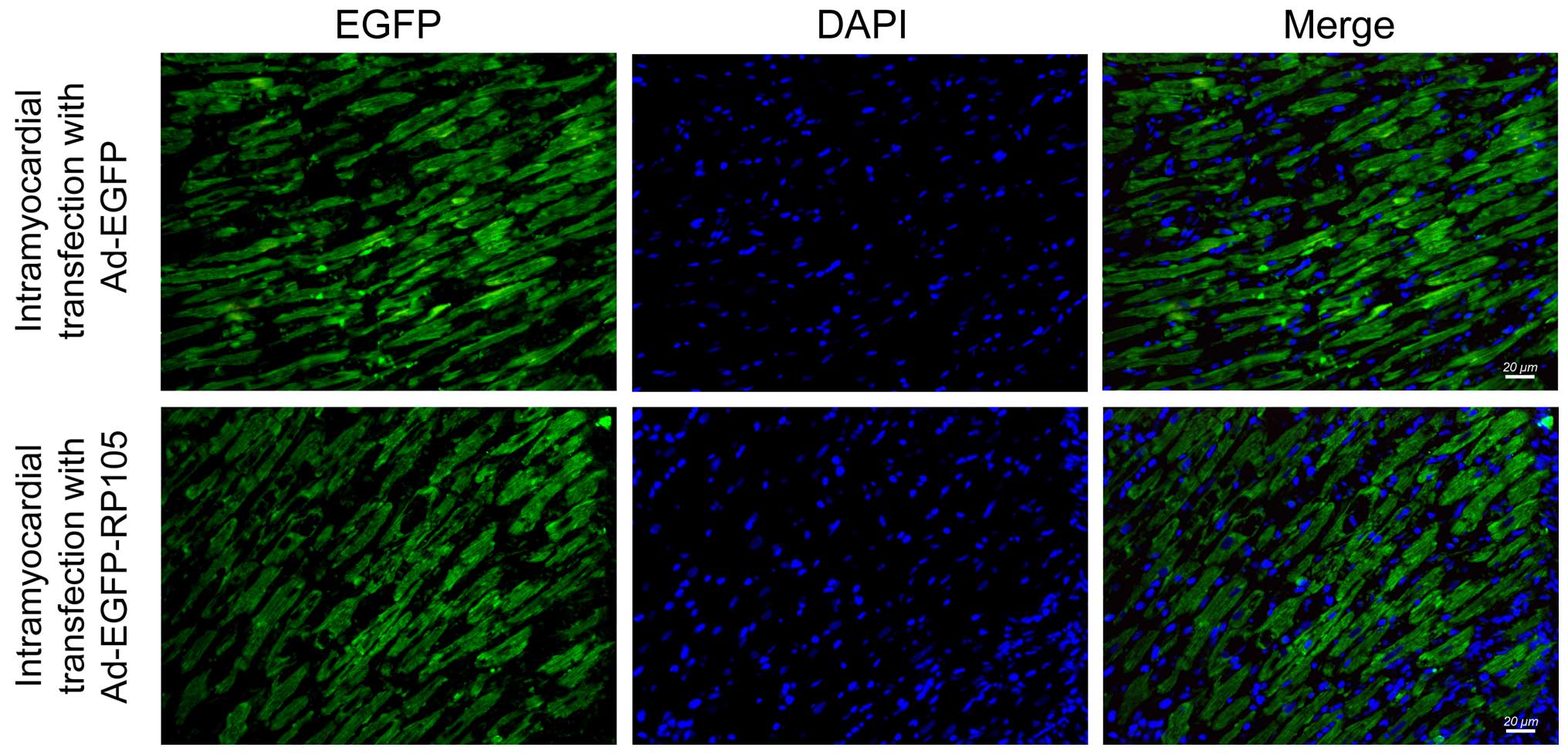
IF microscopy was performed to determine the
efficiency of Ad-EGFP and Ad-EGFP-RP105 transfection into the
myocardium at three days post-transfection. Successful adenovirus
transfection was indicated by high EGFP (green) fluorescence in the
Ad-EGFP- or Ad-EGFP-RP105-transferred myocardium (Fig. 1). However, the EGFP signal in the
myocardium treated with saline or no injection was not detectable
(SO or I/R groups, respectively) (images not shown).
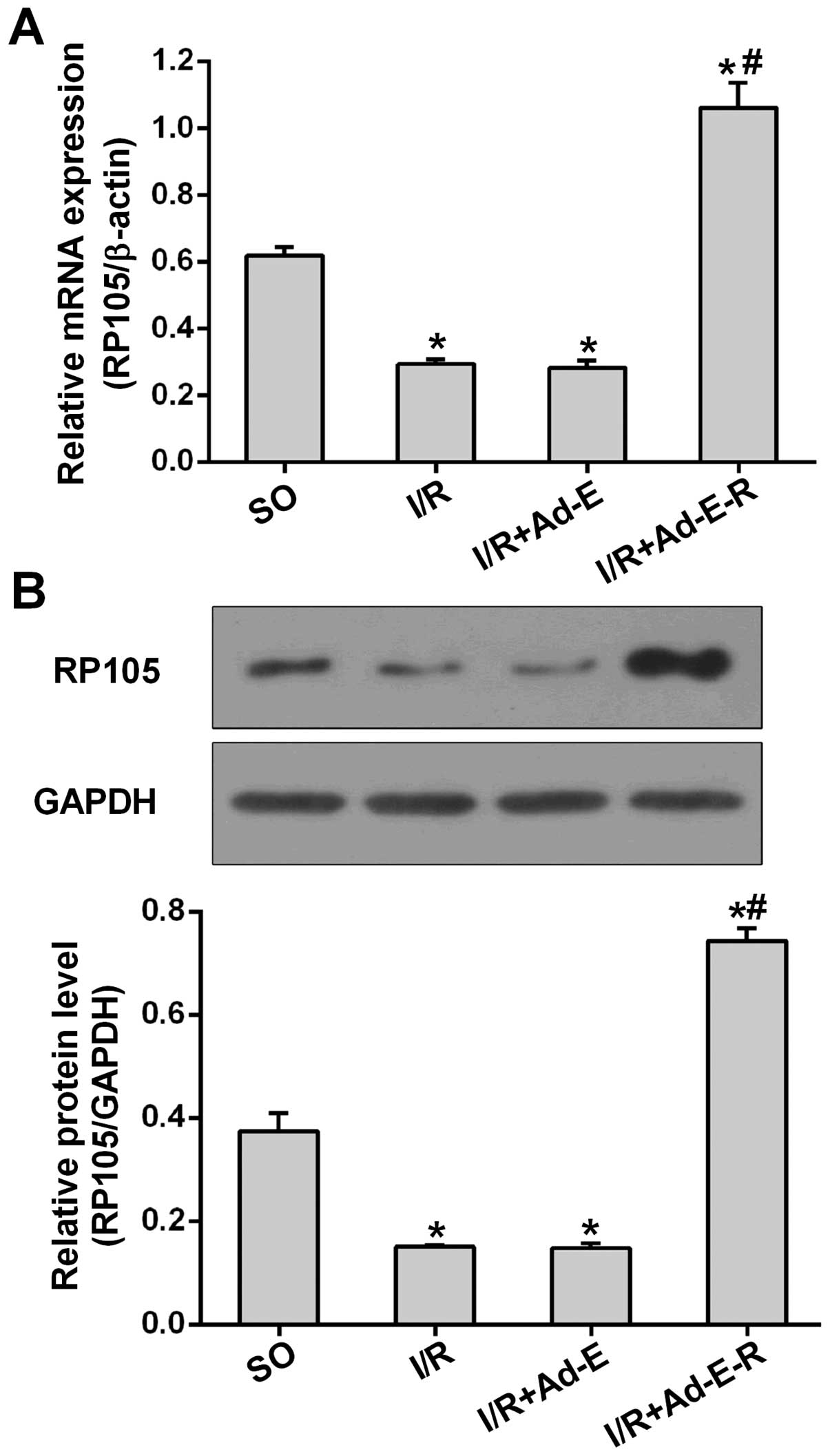
Ad-EGFP-RP105 transfection in vivo
increases RP105 expression
To determine the anti-apoptotic and anti-autophagic
action of RP105 during I/R injury, we performed the MIRI surgical
procedure after adenovirus transfection as described above. As
shown in Fig. 2A, the mRNA
expression of RP105 was markedly downregulated 2.10-fold in
comparison with the SO group. However, the transduction of
Ad-EGFP-RP105 into the myocardium prior to I/R markedly increased
the mRNA expression of RP105 by 73.38% (p<0.05 vs. I/R + Ad-E)
(Fig. 2A). A similar pattern of
RP105 protein expression was analyzed by western blot analysis.
Compared with Ad-EGFP transduction, Ad-EGFP-RP105 transfection
significantly increased the RP105 protein level after MIRI
(0.74±0.03 in the I/R + Ad-E-R group vs. 0.14±0.01 in the I/R +
Ad-E group, p<0.05) (Fig. 2B).
In addition, the mRNA and protein levels of RP105 showed no
statistical difference between the I/R and I/R + Ad-E groups.
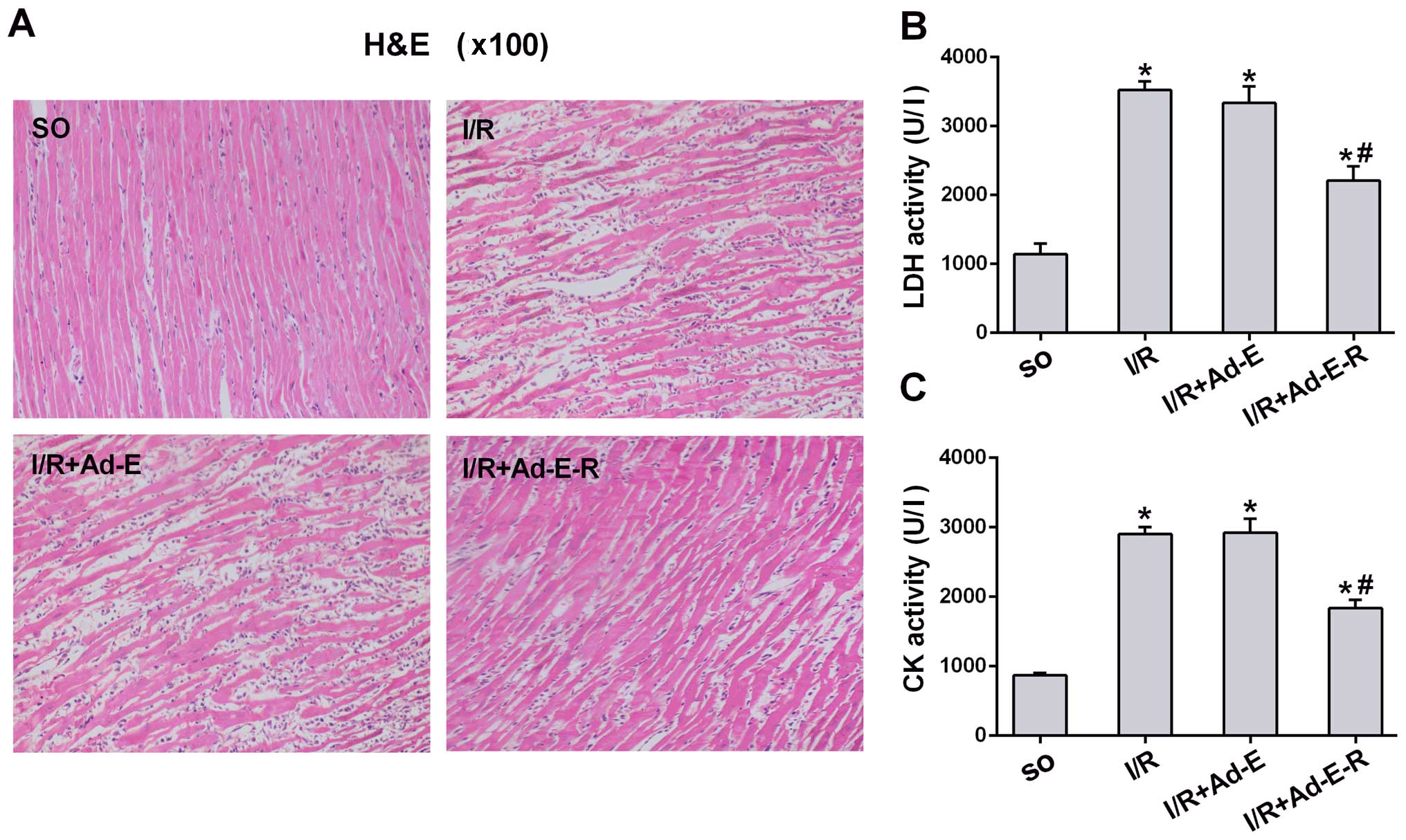
RP105 attenuates MIRI-induced
pathological damage and elevations in myocardial enzyme
activity
To determine whether antagonizing TLR4 with RP105
alleviates myocardial damage, myocardial histopathological analysis
was performed as well as measurements of the activity of serum LDH
and CK as the indices of injury in MIRI. H&E staining (×100
magnification) revealed that the structure of the myocardial
tissues was completely preserved and remained organized in the
sham-operated myocardium. However, the myocardial tissue from the
rats in the I/R group demonstrated focal destruction of myocardial
fibers with erythrocyte and neutrophil infiltration, enlarged
intercellular spaces, extensive edema and myocyte necrosis.
Pathological damage was markedly attenuated in the I/R myocardium
transduced with Ad-EGFP-RP105 when compared with the I/R and I/R +
Ad-E groups (Fig. 3A). With
regard to the leakage of myocardial enzymes, only low activity
levels of serum LDH and CK were detected in the SO group. The
activity of LDH and CK in the other three I/R groups were all
markedly increased relative to the SO group (Fig. 3B and C). However, the
overexpression of RP105 significantly repressed the elevation of
LDH and CK levels (I/R + Ad-E-R vs. I/R, p<0.05). In addition,
the transduction of Ad-EGFP did not affect the myocardial leakage
of LDH and CK in MIRI (I/R + Ad-E vs. I/R, p>0.05). These
findings suggest that RP105 plays a cardioprotective role in
MIRI.
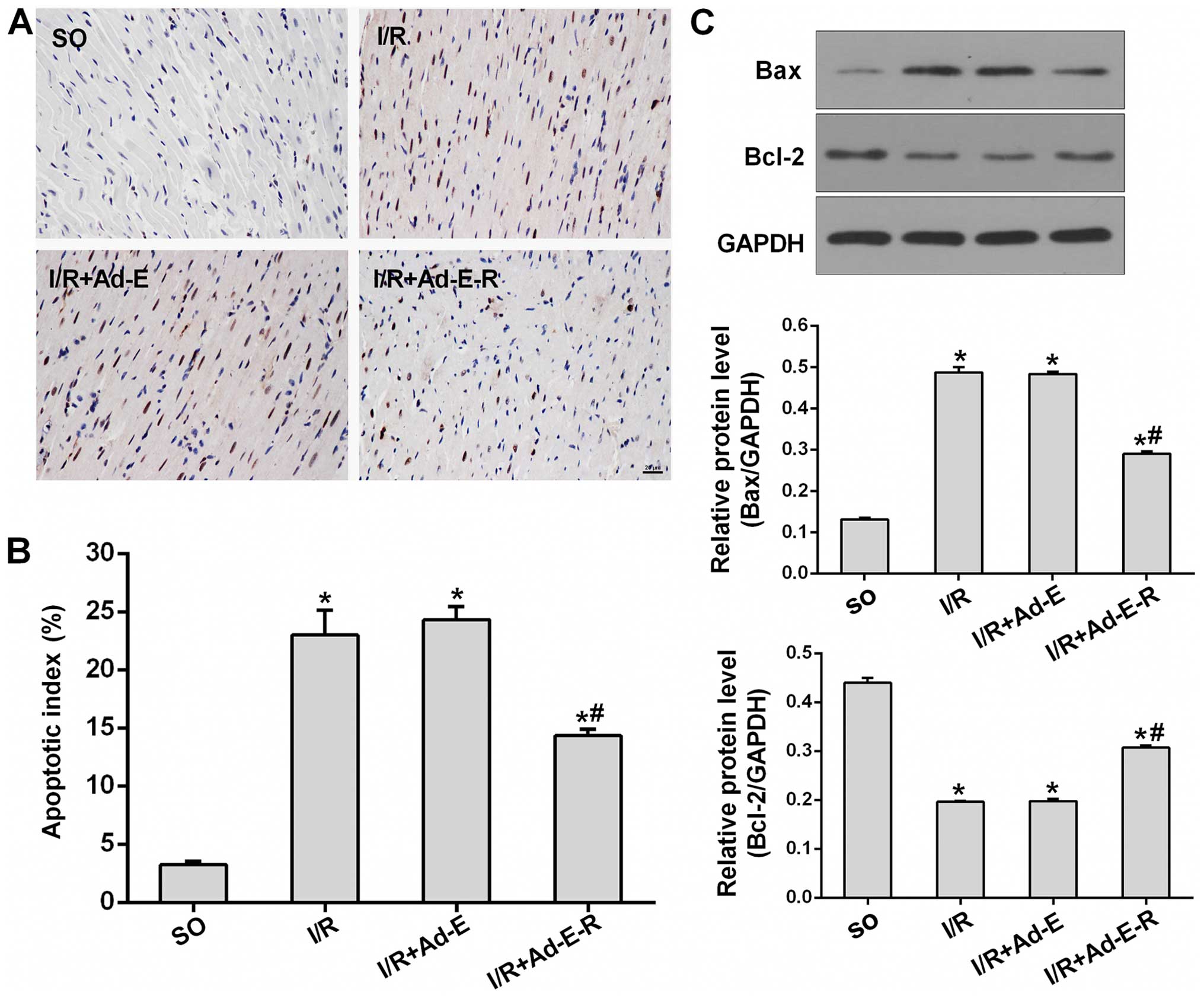
RP105 represses the apoptosis of
cardiomyocytes in MIRI
To determine the anti-apoptotic potency of RP105 in
MIRI, we first detected the number of TUNEL-positive cardiomyocytes
in order to evaluate the apoptosis of cardiomyocytes. The numbers
of TUNEL-positive myocytes in all three I/R groups were
significantly higher than that shown in the SO group (Fig. 4A). However, the overexpression of
RP105 markedly reduced the AI compared with that in the
Ad-EGFP-transduced tissues (14.33±0.59% in the I/R + Ad-E-R group
vs. 24.32±1.13% in the I/R + Ad-E group, p<0.05) (Fig. 4B). In agreement with the results
of the TUNEL assay, RP105 overexpression also led to the limitation
of apoptotic activation in MIRI. This was evidenced by a 39.92%
reduction in the levels of the pro-apoptotic Bax protein, as well
as inversely a 1.55-fold upregulation of the levels of the
anti-apoptotic Bcl-2 protein relative to that in the
Ad-EGFP-transduced tissues (Fig. 4
C; p<0.05). Additionally, Ad-EGFP transduction had no
significant effect on the percentage of TUNEL-positive cells and
apoptotic activation (I/R + Ad-E group vs. I/R group,
p>0.05).
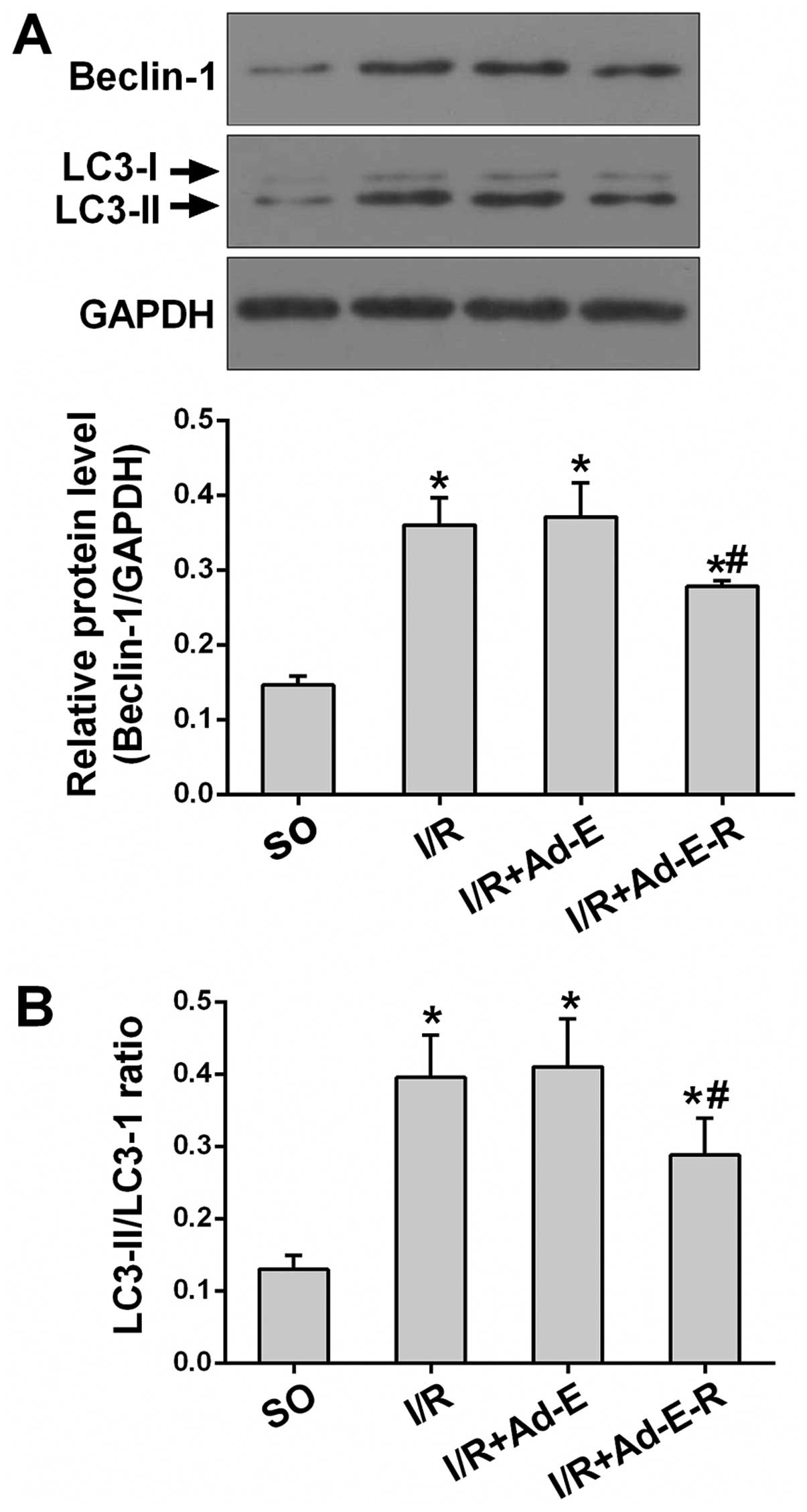
RP105 suppresses autophagic signaling
during MIRI
Beclin-1 and LC3 act as two indispensable mediators
which regulate autophagic signaling. To determine whether
antagonizing the TLR4 pathway with RP105 attenuates the autophagic
process in I/R injury, we detected alternations in Beclin-1 and LC3
(LC3-II and LC3-I) protein expression by western blot analysis
(Fig. 5A and B). As shown in
Fig. 5, only low levels of
Beclin-1 protein and a low LC3-II/LC3-I ratio were measured in the
SO group. Myocardium samples from all three I/R groups demonstrated
elevated levels of Beclin-1 protein and the LC3-II/LC3-I ratio
relative to the SO group. However, overexpression of RP105,
markedly reduced the levels of Beclin-1 and the LC3-II/LC3-I ratio
by 24.98 and 29.73%, respectively (I/R + Ad-E-R group vs. I/R +
Ad-E group, p<0.05) (Fig. 5A and
B). Ad-EGFP transduction had no significant effect on the
levels of autophagic signaling (I/R + Ad-E group vs. I/R group,
p>0.05).
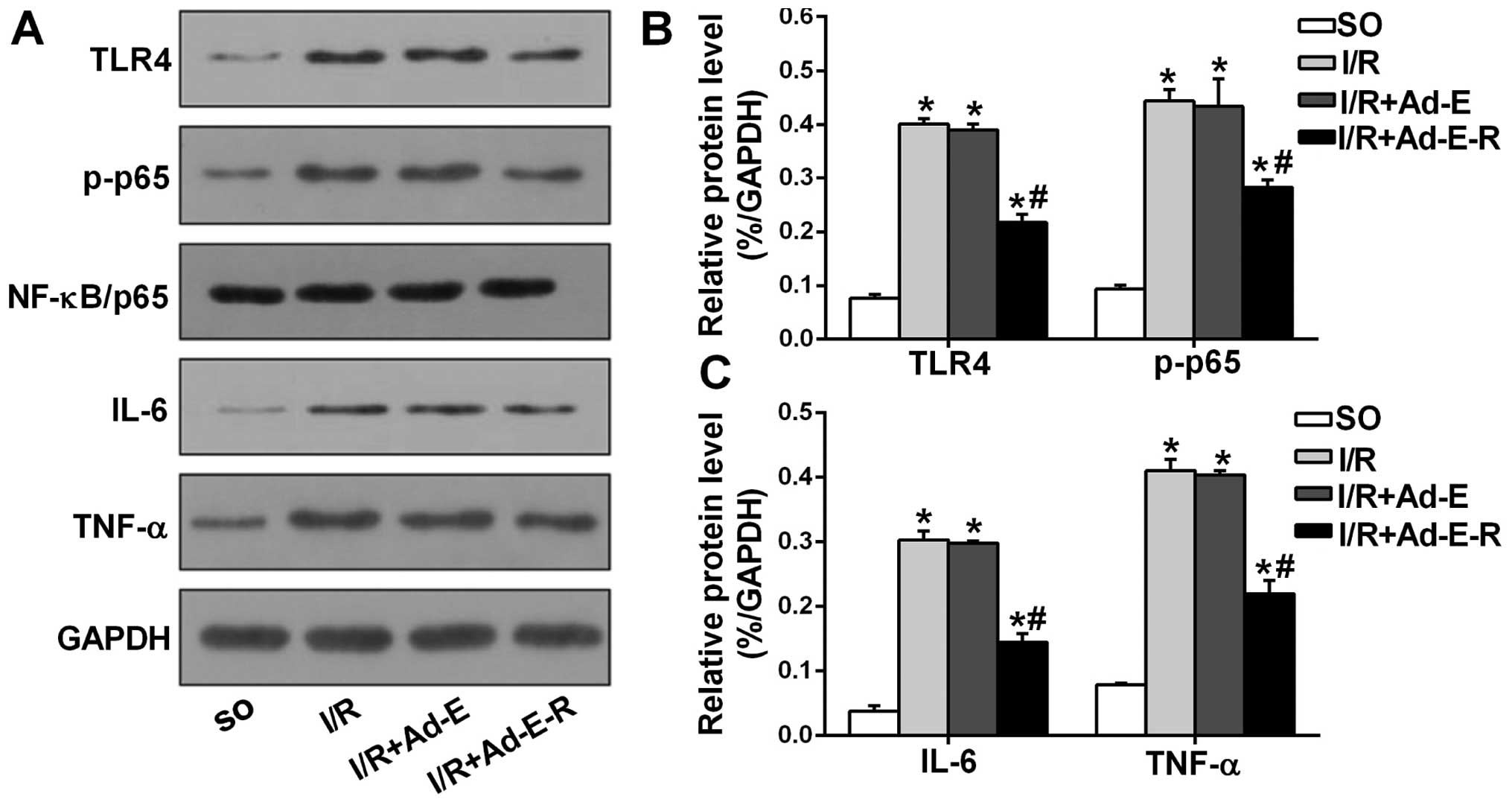
RP105 reduces the expression of TLR4,
NF-κB, IL-6 and TNF-α in MIRI
To determine whether RP105 modulates the activity of
the transcription factor NF-κB and the production of pro-apoptotic
cytokines (IL-6 and TNF-α) through antagonizing TLR4, we detected
the levels of TLR4, NF-κB/p65, p-NF-κB/p65 (p-p65), IL-6 and TNF-α
by western blot analysis (Fig. 6
A–C). Compared with the SO group, the rat myocardium from all
three other I/R groups demonstrated increased protein levels of
TLR4, p-p65, IL-6 and TNF-α. However, RP105 overexpression markedly
suppressed the elevated levels of TLR4, p-p65, IL-6 and TNF-α by
44.27, 34.77, 51.51 and 45.63%, respectively (I/R + Ad-E-R group
vs. I-R + Ad-E group, p<0.05), and exerted no effect on the
total p65 protein level. Ad-EGFP transduction had no significant
effect on the expression levels of those proteins (I/R + Ad-E group
vs. I/R group, p>0.05).
Discussion
Accumulating evidence indicates that apoptosis- and
autophagy-associated myocyte death play key roles in the induction
of MIRI (9,19,27). Our present study demonstrated that
the overexpression of RP105 via a recombinant adenoviral vector is
a promising cardioprotective strategy to induce anti-apoptotic and
anti-autophagic effects in MIRI in vivo. Firstly, we showed
that intramyocardial injection with adenoviral vectors prior to the
I/R procedure successfully led to the overexpression of RP105 in
rat hearts. Secondly, we proved that the overexpression of RP105
significantly contributed to fewer numbers of TUNEL-positive
cardiomyocytes and inhibited apoptotic and autophagic signaling in
comparison with rats subjected to saline or Ad-EGFP injection in
MIRI. These outcomes were accompanied by the attenuation of I/R
injury-induced myocardial pathological damage and reduced leakage
of LDH and CK. Finally, mechanistic studies demonstrated that the
overexpression of RP105 markedly inhibited the activation of the
TLR4/NF-κB pathway, and reduced the expression of pro-apoptotic
cytokines IL-6 and TNF-α. Taken together, these findings strongly
confirm a cardioprotective effect of RP105, as it protects the
myocardium against apoptosis and autophagy in MIRI.
Previous studies have shown that TLR4 plays a
critical role as a pivotal sensor in inducing cell apoptosis during
MIRI (19,20). By contrast, TLR4 deficiency
markedly attenuates I/R-induced myocardial apoptosis (7). However, limited information is
available regarding the molecular mechanism underlying
TLR4-mediated intracellular signaling in I/R-induced myocardial
apoptosis in vivo. The transcription factor NF-κB acts as
the principal downstream signaling target of TLR4 (28). The phosphorylation of p65, an
important subunit of NF-κB, plays a central role in promoting
target gene expression, including the cytokines IL-6 and TNF-α,
which are the main cytokines involved in the initiation of
apoptosis (18,29). Shen et al have confirmed
that the activity of TLR4/NF-κB promoted the production of IL-6 and
TNF-α, and subsequently led to the enhancement of the apoptotic
cascade in the setting of hepatic I/R injury (18). Herein, our data showed, for the
first time to the best of our knowledge, that the TLR4,
p-NF-κB/p65, and IL-6/TNF-α signaling pathways were upregulated in
MIRI, and therfore promoted apoptosis. These results, taken
together with other findings (19,29), regarding the TLR4/NF-κB-IL-6
(TNF-α)-mediated apoptotic mechanism broaden our understanding of
the multiple biological functions of TLR4, beyond being a canonical
inflammatory sensor in MIRI.
Another such response that is modulated through
TLR4-mediated signaling which may play significant role in the
pathogenesis of MIRI is autophagy. Autophagy is an evolutionarily
conserved process of cellular catabolism, mainly modulated by two
indispensible regulatory proteins, Beclin-1 and LC3. Previous
findings have shown that autophagy is regulated by a TLR4-mediated
mechanism, and further aggravates hepatic I/R injury (18). It has been observed that NF-κB is
mainly activated by TLR4. Notably, independent of its
pro-inflammatory and pro-apoptotic effects, NF-κB also acts as an
important mediator in regulating autophagy. Zeng et al
demonstrated that the phosphorylation of NF-κB/p65 aggravates
myocardial damage through promoting Beclin-1-modulated autophagy in
I/R injury (27). The specific
repression of p-NF-κB/p65 expression was shown to reduce Beclin-1
expression, and subsequently limit autophagy in MIRI (27). In the present study, we showed
that the activity of the TLR4/p-NF-κB/p65 signaling pathway
strongly elevated autophagic signaling in MIRI. The new
contributing factor of TLR4-mediated autophagy further widens its
pathogenic action, and especially confers a potent therapeutic
approach for reducing MIRI.
As autophagy and apoptosis are both induced by the
activity of TLR4 signaling pathway, we hypothesized that selective
inhibition of TLR4 and the intercellular signaling pathway may be a
valuable approach for the treatment of MIRI. RP105 is a key
inhibitor of TLR4-mediated signaling, through which it attenuates
pathophysiological processes in various cardiovascular disease
settings such as myocardial infarction, atherosclerosis and
post-interventional vascular remodeling (22–24,30). Previously, we showed that RP015
protected the myocardium against I/R injury, and the
cardioprotective effect was attributed to inhibition of apoptosis
(25). However, the molecular
mechanism underlying the inhibitory effect of RP105 on the
apoptosis of myocytes during I/R injury remains largely unknown.
The novel contribution of RP105 in regulating autophagy also
remains unrecognized. In the present study, we validated that RP105
overexpression alleviated myocardial apoptosis induced by I/R
injury. The effect of RP105 on the suppression of autophagic
process was also expounded for the first time, to the best of our
knowledge. We demonstrated that the potential mechanisms of
protection closely correlated with the inactivation of the
TLR4/NF-κB pathway. Our findings therefore demonstrated the
anti-apoptotic and anti-autophagic effects of RP105 and suggested a
possible mechanism underlying these effects in MIRI. Notably,
previous findings manifested the dichotomous role of RP105 in
mediating TLR4 (31), although
recently it has been validated that RP105 deficiency significantly
aggravated cardiac dysfunction by amplifying the TLR4 pathway in
AMI (23). In light of the
above-mentioned observations and the evidence obtained from our
previous study, we proposed that RP105 may be a therapeutic target
of value in the treatment of MIRI by negatively mediating the TLR4
pathway. Further studies examining the crosstalk between autophagic
and apoptotic functions of RP105 (in terms of how one affects the
other or to determine whether they are independently linked) are
warranted in order to determine how TLR4 signaling exerts potential
therapeutic effects in the setting of MIRI.
Certain common factors and components coordinate and
modulate the apoptotic and autophagic pathways (3,16).
The activity of the apoptotic cascade may induce autophagy, whereas
signals that reduce apoptosis also limit autophagy (32). For instance, anti-apoptotic Bcl-2
was shown to interact with the BH3 domain of Beclin-1 in order to
modulate apoptosis and autophagy simultaneously and also maintained
the inactivation of autophagy (33,34). A recent study demonstrated that
the upregulation of Bcl-2 increased the interaction between Bcl-2
and Beclin-1, thereby leading to the downregulation of autophagy in
the setting of hepatic I/R injury (18). Herein, we confirmed that RP105
attenuated myocardial autophagy through inhibiting the TLR4/NF-κB
pathway and therefore downregulated Beclin-1 in MIRI. On the other
hand, we also speculated that the overexpression of RP105 followed
by the increased expression of Bcl-2 may play an important role in
inhibition of autophagy due to the enhanced binding between Beclin1
and Bcl-2.
These results have revealed the role of the
TLR4/NF-κB signaling pathway in inducing apoptosis and autophagy
and therefore shown that modulating RP105 may be an effective
strategy in ameliorating MIRI. However, further investigations into
the precise mechanism that links the inactivation of the
TLR4-mediated signaling pathway with inhibition of the apoptotic
cascade and autophagic signaling are warranted. Moreover, with
regard to the viewpoint that the contribution of autophagy in MIRI
is subject to controversy (14),
further research is still required in order to validate whether
RP105 is a potential anti-autophagic target, and more specifically,
for the attenuation of MIRI. In conclusion, our findings may have
identified a novel method for limiting I/R-induced myocardial
apoptosis and autophagy, provided innovative cardioprotective
insights as well as a potential therapeutic target for the
treatment of MIRI.
Acknowledgments
The present study was supported by the National
Natural Science Foundation of China (no. 81200156, 81200088 and
81470387), and the Fundamental Research Funds for the Central
Universities (no. 20120141120079).
Abbreviations:
|
TLR4
|
Toll-like receptor 4
|
|
RP105
|
radioprotective 105 kDa protein
|
|
MIRI
|
myocardial ischemia/reperfusion
injury
|
|
AMI
|
acute myocardial infarction
|
|
LC3
|
light chain 3
|
|
TIR
|
Toll-IL-1 receptor
|
|
LAD
|
left anterior descending
|
References
|
1
|
Zhou X, Chen M, Wang S, Yu L and Jiang H:
MG53 protein: a promising novel therapeutic target for myocardial
ischemia reperfusion injury. Int J Cardiol. 199:424–425. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Han XJ, He D, Xu LJ, Chen M, Wang YQ, Feng
JG, Wei MJ, Hong T and Jiang LP: Knockdown of connexin 43
attenuates balloon injury-induced vascular restenosis through the
inhibition of the proliferation and migration of vascular smooth
muscle cells. Int J Mol Med. 36:1361–1368. 2015.PubMed/NCBI
|
|
3
|
Thapalia BA, Zhou Z and Lin X: Autophagy,
a process within reperfusion injury: an update. Int J Clin Exp
Pathol. 7:8322–8341. 2014.
|
|
4
|
Lin Y, Chen L, Li W and Fang J: Role of
high-mobility group box-1 in myocardial ischemia/reperfusion injury
and the effect of ethyl pyruvate. Exp Ther Med. 9:1537–1541.
2015.PubMed/NCBI
|
|
5
|
Yao T, Ying X, Zhao Y, Yuan A, He Q, Tong
H, Ding S, Liu J, Peng X, Gao E, et al: Vitamin D receptor
activation protects against myocardial reperfusion injury through
inhibition of apoptosis and modulation of autophagy. Antioxid Redox
Signal. 22:633–650. 2015. View Article : Google Scholar :
|
|
6
|
Barry SP, Ounzain S, McCormick J,
Scarabelli TM, Chen-Scarabelli C, Saravolatz LI, Faggian G,
Mazzucco A, Suzuki H, Thiemermann C, et al: Enhanced IL-17
signalling following myocardial ischaemia/reperfusion injury. Int J
Cardiol. 163:326–334. 2013. View Article : Google Scholar :
|
|
7
|
Ding HS, Yang J, Chen P, Yang J, Bo SQ,
Ding JW and Yu QQ: The HMGB1-TLR4 axis contributes to myocardial
ischemia/reperfusion injury via regulation of cardiomyocyte
apoptosis. Gene. 527:389–393. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang B, Zhong S, Zheng F, Zhang Y, Gao F,
Chen Y, Lu B, Xu H and Shi G: N-n-butyl haloperidol iodide protects
cardiomyocytes against hypoxia/reoxygenation injury by inhibiting
autophagy. Oncotarget. 6:24709–24721. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Murphy E and Steenbergen C: Mechanisms
underlying acute protection from cardiac ischemia-reperfusion
injury. Physiol Rev. 88:581–609. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wu H, Che X, Zheng Q, Wu A, Pan K, Shao A,
Wu Q, Zhang J and Hong Y: Caspases: a molecular switch node in the
crosstalk between autophagy and apoptosis. Int J Biol Sci.
10:1072–1083. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kalogeris T, Baines CP, Krenz M and
Korthuis RJ: Cell biology of ischemia/reperfusion injury. Int Rev
Cell Mol Biol. 298:229–317. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Xie M, Kong Y, Tan W, May H, Battiprolu
PK, Pedrozo Z, Wang ZV, Morales C, Luo X, Cho G, et al: Histone
deacetylase inhibition blunts ischemia/reperfusion injury by
inducing cardiomyocyte autophagy. Circulation. 129:1139–1151. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gustafsson ÅB and Gottlieb RA: Eat your
heart out: role of autophagy in myocardial ischemia/reperfusion.
Autophagy. 4:416–421. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Xu W, Jiang H, Hu X and Fu W: Effects of
high-mobility group box 1 on the expression of Beclin-1 and LC3
proteins following hypoxia and reoxygenation injury in rat
cardiomyocytes. Int J Clin Exp Med. 7:5353–5357. 2014.
|
|
15
|
Ke J, Yao B, Li T, Cui S and Ding H: A2
adenosine receptor-mediated cardioprotection against reperfusion
injury in rat hearts is associated with autophagy downregulation. J
Cardiovasc Pharmacol. 66:25–34. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Nikoletopoulou V, Markaki M, Palikaras K
and Tavernarakis N: Crosstalk between apoptosis, necrosis and
autophagy. Biochim Biophys Acta. 1833:3448–3459. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Xu J, Qin X, Cai X, Yang L, Xing Y, Li J,
Zhang L, Tang Y, Liu J, Zhang X and Gao F: Mitochondrial JNK
activation triggers autophagy and apoptosis and aggravates
myocardial injury following ischemia/reperfusion. Biochim Biophys
Acta. 1852:262–270. 2015. View Article : Google Scholar
|
|
18
|
Shen M, Lu J, Dai W, Wang F, Xu L, Chen K,
He L, Cheng P, Zhang Y, Wang C, et al: Ethyl pyruvate ameliorates
hepatic ischemia-reperfusion injury by inhibiting intrinsic pathway
of apoptosis and autophagy. Mediators Inflamm. 2013:4615362013.
View Article : Google Scholar
|
|
19
|
Zhao Y, Xu Y, Zhang J and Ji T:
Cardioprotective effect of carvedilol: inhibition of apoptosis in
H9c2 cardiomyocytes via the TLR4/NF-κB pathway following
ischemia/reperfusion injury. Exp Ther Med. 8:1092–1096.
2014.PubMed/NCBI
|
|
20
|
Xu Y, Jagannath C, Liu XD, Sharafkhaneh A,
Kolodziejska KE and Eissa NT: Toll-like receptor 4 is a sensor for
autophagy associated with innate immunity. Immunity. 27:135–144.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Porakishvili N, Vispute K, Steele AJ,
Rajakaruna N, Kulikova N, Tsertsvadze T, Nathwani A, Damle RN,
Clark EA, Rai KR, et al: Rewiring of sIgM-mediated intracellular
signaling through the CD180 Toll-like receptor. Mol Med. 21:46–57.
2015. View Article : Google Scholar :
|
|
22
|
Wezel A, van der Velden D, Maassen JM,
Lagraauw HM, de Vries MR, Karper JC, Kuiper J, Bot I and Quax PH:
RP105 deficiency attenuates early atherosclerosis via decreased
monocyte influx in a CCR2 dependent manner. Atherosclerosis.
238:132–139. 2015. View Article : Google Scholar
|
|
23
|
Louwe MC, Karper JC, de Vries MR, Nossent
AY, Bastiaansen AJ, van der Hoorn JW, Willems van Dijk K, Rensen
PC, Steendijk P, Smit JW and Quax PH: RP105 deficiency aggravates
cardiac dysfunction after myocardial infarction in mice. Int J
Cardiol. 176:788–793. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Karper JC, Ewing MM, de Vries MR, de Jager
SC, Peters EA, de Boer HC, van Zonneveld AJ, Kuiper J, Huizinga EG,
Brondijk TH, et al: TLR accessory molecule RP105 (CD180) is
involved in post-interventional vascular remodeling and soluble
RP105 modulates neointima formation. PLoS One. 8:e679232013.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yang J, Guo X, Yang J, Ding JW, Li S, Yang
R, Fan ZX and Yang CJ: RP105 protects against apoptosis in
ischemia/reperfusion-induced myocardial damage in rats by
suppressing TLR4-mediated signaling pathways. Cell Physiol Biochem.
36:2137–2148. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yang J, Jiang H, Chen SS, Chen J, Li WQ,
Xu SK and Wang JC: Lentivirus-mediated RNAi targeting CREB binding
protein attenuates neointimal formation and promotes
re-endothelialization in balloon injured rat carotid artery. Cell
Physiol Biochem. 26:441–448. 2010. View Article : Google Scholar
|
|
27
|
Zeng M, Wei X, Wu Z, Li W, Li B, Zhen Y,
Chen J, Wang P and Fei Y: NF-κB-mediated induction of autophagy in
cardiac ischemia/reperfusion injury. Biochem Biophys Res Commun.
436:180–185. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Shyu KG, Wang BW, Lin CM and Chang H:
Cyclic stretch enhances the expression of toll-like receptor 4 gene
in cultured cardiomyocytes via p38 MAP kinase and NF-kappaB
pathway. J Biomed Sci. 17:152010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Eltzschig HK and Eckle T: Ischemia and
reperfusion - from mechanism to translation. Nat Med. 17:1391–1401.
2011. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Divanovic S, Trompette A, Atabani SF,
Madan R, Golenbock DT, Visintin A, Finberg RW, Tarakhovsky A, Vogel
SN, Belkaid Y, et al: Negative regulation of Toll-like receptor 4
signaling by the Toll-like receptor homolog RP105. Nat Immunol.
6:571–578. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yazawa N, Fujimoto M, Sato S, Miyake K,
Asano N, Nagai Y, Takeuchi O, Takeda K, Okochi H, Akira S, et al:
CD19 regulates innate immunity by the toll-like receptor RP105
signaling in B lymphocytes. Blood. 102:1374–1380. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hsieh YC, Athar M and Chaudry IH: When
apoptosis meets autophagy: deciding cell fate after trauma and
sepsis. Trends Mol Med. 15:129–138. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yang Y, Gao K, Hu Z, Li W, Davies H, Ling
S, Rudd JA and Fang M: Autophagy upregulation and apoptosis
downregulation in DAHP and triptolide treated cerebral ischemia.
Mediators Inflamm. 2015:1201982015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Maejima Y, Kyoi S, Zhai P, Liu T, Li H,
Ivessa A, Sciarretta S, Del Re DP, Zablocki DK, Hsu CP, et al: Mst1
inhibits autophagy by promoting Beclin1-Bcl-2 interaction. Nat Med.
19:1478–1488. 2013. View Article : Google Scholar : PubMed/NCBI
|