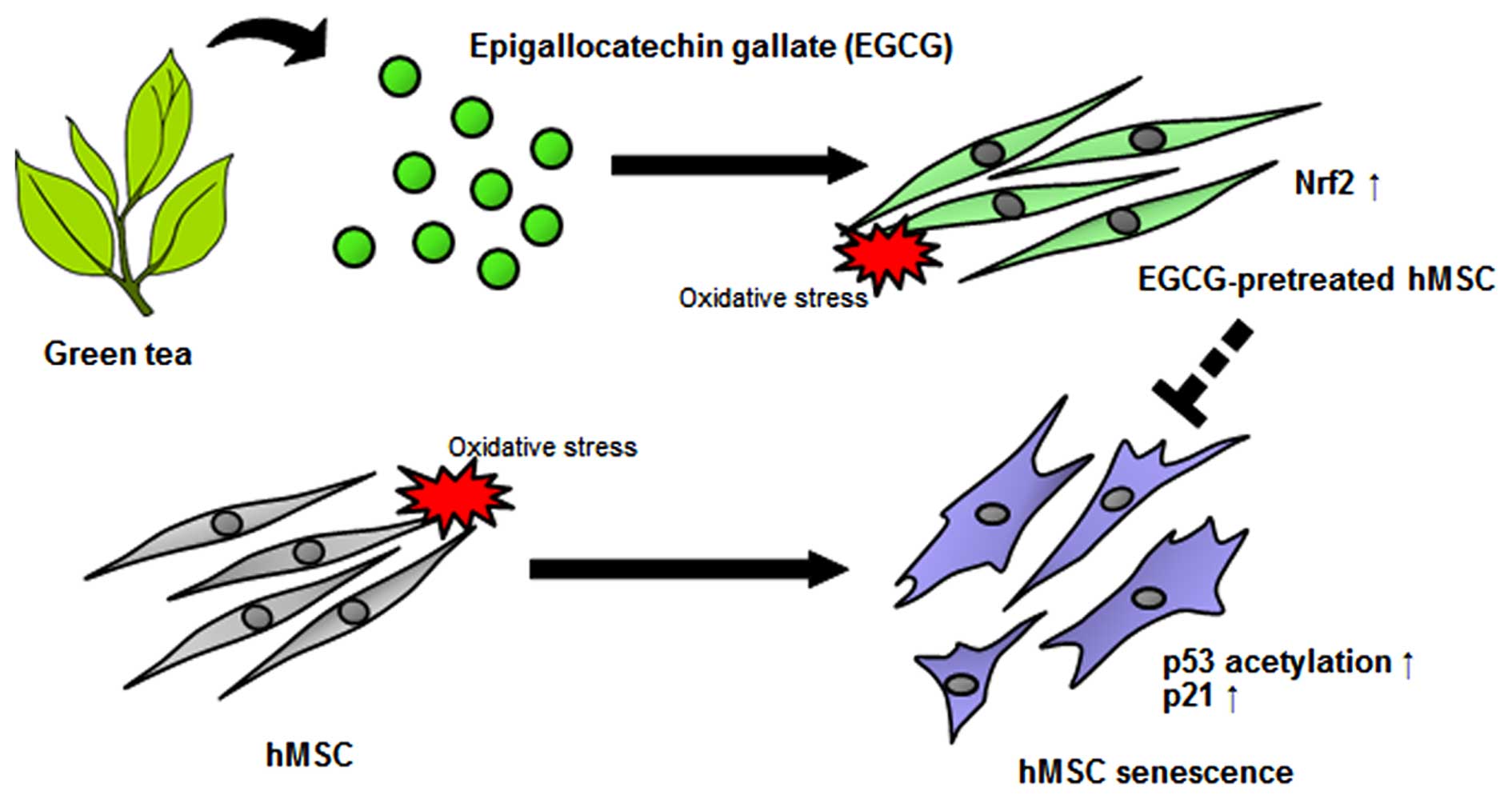
Introduction
Bone marrow-derived human mesenchymal stem cells
(hMSCs) are a desirable cell source for cell-based therapy owing to
their high plasticity, immune privileged status and ease of
preparation, as well as a lack of ethical barriers to their use.
They also have high self-renewal capacity with sustained
proliferation in vitro (1,2).
However, obtaining the large numbers of cells required for
therapeutic applications is often problematic as hMSCs are subject
to the Hayflick limit, a finite proliferation capacity in
vitro and replicative senescence after long-term culture
(3–5). Senescent cells have shown reduced
multipotency, clonogenicity and subsequent arrest of proliferation,
thus limiting the regenerative potential of hMSCs necessary for the
desired therapeutic effects (5).
Cellular senescence is characterized by irreversible
cell cycle arrest, despite continued metabolic activity and
viability. Senescence is caused by inadequate culture conditions,
such as culture shock or cellular stress (3,4).
The stress-induced premature senescence (SIPS) of human stem cells
may be induced by subcytotoxic stress (H2O2,
histone deacetylase inhibitors and radiation) (5,6).
Oxidative stress, mediated by reactive oxygen
species (ROS) including hydrogen peroxide
(H2O2), superoxide anion radical, hydroxyl
radical and peroxide, plays a crucial role in the induction of SIPS
(3,4). Sublethal concentrations of
H2O2 may damage cellular components including
DNA, which leads to low metabolic activity and cell cycle arrest
through the activation of either the p53/p21 or the p16/pRb pathway
(7). Notably, p53 acetylation,
which is induced by Sirt1, the human homolog of yeast SIR2, has
been proposed to promote senescence (8–11).
Acetylation of p53 is a translational modification that results in
the activation of p53. Cellular senescence was observed in
serially-passaged and H2O2-treated human
dermal fibroblast cells and acetyl-p53 levels were markedly
increased compared with phosphorylated p53 levels (12). These findings suggest an
association between oxidative stress-mediated senescence and p53
acetylation.
Polyphenols, or polyphenolic compounds, are widely
distributed in nature. Polyphenols, such as the green tea
polyphenol epigallocatechin-3-gallate (EGCG), have been
demonstrated to exhibit various biological properties, including
DNA damage protection and free radical scavenging (13). Furthermore, polyphenols are
pharmacologically safe compounds in humans (14). In addition to the ability to act
as a neutralizing agent of excessive ROS, EGCG exerts antioxidant,
anti-inflammatory and anti-tumorigenic effects (15). Recently, EGCG has been shown to
suppress H2O2-mediated apoptotic cell death
in hMSCs (16). It is well known
that EGCG exerts an antioxidant effect by activating the nuclear
factor-erythroid 2-related factor 2 (Nrf2) signaling pathway, which
is involved in the cellular antioxidant defense system (17). Nrf2 activation is closely
regulated by Kelch-like ECH-associated protein 1 (Keap1), a
substrate adaptor for Cul3-based E3 ligase, which targets Nrf2 for
proteasomal degradation (18). In
response to oxidative stress, Nrf2 upregulates the expression of
antioxidant and detoxifying genes by binding to antioxidant
response elements (AREs) in the promoter region of the encoding
genes (19,20).
The purpose of this study was to examine the novel
molecular mechanisms underlying the anti-senescent effect of EGCG
in H2O2-exposed hMSCs. Our data demonstrated
that EGCG reversed H2O2-induced oxidative
stress by downregulating the p53-p21 signaling pathway and
upregulating Nrf2 expression. Nrf2-knockdown hMSCs showed
significantly increased protein levels of acetyl-p53 and p21
following EGCG pre-treatment and H2O2
exposure, which suggests a potential role for Nrf2 in p53/p21
regulation to thereby prevent oxidative stress-induced cellular
senescence in hMSCs.
Materials and methods
Culture of hMSCs
Adult bone marrow-derived hMSCs were purchased from
Cambrex (Walkersville, MD, USA). hMSCs (passages 4–10) were
cultured in Dulbecco's modified Eagle's medium (DMEM) low glucose
containing 10% fetal bovine serum (FBS) (both from Gibco, Grand
Island, NY, USA) at 37°C with 5% CO2.
EGCG treatment and exposure of cells to
H2O2
EGCG and H2O2 were purchased
from Sigma-Aldrich (St. Louis, MO, USA). To define the optimal
concentrations for use in subsequent experiments, hMSCs were
pre-incubated with different amounts of EGCG (50 and 100 μM)
for 6 h and then the cells were exposed to 200 μM
H2O2 (diluted in DMEM supplemented with 10%
FBS) for 2 h. The cells were washed twice with DMEM to remove
excess H2O2 and re-incubated in fresh
complete medium for 24 h to prevent cell death and allow for the
observation of senescent characteristics
Cellular senescence assay
The activity of senescence-associated
β-galactosidase (SAβ-gal), a marker of senescence, was analyzed in
hMSCs using a cellular senescence assay kit (EMD Millipore,
Billerica, MA, USA) according to the manufacturer's instructions.
Briefly, the medium was aspirated and the cells were washed once
with phosphate-buffered saline (PBS; pH 6.0). After fixing the
cells with 1X fixing solution at room temperature for 10 min, the
cells were washed again with PBS and incubated without light for at
least 4 h with prepared SAβ-gal detection solution at 37°C without
CO2. The percentage of senescence-stained cells was
obtained by counting the number of blue-stained cells and the total
number of cells per field under the microscope (CKX41; Olympus,
Tokyo, Japan; 100–200 cells in four random fields).
Cell viability assay
Cell viability was analyzed using the
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
method. Briefly, hMSCs were cultured in 24-well tissue culture
plates and exposed to 200 μM H2O2 for
2 h. After 24 h, the cells were stained with 1 mg/ml MTT
(Sigma-Aldrich). The media were then carefully aspirated and 150
μl dimethyl sulfoxide (DMSO) was added to solubilize the
colored formazan product. The optical density was read at 554 nm
using a microplate reader (Floustar Optima; BMG Labtech, Ortenberg,
Germany).
Western blot analysis
The cells were washed twice with cold PBS and lysed
with RIPA buffer [50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% NP-40,
0.25% sodium deoxycholate, 0.2 mg/ml leupeptin, 0.2 mg/ml
aprotinin, 0.1 M phenylmethylsulfonylfluoride (PMSF), 1 mM
Na3VO4 and 0.5 M NaF]. The lysates were
centrifuged at 13,500 × g for 15 min at 4°C and the supernatants
were loaded on to 12% sodium dodecyl sulfate-polyacrylamide gel
electrophoresis (SDS-PAGE) gels. The following primary antibodies
were used: rabbit anti-p53 (1:1,000; sc-6243; Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA); rabbit anti-acetyl p53
(1:1,000; 06-758; Upstate Biotechnology, Lake Placid, NY, USA);
mouse anti-p21 (1:2,000; sc-6246) and rabbit anti-Nrf2 (1:1,000;
SC-722) (both from Santa Cruz Biotechnology, Inc.); mouse
anti-α-tubulin (1:5,000; T5168; Sigma-Aldrich) and goat anti-lamin
B (1:2,000; sc-6216; Santa Cruz Biotechnology, Inc.). Primary
antibodies were detected using horseradish peroxidase-conjugated
goat anti-mouse (A2554), -rabbit (A0545) (Sigma-Aldrich), or donkey
anti-goat secondary antibodies (sc-2020; Santa Cruz Biotechnology,
Inc.) and visualized using an enhanced chemiluminescence detection
system (Thermo Fisher Scientific, Rockford, IL, USA).
Subcellular fractionation
To obtain nuclear and cytoplasmic fractions, the
cells were harvested and suspended in ice-cold cytoplasmic lysis
buffer (10 mM HEPES, pH 7.9, 10 mM KCl, 1.5 mM MgCl2,
0.5 mM DTT and 0.2 mM PMSF) on ice for 15 min. The suspensions were
then centrifuged at 13,500 × g for 10 min at 4°C and the
supernatants were saved as the cytoplasmic fractions. The pellets
were resuspended in nuclear lysis buffer (20 mM HEPES, pH 7.9, 20%
glycerol, 0.4 M NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5 mM
DTT and 0.2 mM PMSF) and incubated on ice for 40 min with
occasional gentle shaking. The suspensions were then centrifuged at
13,500 × g for 15 min and the supernatants were used as nuclear
fractions. Quantification of the results of western blot analysis
was performed using ImageJ software (NIH, Bethesda, MD, USA).
Immunocytochemistry
The hMSCs were pre-incubated with 100 μM EGCG
for 6 h, fixed in PBS containing 4% PFA and incubated overnight at
4°C with rabbit anti-Nrf2 (1:100). Alexa Fluor 546 anti-rabbit IgG
(Invitrogen, Carlsbad, CA, USA) was used as a secondary antibody.
The cells were counterstained with 100 ng/ml
4,6-diamidino-2-phenylindole (DAPI) (Santa Cruz Biotechnology,
Inc.) for nuclear staining and visualized using a confocal laser
scanning microscope (FV300; Olympus).
Transfection of small interfering RNA
(siRNA)
Human Nrf2-specific siRNA oligo nucleotides
(SMARTpool) were purchased from Dharmacon (Lafayette, CO, USA). The
following target specific siRNA sequences were used:
5′-UAAAGUGGCUGCUCAGAAU-3′; 5′-GAGUUACAGUGUCUUAAUA-3′;
5′-UGGAGUAAGUCGAGAAGUA-3′; and 5′-CACCUUAUAUCUCGAAGUU-3′.
Non-targeting scrambled 20–25 nt siRNA oligonucleotides (Santa Cruz
Biotechnology, Inc.) were used as a control. Transient
transfections were performed using DharmaFECT 3 transfection
reagent (Dharmacon) according to the manufacturer's instructions.
Briefly, siRNA/lipid complexes were added to the wells at a final
concentration of 100 nM siRNA and 1 μl/well of DharmaFECT 3.
Nrf2 gene expression was determined at 48 h after transfection.
Statistical analysis
Statistical analysis was performed by analysis of
variance (ANOVA) followed by a post hoc Newman-Keuls test. P-values
<0.05 were considered to indicate a statistically significant
difference. All data are presented as the means ± standard error of
mean (SEM).
Results
EGCG pre-treatment reduces cellular
senescence in H2O2- treated hMSCs
The stimulation of cells with exogenous ROS
activates various signaling pathways that result in DNA damage,
cellular senescence and apoptosis (3). In order to examine the effects of
H2O2 exposure on cellular senescence, the
hMSCs were exposed to 200 μM H2O2
diluted in DMEM supplemented with 10% FBS for 2 h, in order to
allow the observation of senescent characteristics without
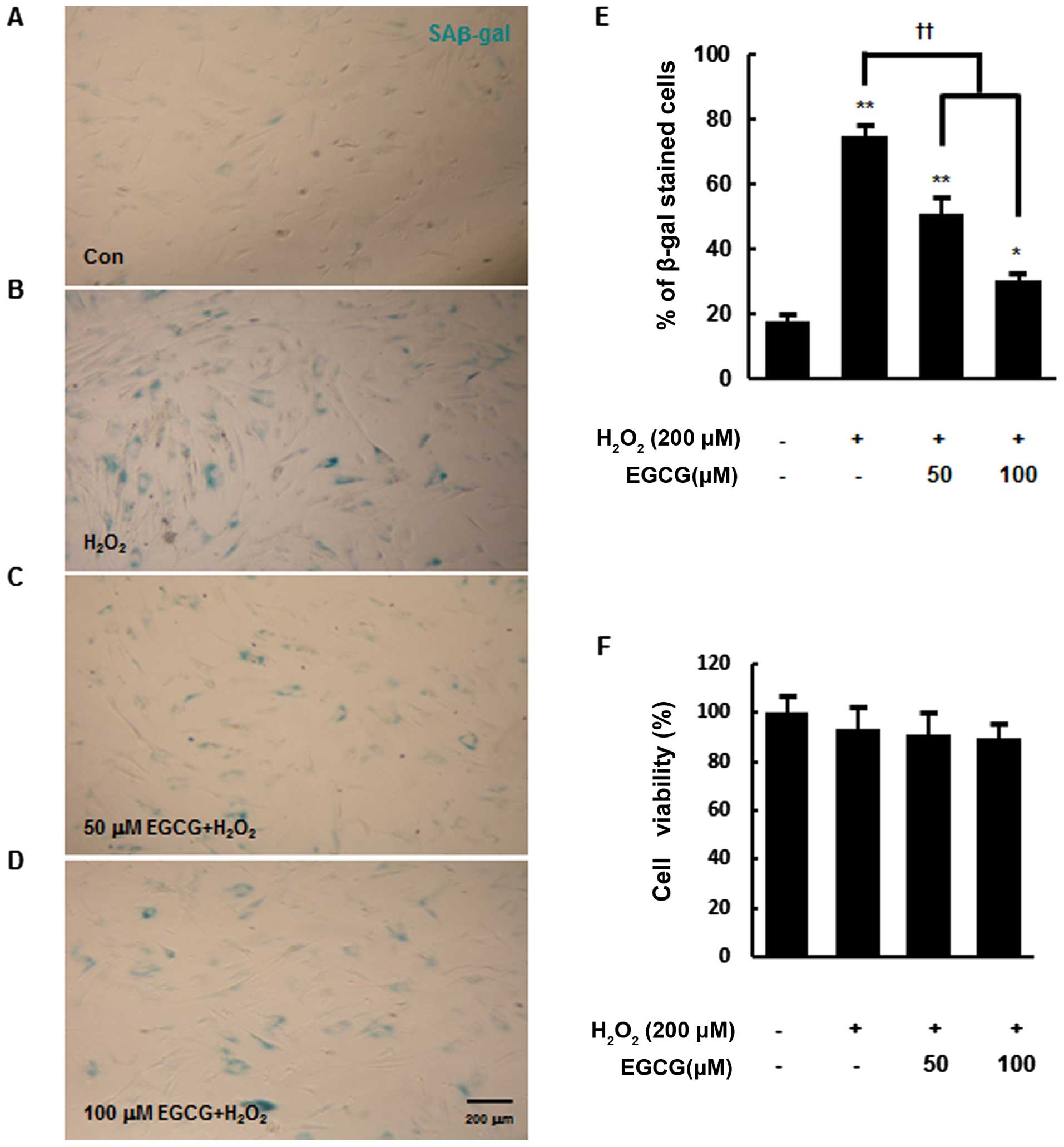
significant cell death. In the present study, the activity of
SAβ-gal was measured by SAβ-gal staining at 24 h after
H2O2 exposure. Approximately 75% of
H2O2-exposed hMSCs were positive for SAβ-gal
(blue cytoplasmic stain) (74.6±3.6%), whereas only 20% of the
control cells without H2O2 exposure were
SAβ-gal-positive (P<0.01) (Fig.
1A, B and E). However, the pre-treatment of hMSCs with 50 or
100 μM EGCG for 6 h reduced the percentage of
SAβ-gal-positive cells following H2O2
exposure to 50.7±4.8 and 30.4±1.9%, respectively (P<0.01)
(Fig. 1C–E). Taken together,
these results suggest that cellular senescence in hMSCs is
accelerated by H2O2 exposure and EGCG
pre-treatment reduces this acceleration in a dose-dependent manner.
Furthermore, there were no significant differences in cell death
among the experimental groups, indicating that
H2O2 exposure induced cellular senescence
without causing significant cell death (Fig. 1F).
EGCG pre-treatment reduces
H2O2-induced increases in acetylated p53 and
p21 protein levels in hMSCs
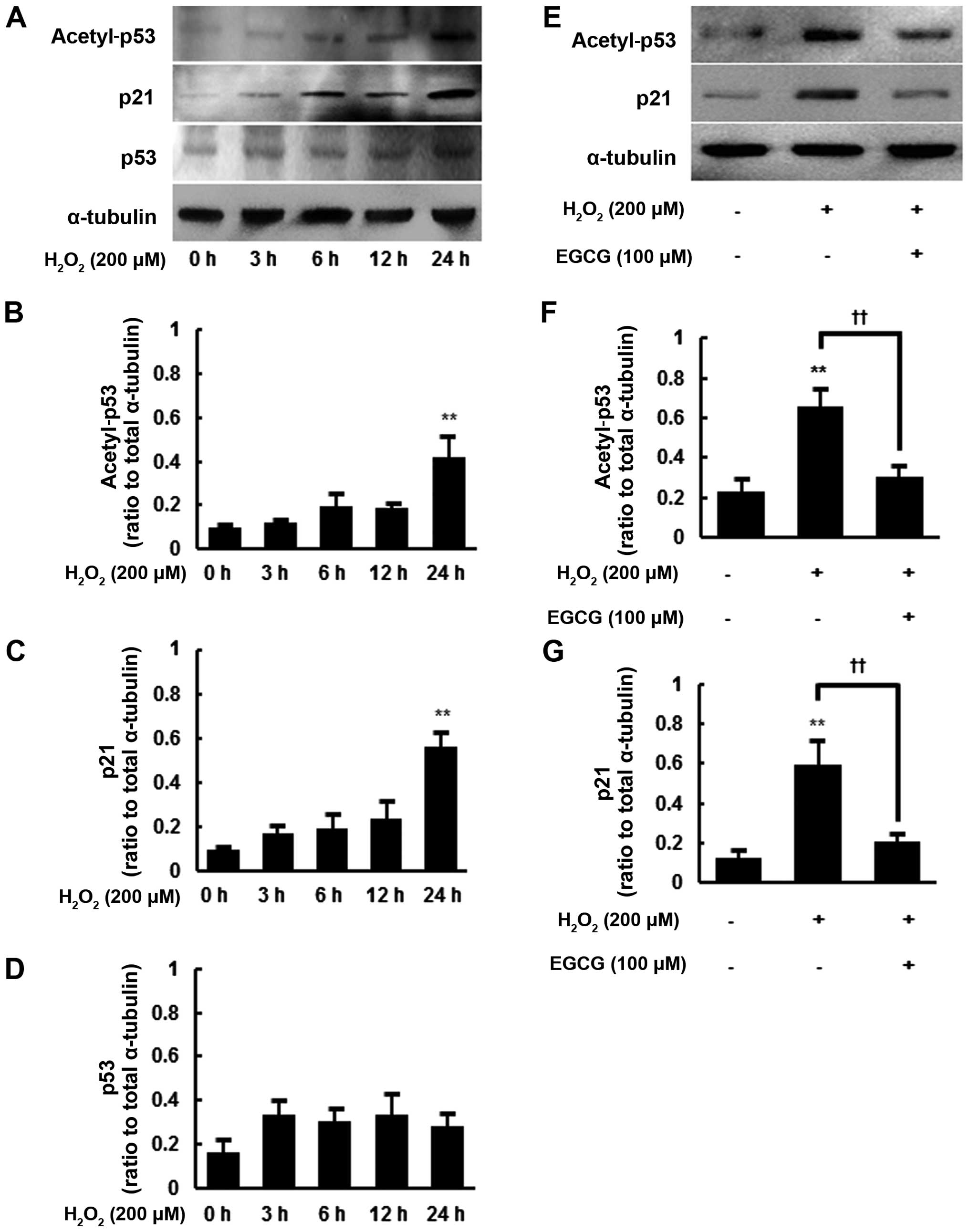
To further evaluate
H2O2-induced changes in senescent cells, we
next examined the protein levels of acetyl-p53, p53 and p21 in
hMSCs at different times following 200 μM
H2O2 exposure. The expression of p53 and p21
is known to correlate with senescence in human primary cells and
p53 acetylation has been shown to strongly promote cellular
senescence (8,12). Consistent with the findings of
previous studies, there were senescence-associated increases in the
protein levels of acetyl-p53, p21 and p53 following 200 μM
H2O2 exposure (Fig. 2A–D). Particularly after 24 h of
H2O2 exposure, the protein levels of
acetyl-p53 and p21 were significantly increased by up to 4.4- and
5.9-fold, respectively, compared with the controls, (P<0.01)
(Fig. 2B and C). Despite an
increasing trend in total p53 protein levels following
H2O2 exposure, the increase did not reach
statistical significance (Fig.
2D).
Thus, we decided to examine the effect of EGCG
pretreatment on acetyl-p53 and p21 protein levels in hMSCs after 24
h of H2O2 exposure. As previously shown,
there were significant increases in acetyl-p53 and p21 protein
levels at 24 h after 200 μM H2O2
exposure (P<0.01) (Fig. 2E–G).
However, EGCG pre-treatment (100 μM) significantly decreased
the protein levels of acetyl-p53 and p21 in the
H2O2-exposed hMSCs by 46.3±8.1 and 35.1±6.5%
(P<0.01), respectively, compared with the cells given no EGCG
pretreatment (Fig. 2E–G).
EGCG induces nuclear translocation of
Nrf2 in hMSCs
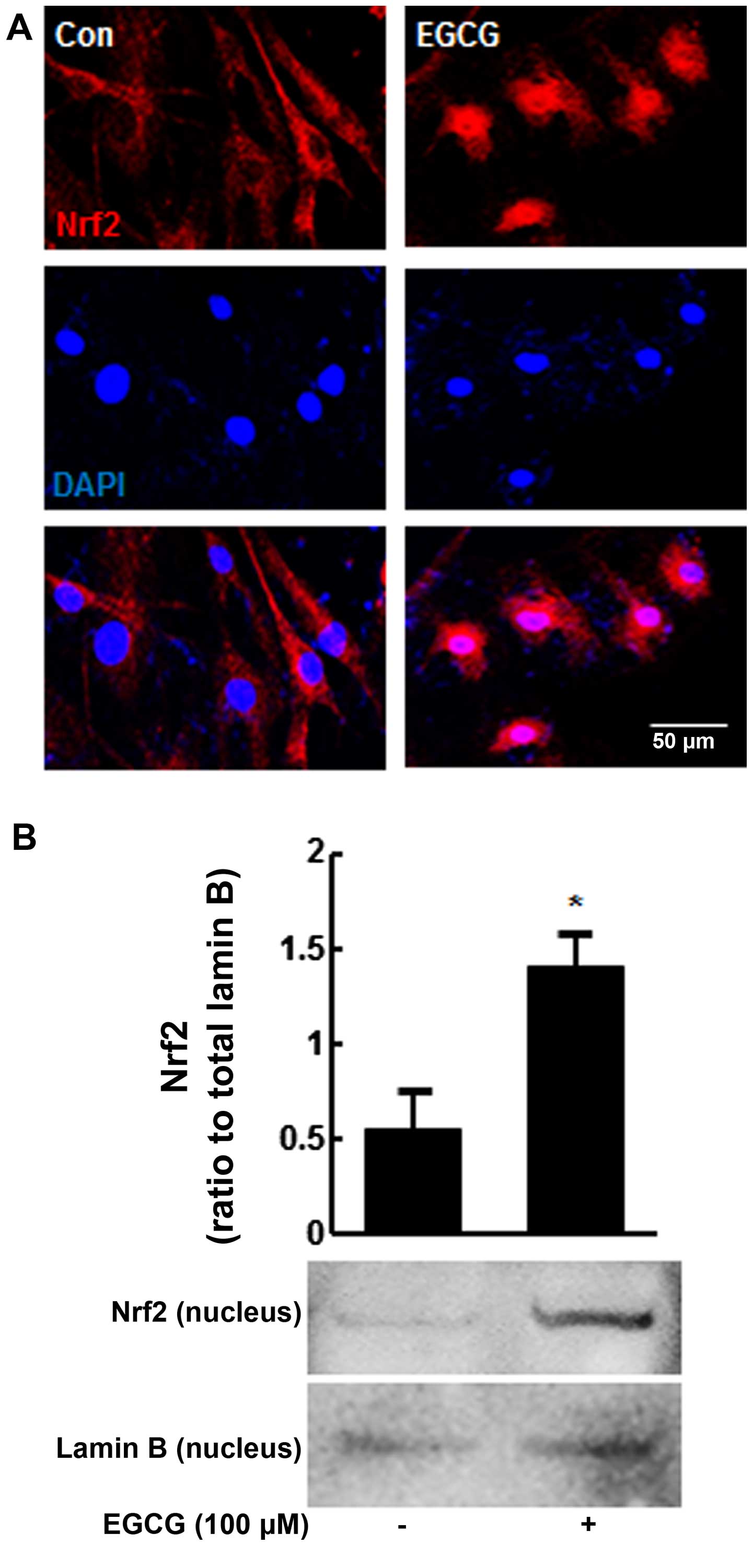
To determine whether the suppression of cellular
senescence by EGCG in H2O2-exposed hMSCs is
associated with Nrf2 activation, we performed double-labeling
experiments with anti-Nrf2 antibody and DAPI after 6 h of EGCG
treatment (100 μM). Nrf2 was mostly found to be localized in
the cytoplasm in the untreated cells (Fig. 3A, left panel). However, marked
translocation of Nrf2 to the nuclei was observed after 6 h of EGCG
treatment, although some Nrf2 remained in the cytoplasm (Fig. 3A, right panel). In addition,
nuclear fractions were subjected to western blot analysis, showing
that pre-treatment with EGCG increased nuclear Nrf2 protein levels
2.5-fold compared with the untreated cells (P<0.05) (Fig. 3B).
EGCG pre-treatment suppresses
H2O2-induced cellular senescence and the
expression of acetyl-p53 and p21 in hMSCs through Nrf2
activation
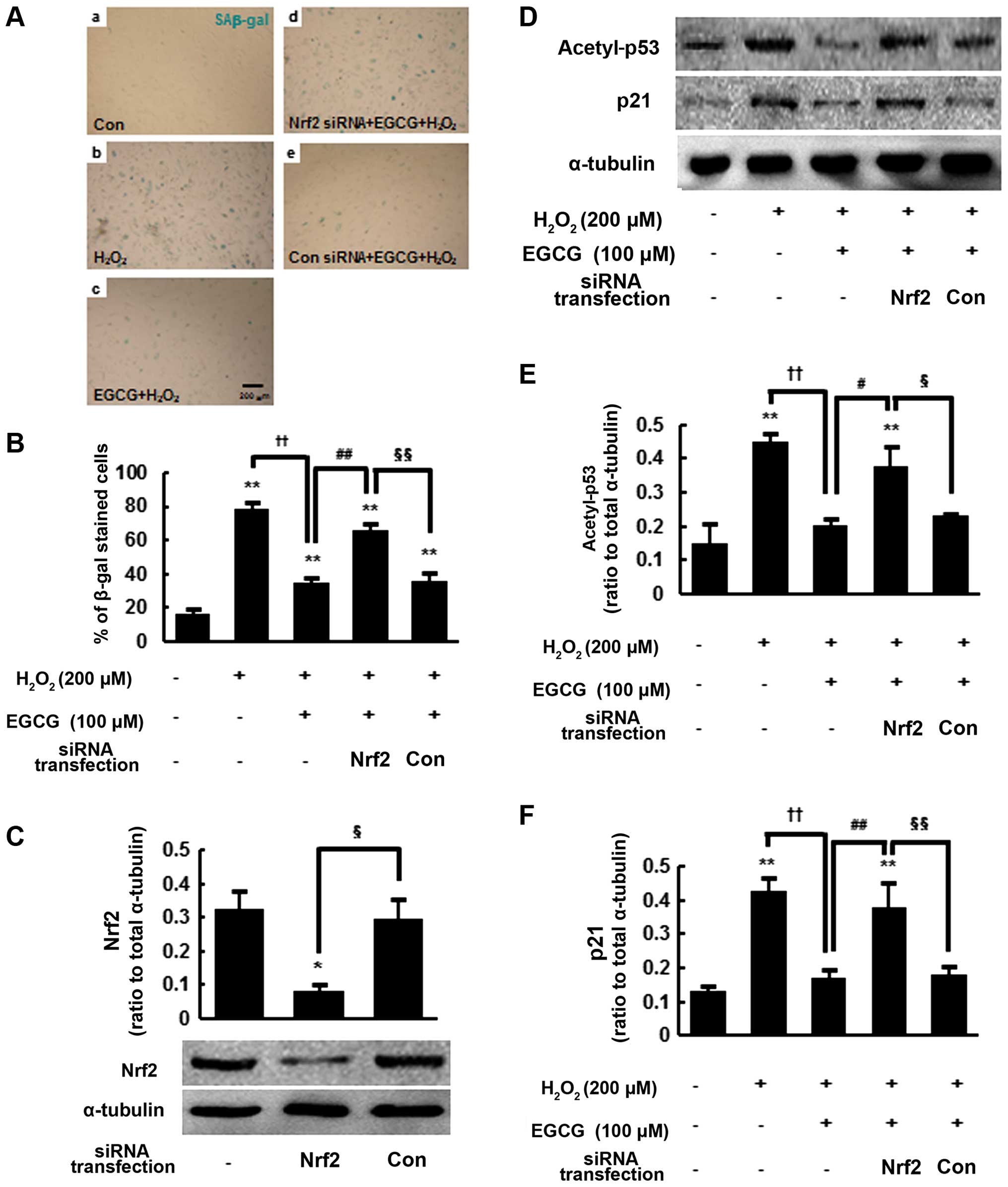
We hypothesized that Nrf2 activation may play an
important role in the anti-senescence effects of EGCG. To test this
hypothesis, we performed SAβ-gal staining at 48 h after
siRNA-mediated Nrf2 knockdown or control siRNA transfection
(Fig. 4A). As previously shown in
Fig. 1, the percentage of
SAβ-gal-positive cells in the 100 μM
EGCG-pretreated/H2O2-exposed group was
significantly reduced (35.1±2.5%) compared with the
H2O2-exposed cells without pre-treatment
(78.4±3.7%) (P<0.01) (Fig. 4A,
panels b and c and 4B). However, EGCG-pre-treated and
H2O2-exposed/Nrf2-siRNA-transfected cells
exhibited increased positive staining for SAβ-gal (65.6±3.9%)
(P<0.01), which is similar to that of the
H2O2-exposed cells (Fig. 4A, panels b and d and 4B). By
contrast,
EGCG-pre-treated/H2O2-exposed/control-siRNA-transfected
cells stained positive at a significantly lower rate of 36±4.2%,
which is similar to that of the
EGCG-pre-treated/H2O2-treated cells (Fig. 4A, panels c and e and 4B). We
confirmed that Nrf2 protein levels were reduced to 30±5.4% at 48 h
after Nrf2 siRNA transfection compared with the control siRNA
(P<0.05) (Fig. 4C). These
results suggest that Nrf2 may play an important role in the
anti-senescence activity of EGCG.
We next examined acetyl-p53 and p21 protein levels
in Nrf2-knockdown hMSCs. As previously shown (Fig. 2E–G), acetyl-p53 and p21 protein
levels were significantly reduced by 44.6±3.7 and 39.7±5.4%,
respectively, in the
EGCG-pretreated/H2O2-exposed cells compared
with the H2O2-exposed cells (P<0.01)
(Fig. 4D–F). However, at 48 h
after Nrf2-siRNA transfection, acetyl-p53 and p21 protein levels
were significantly increased in the
EGCG-pre-treated/H2O2-exposed cells. The
protein levels of acetyl-p53 and p21 were similar to those in the
H2O2-exposed cells (Fig. 4D–F). By contrast, control siRNA
transfection did not change the acetyl-p53 and p21 protein levels
in the EGCG-pre-treated/H2O2-exposed cells.
Taken together, these results indicate that Nrf2 activation by EGCG
pre-treatment suppresses H2O2-induced
cellular senescence and the expression of acetyl-p53 and p21 in
hMSCs (Fig. 5).
Discussion
The therapeutic applications of hMSCs are often
limited by various factors, including senescence caused by the
inadequate culture conditions that affect their capacity for
self-renewal and differentiation (3–5).
Therefore, modulating hMSCs to block oxidative stress-induced
cellular senescence may improve their clinical utility. Oxidative
stress has been shown to induce cellular senescence as previously
observed in human primary cells and hMSCs (6,12,21). In the present study, we also
observed a significant increase in the number of SAβ-gal-positive
hMSCs following H2O2 exposure, which induces
cellular senescence by generating intracellular ROS (3,4).
EGCG, a polyphenol, is a strong neutralizing agent
of excessive ROS and induces Nrf2 expression (17). Nrf2 plays an important role in the
cellular antioxidant defense system by activating the expression of
antioxidant and detoxifying genes, such as superoxide dismutase,
heme oxygenase 1, and glutathione S-transferases. These genes have
been shown to protect cells against oxidative stress caused by ROS
by restoring redox homeostasis and inhibiting oxidative damage
(20). A recent study has
reported that EGCG suppressed H2O2-mediated
oxidative stress in hMSCs (16).
Consistently, our results also demonstrated that EGCG prevented
H2O2-induced senescence in hMSCs.
ARE-mediated antioxidant gene expression is a widely
accepted model for the activity of EGCG (20). In general, the serine/threonine
residues of Nrf2 are phosphorylated by protein kinases such as
PI3K, ERK, p38 and JNK thereby enhancing the nuclear translocation
of Nrf2 and subsequent ARE binding. Oxidized or other reactive
forms of EGCG conjugate with glutathione (GSH) and decrease
cellular GSH concentrations, which leads to a disruption of the
redox state and the activation of upstream protein kinases,
triggering Nrf2 phosphorylation. It is also plausible that EGCG may
oxidize or modify specific cysteine thiol groups in Keap1 that
allow the nuclear translocation of Nrf2. We observed the marked
translocation of Nrf2 into the nuclei after EGCG treatment
(Fig. 3). Both of these are
plausible mechanisms for EGCG-induced Nrf2 activation as
electrophilic agents or compounds have been reported to interact
with cysteine residues directly and stimulate Nrf2 dissociation
(22).
p53 acetylation has been shown to promote cellular
senescence in addition to activating growth suppressive genes
(23,24). The first confirmed downstream
target of p53, p21, is an essential regulator of p53-dependent cell
cycle arrest which leads to cell cycle arrest in response to DNA
damage. As a cyclin-dependent kinase inhibitor, p21 regulates the
function of cyclin D1/CDK4 and cyclin E/CDK2 complexes and induces
the accumulation of hypophosphorylated Rb, which leads to Rb
binding with E2F transcription factors, resulting in cell cycle
arrest (25,26). In addition, previous studies have
shown that p21 is a key regulator of cellular senescence in human
primary cells (27,28).
Recent studies have challenged the known paradigm of
Nrf2. The inhibition of Nrf2 by caveolin-1, a structural protein of
caveolae, reduces its cellular antioxidant response following
H2O2 exposure (29). The inhibition of Nrf2 also
suppresses the expression of murine double minute (Mdm2), an
oncogene which promotes p53 degradation, resulting in p53 pathway
activation (30). In addition to
the Keap1-Nrf2 complex formation, caveolin-1 and/or Mdm2 may be
candidates responsible for modulating p53 acetylation and p21
activation in hMSCs in response to oxidative stress. However,
further studies are warranted in order to elucidate the
physiological relevance of these mechanisms.
In conclusion, our results are consistent with the
hypothesis that Nrf2 activation inhibits oxidative stress in cells.
The upregulation of Nrf2 by EGCG prevented oxidative stress-induced
cellular senescence through the downregulation of p53 acetylation
and p21 in hMSCs. These findings demonstrate that EGCG is capable
of increasing Nrf2 activation in hMSCs and suggest a novel approach
for preventing the oxidative stress-induced cellular senescence of
human stem cells.
Acknowledgments
The present study was supported by the Basic Science
Research Program through the National Research Foundation of Korea
(M-SC, NRF-2015R1D1A1A01056950) funded by the Ministry of
Education, and by a grant from the Korean Health Technology R&D
Project (M-SC, A120476), Ministry of Health and Welfare, Republic
of Korea.
References
|
1
|
Pittenger MF1, Mackay AM, Beck SC, Jaiswal
RK, Douglas R, Mosca JD, Moorman MA, Simonetti DW, Craig S and
Marshak DR: Multilineage potential of adult human mesenchymal stem
cells. Science. 284:143–147. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sherman LS, Munoz J, Patel SA, Dave MA,
Paige I and Rameshwar P: Moving from the laboratory bench to
patients' bedside: considerations for effective therapy with stem
cells. Clin Transl Sci. 4:380–386. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kuilman T, Michaloglou C, Mooi WJ and
Peeper DS: The essence of senescence. Genes Dev. 24:2463–2479.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
López-Otín C, Blasco MA, Partridge L,
Serrano M and Kroemer G: The hallmarks of aging. Cell.
153:1194–1217. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Oh J, Lee YD and Wagers AJ: Stem cell
aging: mechanisms, regulators and therapeutic opportunities. Nat
Med. 20:870–880. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Brandl A, Meyer M, Bechmann V, Nerlich M
and Angele P: Oxidative stress induces senescence in human
mesenchymal stem cells. Exp Cell Res. 317:1541–1547. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Campisi J and d'Adda di Fagagna F:
Cellular senescence: when bad things happen to good cells. Nat Rev
Mol Cell Biol. 8:729–740. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kume S, Haneda M, Kanasaki K, Sugimoto T,
Araki S, Isono M, Isshiki K, Uzu T, Kashiwagi A and Koya D: Silent
information regulator 2 (SIRT1) attenuates oxidative stress-induced
mesangial cell apoptosis via p53 deacetylation. Free Radic Biol
Med. 40:2175–2182. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Langley E, Pearson M, Faretta M, Bauer UM,
Frye RA, Minucci S, Pelicci PG and Kouzarides T: Human SIR2
deacetylates p53 and antagonizes PML/p53-induced cellular
senescence. EMBO J. 21:2383–2396. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Luo J, Nikolaev AY, Imai S, Chen D, Su F,
Shiloh A, Guarente L and Gu W: Negative control of p53 by Sir2α
promotes cell survival under stress. Cell. 107:137–148. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Vaziri H, Dessain SK, Ng Eaton E, Imai SI,
Frye RA, Pandita TK, Guarente L and Weinberg RA: hSIR2(SIRT1)
functions as an NAD-dependent p53 deacetylase. Cell. 107:149–159.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Han DW, Lee MH, Kim B, Lee JJ, Hyon SH and
Park JC: Preventive effects of epigallocatechin-3-O-gallate against
replicative senescence associated with p53 acetylation in human
dermal fibroblasts. Oxid Med Cell Longev. 2012:8506842012.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Anderson RF, Fisher LJ, Hara Y, Harris T,
Mak WB, Melton LD and Packer JE: Green tea catechins partially
protect DNA from (.)OH radical-induced strand breaks and base
damage through fast chemical repair of DNA radicals.
Carcinogenesis. 22:1189–1193. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chow HH, Cai Y, Alberts DS, Hakim I, Dorr
R, Shahi F, Crowell JA, Yang CS and Hara Y: Phase I pharmacokinetic
study of tea polyphenols following single-dose administration of
epigallocatechin gallate and polyphenon E. Cancer Epidemiol
Biomarkers Prev. 10:53–58. 2001.PubMed/NCBI
|
|
15
|
Higdon JV and Frei B: Tea catechins and
polyphenols: health effects, metabolism, and antioxidant functions.
Crit Rev Food Sci Nutr. 43:89–143. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yagi H, Tan J and Tuan RS: Polyphenols
suppress hydrogen peroxide-induced oxidative stress in human
bone-marrow derived mesenchymal stem cells. J Cell Biochem.
114:1163–1173. 2013. View Article : Google Scholar
|
|
17
|
Surh YJ, Kundu JK, Na HK and Lee JS:
Redox-sensitive transcription factors as prime targets for
chemoprevention with anti-inflammatory and antioxidative
phytochemicals. J Nutr. 135(Suppl 12): 2993S–3001S. 2005.PubMed/NCBI
|
|
18
|
Hayes JD and McMahon M: NRF2 and KEAP1
mutations: permanent activation of an adaptive response in cancer.
Trends Biochem Sci. 34:176–188. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Itoh K, Wakabayashi N, Katoh Y, Ishii T,
Igarashi K, Engel JD and Yamamoto M: Keap1 represses nuclear
activation of antioxidant responsive elements by Nrf2 through
binding to the amino-terminal Neh2 domain. Genes Dev. 13:76–86.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Nguyen T, Nioi P and Pickett CB: The
Nrf2-antioxidant response element signaling pathway and its
activation by oxidative stress. J Biol Chem. 284:13291–13295. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Burova E, Borodkina A, Shatrova A and
Nikolsky N: Sublethal oxidative stress induces the premature
senescence of human mesenchymal stem cells derived from
endometrium. Oxid Med Cell Longev. 2013:4749312013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dinkova-Kostova AT, Holtzclaw WD and
Wakabayashi N: Keap1, the sensor for electrophiles and oxidants
that regulates the phase 2 response, is a zinc metalloprotein.
Biochemistry. 44:6889–6899. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bond J, Haughton M, Blaydes J, Gire V,
Wynford-Thomas D and Wyllie F: Evidence that transcriptional
activation by p53 plays a direct role in the induction of cellular
senescence. Oncogene. 13:2097–2104. 1996.PubMed/NCBI
|
|
24
|
Luo J, Li M, Tang Y, Laszkowska M, Roeder
RG and Gu W: Acetylation of p53 augments its site-specific DNA
binding both in vitro and in vivo. Proc Natl Acad Sci USA.
101:2259–2264. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
el-Deiry WS, Tokino T, Velculescu VE, Levy
DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW and
Vogelstein B: WAF1, a potential mediator of p53 tumor suppression.
Cell. 75:817–825. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Harper JW, Adami GR, Wei N, Keyomarsi K
and Elledge SJ: The p21 Cdk-interacting protein Cip1 is a potent
inhibitor of G1 cyclin-dependent kinases. Cell. 75:805–816. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Brown JP, Wei W and Sedivy JM: Bypass of
senescence after disruption of p21CIP1/WAF1 gene in normal diploid
human fibroblasts. Science. 277:831–834. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Herbig U, Wei W, Dutriaux A, Jobling WA
and Sedivy JM: Real-time imaging of transcriptional activation in
live cells reveals rapid up-regulation of the cyclin-dependent
kinase inhibitor gene CDKN1A in replicative cellular senescence.
Aging Cell. 2:295–304. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Volonte D, Liu Z, Musille PM, Stoppani E,
Wakabayashi N, Di YP, Lisanti MP, Kensler TW and Galbiati F:
Inhibition of nuclear factor-erythroid 2-related factor (Nrf2) by
caveolin-1 promotes stress-induced premature senescence. Mol Biol
Cell. 24:1852–1862. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
You A, Nam CW, Wakabayashi N, Yamamoto M,
Kensler TW and Kwak MK: Transcription factor Nrf2 maintains the
basal expression of Mdm2: an implication of the regulation of p53
signaling by Nrf2. Arch Biochem Biophys. 507:356–364. 2011.
View Article : Google Scholar : PubMed/NCBI
|