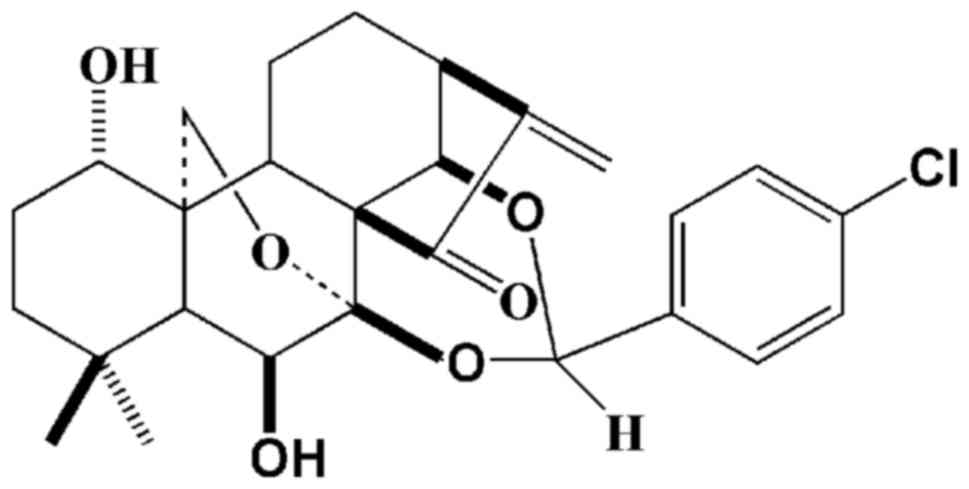
Introduction
Chemotherapy is one of the main therapeutic
strategies in the treatment of cancer. However, the success of
chemotherapy depends on the sensitivity of the tumor to the
anti-neoplastic agent (1). Cancer
cells can develop resistance to the anticancer drugs used during
cancer therapy (2). The drug
resistance properties of the tumor greatly limit the effectiveness
of chemotherapeutic drugs, resulting in the failure of chemotherapy
and the recurrence of the tumor. Furthermore, cancer cells exposed
to one drug may exhibit cross-resistance to other drugs that have
different structures and functions. This phenomenon is known as
multidrug resistance (MDR) and can be intrinsic or acquired
following chemotherapy using a cytotoxic agent (3,4).
Resistance to anticancer drugs is a major obstacle for successful
chemotherapy. Thus, the exploration of new drugs and strategies
with which to combat drug resistance is of utmost importance.
Paclitaxel (PTX) is frequently used in all clinical
chemotherapy protocols and is an important drug for the treatment
of esophageal carcinoma; however, resistance has been observed in
clinical practice. Resistance to PTX has been attributed to several
mechanisms, including the overexpression of the P-gp efflux pump
and resistance to apoptosis, among others (5,6).
Consequently, these mechanisms force cells to dysregulate
apoptosis, which facilitates tumor progression. The majority of
chemotherapeutic drugs kill cancer cells via the induction of
apoptosis; however, cancer cells can escape apoptosis or antagonize
apoptosis. Some scientists believe that cells with the MDR
characteristic often exhibit resistance to apoptosis, and
resistance to apoptosis may be one of the essential characteristics
of MDR (4,7,8).
Isodon rubescens, a Chinese herb, has been
used as a folk botanical medicine in China for the treatment of
cancer and inflammatory diseases for many years. Oridonin, an
active diterpenoid compound found in Isodon rubescens, has
been widely used in the treatment of human diseases ranging from
inflammation to cancer (9,10).
Jesridonin (JD; chemical structure shown in Fig. 1) is a diterpenoid compound that
was obtained via the structural modification of oridonin. Recently,
in a previous study, we demonstrated that JD exhibited strong
antitumor activity in EC109 human esophageal carcinoma cells, both
in vitro and in vivo, and demonstrated no adverse
effects on major organs in nude mice (11). However, the anti-tumor activity of
JD in paclitaxel-resistant cells remains unclear. To overcome the
obstacles related to drug resistance and to improve the clinical
outcomes of patients with esophageal squamous carcinoma (ESCC), in
this study, we examined the effects of JD on the PTX-resistant
EC109 human cancer cell line, EC109/Taxol, with the ultimate goal
of identifying a novel therapy for paclitaxel-resistant ESCC.
Materials and methods
Reagents
Propidium iodide (PI), dimethyl sulfoxide (DMSO),
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
and bisbenzimide Hoechst 33258 were from Sigma (St. Louis, MO,
USA). RNase A and the bicinchoninic acid (BCA) protein assay kit
were from Solarbio (Beijing, China). The rabbit monoclonal
antibodies against p53 (sc-393031), caspase-9 (sc-8355) and
caspase-3 (sc-65497) are the products of Santa Cruz Biotechnology,
Inc. (Santa Cruz, CA, USA). Rabbit monoclonal antibodies against
Bcl-2 (ZA-0536) and Bax (TA337090) were from Zhongshan Goldenbridge
Biotechnology, Co., Ltd. (Beijing, China). Primary antibody against
p53 upregulated modulator of apoptosis (PUMA; E1A5173) was from
Nanjing EnoGene Biotechnology (Nanjing, China). Primary antibody
against cleaved caspase-3 (#9661) and cleaved caspase-9 (#9501) was
purchased from Cell Signaling Technology (Danvers, MA, USA).
Peroxidase-conjugated affinipure goat anti-rabbit IgG (ZB-2301) and
anti-mouse IgG (ZB-2305) IgG as secondary antibodies were from
Zhongshan Goldenbridge Biotechnology, Co., Ltd.
Cell culture and compounds
The EC109 cell line was obtained from the Chinese
Academy of Sciences (Shanghai, China). The cells were cultured in
RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS)
(both from HyClone, Beijing, China), 100 U/ml of penicillin and 100
µg/ml of streptomycin at 37°C in a humidified air atmosphere
containing 5% CO2. The resistant cell line (EC109/Taxol)
was developed in vitro by the intermittent exposure of the
human ESCC cell line, EC109, to a high concentration of PTX, with a
stepwise increase in concentration over a period of 6 months. These
cells were developed, cultured and maintained as previously
described (8).
JD was obtained from the New Drug Research and
Development Center of Zhengzhou University. JD is a 7,14-acetal
derivative of Oridonin (a natural antitumor compound isolated from
Isodon rubescens). The chemical structure was confirmed
using NMR, MS and IR data (Fig.
1). For the in vitro assay, JD was dissolved in DMSO and
stored at −20°C. The concentration of DMSO in the culture medium
was under 1% (v/v) and had no intrinsic effect on cell
proliferation.
Animals
A total of 15 female 6-week-old BALB/c nu/nu mice
weighing 18–19 g each were purchased from Hunan Slack King of
Laboratory Animal, Co., Ltd. (Hunan, China) and maintained under
specific pathogen-free (SPF) conditions at 25°C in an atmosphere
with 50% humidity for the experiments. Lighting was operated
automatically on a 12-h light/dark cycle. The mice were raised in a
sterile environment and received adequate water and food.
Throughout the trial period, all experiments strictly followed
institutional guidelines and were approved by the Experimental
Animal Care Committee of Zhengzhou University (approval no.
SPS140302).
Cytotoxic activity assays
The cells (8×103 cells/well) were
inoculated into each well in 96-well plates (Nest Biotechnology
Co., Ltd., Wuxi, Jiangsu, China) in 100 µl of culture
medium. Following an overnight incubation (37°C with 5%
CO2), the medium was removed and replaced with various
concentrations of JD. The plates were incubated at 37°C in 5%
CO2 for 24, 48 and 72 h. Thereafter, 20 µl of MTT
solution (5 mg/ml) were added to each well, and the plate was
incubated for an additional 4 h at 37°C. The resulting formazan
crystals were dissolved in 150 µl of DMSO following
aspiration of the culture medium. The plates were read at 490 nm
using a microplate reader (BioTek Instruments, Inc., Winooski, VT,
USA). The 50% inhibitory concentration (IC50) represents
the concentration of the modulators that was required for 50%
inhibition.
Cell proliferation assay
The cells were seeded at 1.4×104
cells/well (in 24-well plates). Following an overnight incubation,
the medium was removed and replaced by 10 µM of JD. Cells
from 3 wells were trypsinized and separately counted at the time
points of 24, 48, 72, 96, 120, 144 and 168 h following treatment
with JD.
Colony formation assay
A colony formation assay was performed to determine
the effects of JD treatment on the colony-forming ability of
EC109/Taxol cells. Briefly, the cells were seeded into 6-well
plates at 1×103 cells/well. Following an overnight
incubation, the medium was removed and replaced by various
concentrations of JD. Following incubation for 7 days, the cells
were then fixed with 95% ethanol for 30 min and stained with 0.5%
crystal violet for 30 min. Cell colonies (≥50 cells) were counted
under an inverted light microscope (BDS15010071, Aote light
microscope, Chongqing, China). The colony formation rate was
calculated as follows: (colony counts)/(cells inoculated)
×100%.
Assessment of cancer growth in mice
The evaluation of JD activity was also carried out
on nude mice bearing EC109/Taxol xenografts. The tumor model was
established as previously described (8) by subcutaneously inoculating
EC109/Taxol cells (1×107/mice) into the left armpits of
the mice. The tumors were allowed to reach an average size of
100–150 mm3 (as estimated by Vernier caliper
measurement), and the mice bearing the EC109/Taxol cells were then
randomly divided into 3 groups. The 3 groups of mice were treated
as follows: the mice in group C were the free control, the mice in
group T1 were treated with 5 mg/kg of JD, and the mice in group T2
were treated with 10 mg/kg of JD (n=5 mice/group). The drug (the
clathrate of JD was formed with β-cycloamylose and dissolved in
0.9% NaCl) and the vehicle (β-cycloamylose in 0.9% NaCl, 10 mg/kg)
were administered by intravenous tail vein injection every 3 days
for a total of 21 days. At the end of the treatment, the mice were
sacrificed by cervical vertebrae dislocation, and the tumors were
dissected and weighed. During this period, the body weights of the
animals were recorded every 3 days, and the tumor sizes were
measured by determining 2 perpendicular dimensions at the same
time. The volume (V) of each tumor was then calculated using the
following formula:
V=axb2x1/2
where length (a, mm) is the longest diameter and width (b, mm) is
the shortest diameter perpendicular to the length (,). The calculated tumor volumes were
expressed in percentages as relative tumor volume (RTV) using the
following equation ():
RTV=Vn/V0
where Vn is the tumor volume at day n of treatment and
V0 represents the initial tumor volume at the onset of
treatment. The inhibition of cancer growth was defined as a ratio
of the tumor weight compared to the vehicle control tumor weight.
The specimens from the EC109/Taxol xenografts were removed for
hematoxylin and eosin (H&E) staining.
H&E staining assay
The specimens of EC109/Taxol xenografts were fixed
in 10% formalin and then processed by the usual technique of
paraffin inclusion. The histological technique of paraffin
inclusion used in this study was performed in the following
sequence: dehydration, clarification, waxing and proper inclusion.
The embedded tissues were cut into 4-µm-thick sections,
affixed to glass slides, deparaffinized in xylene, rehydrated in
ethanol, rinsed in distilled water and then fixed with 4%
formaldehyde. Thereafter, the paraffin sections were stained with
H&E, dehydrated in graded alcohol, cleared in xylene, cemented
with neutral resin. General morphologies were viewed under a light
microscope (347655; Leica Microsystems CMS GmbH, Wetzlar,
Germany).
Nuclear staining by Hoechst 33258
The cells were seeded in a 6-well plate and treated
with JD at the indicated concentrations for 24 h. The cells were
then washed with phosphate-buffered saline (PBS) 3 times followed
by fixation with 4% paraformaldehyde for 15 min, then washed 3
times with PBS. Hoechst 33258 was subsequently added at a final
concentration of 10 µg/ml in the dark. Thirty minutes later,
the cells were carefully washed twice with PBS. The apoptotic cells
were examined and identified according to the condensation and
fragmentation of their nuclei under a fluorescence microscope
(Eclipse TE2000-S; Nikon, Tokyo, Japan).
Cell cycle and apoptosis analysis by flow
cytometry
To determine the effect of JD on the cell cycle,
5.0×106 EC109/Taxol cells in the exponential growth
phase were treated with various concentrations of JD for 24 h.
After an incubation period, the cells were collected, centrifuged
and fixed with ice-cold 70% ethanol at 4°C overnight. The cells
were washed with PBS before staining with PI and then suspended in
staining buffer (50 µg/ml PI, 1% Triton X-100 and 0.5 mg/ml
RNase A in PBS) at 37°C for 30 min in the dark. The cells were
analyzed on an Accuri C6 flow cytometer (Becton-Dickinson, Franklin
Lakes, NJ, USA). The histograms of DNA distribution were shown as a
sum of cell populations at the G0/G1, G2/M or S phase using FlowJo
software.
An Annexin V-fluorescein isothiocyanate kit (Annexin
V- FITC; Nanjing KeyGen Biotech Co., Ltd., Nanjing, China) was used
to stain and identify the percentage of apoptotic cells. Briefly,
the cells were seeded into 6-well plates and treated with 0, 10, 20
and 40 µM of JD for 24 h. The cells were then harvested,
washed with ice-cold PBS and resuspended in binding buffer
containing Annexin V-FITC (0.5 mg/ml) and PI (0.5 mg/ml). Following
15 min of incubation at room temperature in the dark, the stained
cells were analyzed immediately by an Accuri C6 flow cytometer
(Becton-Dickinson). Apoptotic cells were determined by cells that
were both Annexin V-positive and PI-negative.
Protein extraction and western blot
analysis
Western blot analyses were conducted as described
previously (8). The blots were
then probed with each of the following antibodies: anti-p53
antibody (1:1,000), anti-Bcl-2 antibody (1:500), anti-Bax antibody
(1:1,000), anti-PUMA antibody (1:500), anti-procaspase-3 antibody
(1:1,000), anti-cleaved-caspase-3 antibody (1:250) and
anti-procaspase-9 antibody (1:1,000). After washing the membrane
with PBST (PBS, 0.05% Tween-20) 3 times (10 min each), the blots
were subsequently incubated with a goat anti-mouse or anti-rabbit
IgG-HRP secondary antibody (1:10,000) at 37°C for 2 h. The
antibody-reactive bands were revealed by an enhanced
chemiluminescence substrate and were exposed to Kodak radiographic
film.
Statistical analysis
All statistical analyses were carried out using the
SPSS 17.0 statistical packages. The data are expressed as the means
± standard deviation (SD). Continuous variables were analyzed using
the Student's t-test. A value of P<0.05 was considered to
indicate a statistically significant difference, and a value of
P<0.01 a highly statistically significant difference.
Results
Inhibition of cancer cell growth in
vitro
Our initial experiment was carried out to determine
the inhibitory effect of JD on EC109 and EC109/Taxol cells by MTT
assay. The results revealed that JD displayed similar
IC50 values against drug-sensitive EC109 cells and
paclitaxel-resistant EC109/Taxol cells in vitro and
indicated that JD had a potent growth inhibitory effect on both of
these cell lines in a concentration- and time-dependent manner
(Fig. 2A and B and Table I).
 | Table IThe IC50 values of JD on
EC109 and EC109/Taxol cells. |
Table I
The IC50 values of JD on
EC109 and EC109/Taxol cells.
| Time (h) | IC50
(µM)
|
|---|
| EC109 | EC109/Taxol |
|---|
| 24 | 24.354±0.267 | 25.679±2.505 |
| 48 | 14.470±0.518 |
19.829±0.090a |
| 72 | 8.590±0.467 | 6.684±0.420a |
The activity of JD was further evaluated using the
colony formation assay. As shown in Fig. 2C and Table II, JD not only significantly
inhibited the proliferation of EC109/Taxol cells, but also reduced
the colony-forming ability compared with the control groups. The
colony formation assays demonstrated that JD treatment at
concentrations as low as 0.5 µM significantly suppressed the
colony-forming activity of EC109/Taxol cells. According to the
inhibitory effects of JD on the colony-formaing ability of
EC109/Taxol cells (Table II), we
calculated that the IC50 value of JD was 0.87 µM
using SPSS software. The data also indicated that the JD-induced
suppression of colony formation correlated well with the
concentrations of JD, suggesting that JD treatment inhibited the
colony-forming ability of EC109/Taxol cells in a dose-dependent
manner.
| Table IIInhibitory effects of JD on the
colony formation of EC109/Taxol cells. |
Table II
Inhibitory effects of JD on the
colony formation of EC109/Taxol cells.
| Group | Colony formation
rate (%) |
|---|
| EC109/Taxol | 53.30±0.04 |
| 0.25 µM
JD | 47.00±0.04 |
| 0.5 µM
JD | 34.33±0.07a |
| 1 µM JD | 25.24±0.01a |
| 2 µM JD | 11.36±0.02b |
| 4 µM JD | 0.06±0.01b |
In addition, proliferation curves were generated for
the EC109/Taxol and EC109 cells incubated with 10 µM of JD.
The results indicated that 10 µM JD induced significantly
decreased the proliferation of EC109/Taxol and EC109 cells. After
48 h and 72 h of incubation, the numbers (×104) of the
EC109/Taxol (48 h-0.219±0.03, 72 h-0.125±0.01) and EC109 (48
h-0.563±0.02, 72 h-0.025±0.01) cells were only a few. Following 96
h of incubation, the proliferation of the EC109/Taxol and EC109
cells was completely inhibited (Fig.
2D).
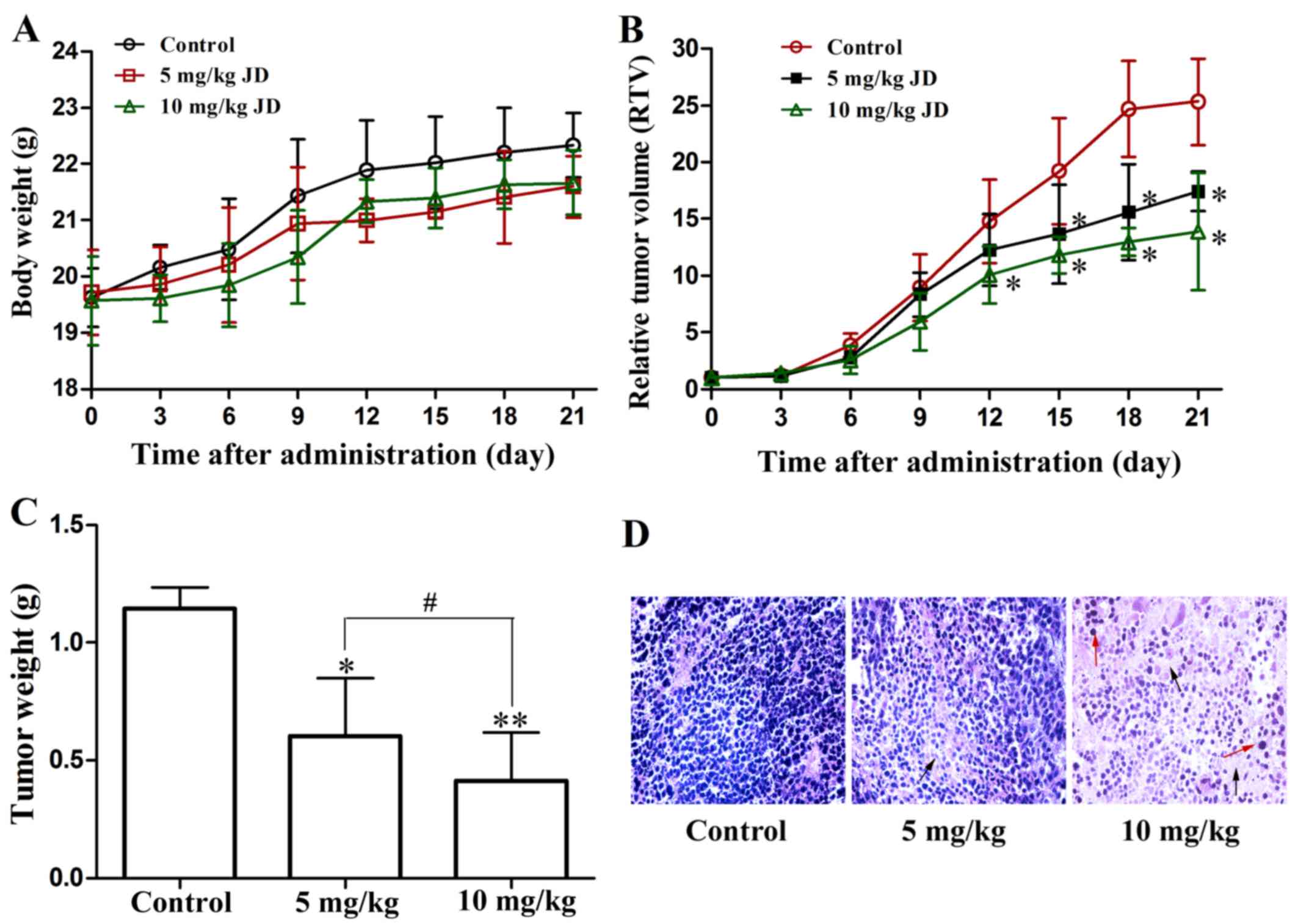
Assessment of cancer growth in mice
To evaluate the in vivo protective effects of
JD, we used the growth of EC109/Taxol cell xenografts in female
nude mice as an in vivo model. Five animals per treatment
group, injected intravenously, were used. No significant difference
was observed in the body weight changes among the different
treatment groups, suggesting that this regimen was safe (Fig. 3A). Compared with the control
group, the group treated with JD (either 5 or 10 mg/kg)
demonstrated a significantly inhibition of tumor growth, both in
terms of tumor size and weight (Fig.
3B and C).
Histological analysis of the H&E-stained tumor
sections from the EC109/Taxol xenografts from the mice treated with
JD demonstrated a marked change in tissue and cell morphology
compared with those from the vehicle control group (Fig. 3D). These changes included a low
cell density and multinucleated cells with condensed chromatin
staining and pyknosis, indicating mitotic catastrophe and
apoptosis.
Effects of JD on the cell cycle
distribution of EC109/Taxol cells
The cellular DNA content of JD-treated cells and
untreated cells was analyzed by flow cytometry to detect changes in
the cell cycle distribution of the EC109/Taxol cells. As shown in
Fig. 4 and Table III, treatment of the EC109/Taxol
cells with JD at 10, 20 and 40 µM for 24 h significantly
increased the G2/M cell population up to 33.95±1.97, 48.29±1.38 and
69.169.10±4.02%, respectively, whereas 25.56±2.02% of the control
cell population was in the G2/M phase. The increase in the G2/M
cell population induced by JD occurred at the expense of a decrease
in the G0/G1 and S cell populations. These results indicated that
the cells treated with JD exhibited a G2/M phase arrest.
| Table IIIPercentage of cells in the different
cell cycle phases. |
Table III
Percentage of cells in the different
cell cycle phases.
| Group | G0/G1 (%) | S (%) | G2/M (%) |
|---|
| EC109/Taxol | 47.96±0.91 | 26.48±1.24 | 25.56±2.02 |
| 10 µM
JD | 43.58±0.87a | 22.65±1.56a | 33.95±1.97a |
| 20 µM
JD | 35.23±2.45a | 15.27±0.96a | 48.29±1.38a |
| 40 µM
JD | 18.61±2.14a | 11.25±0.98a | 69.10±4.02a |
JD induces the apoptosis of EC109/Taxol
cells
Following treatment with JD at the indicated
concentrations for 24 h, morphological changes in EC109/Taxol cells
were assessed using an inverted microscope. DMSO (0.1%) was used as
a control. The EC109/Taxol cells treated with JD exhibited
apoptosis-related morphologies, such as cell shrinkage and rounding
up, cell membrane blebbing and nuclear fragmentation and
condensation (Fig. 5A). In
addition, nuclear morphological changes at the corresponding
concentration were also recorded and qualitatively evaluated by
means of Hoechst 33258 fluorescent staining following incubation
for 24 h. Typical apoptotic markers, including nuclear
fragmentation and nuclear condensation, were detected, particularly
at the highest concentrations of JD (Fig. 5B).
In addition, to determine whether JD treatment
induces apoptosis, cells incubated with increasing concentrations
of JD for 24 h were analyzed using flow cytometry to assess the
diploid DNA content and for phosphatidylserine (PS) exposure to the
cell surface. The percentages of live, early-apoptotic,
late-apoptotic and necrotic cells were calculated using an Accuri
C6 flow cytometer. Treatment of the EC109/Taxol cells for 24 h with
10, 20 and 40 µM of JD resulted in a dose-dependent increase
in the percentage of the apoptotic population up to 6.7±1.80,
36.2±3.60 and 46.1±2.62%, respectively, compared with the control
treated cells (3.6±0.64%) (Fig. 5C
and D).
Western blot analysis
As p53 is a major tumor suppressor, we examined the
role of p53 in JD-mediated growth inhibition. JD treatment resulted
in an increase in the p53 protein levels in the EC109/Taxol cells
(Fig. 6A and B). Apoptosis is a
well-organized cellular event, characterized by the activation of
caspase cascades. Caspases are synthesized as pro-enzymes that must
be activated to perform their functions. Caspase-9 is the major
caspase member that regulates the intrinsic apoptosis pathway,
whereas caspase-3 is one of the critical effector caspases in the
downstream execution of apoptosis (5,15).
As shown in Fig. 6A, F and G,
upon treatment with JD, the procaspase-9 and procaspase-3 levels
decreased, and the cleaved-caspase-9 and cleaved caspase-3 levels
increased, indicating the activation of caspase-9 and
caspase-3.
We also examined the expressions levels of Bax and
Bcl-2 and then analyzed the ratio of Bax/Bcl-2. The results of
western blot analysis revealed that Bcl-2 expression was
significantly decreased and that Bax expression was not altered in
the JD-treated EC109/Taxol cells (Fig. 6A and C). Statistical analysis also
indicated that JD in the range of 10-40 µM increased the
ratio of Bax/Bcl-2 (Fig. 6D).
Moreover, PUMA expression was upregulated following treatment with
JD (Fig. 6A and E).
Discussion
The EC109/Taxol cells, a paclitaxel-resistant cell
line expressing high levels of P-gp, were generated from the
drug-sensitive parental EC109 cells by the intermittent exposure to
a high concentration of PTX in stepwise time increments over a
period of 6 months. The PTX resistance of the EC109/Taxol cell line
was approximately 67.2-fold that of the EC109 cell line. In
addition, the EC109/Taxol cells exhibited a greater
cross-resistance to 5-fluorouracil (5-FU), cisplatin (CDDP) and
epirubicin (EPI) than its parental EC109 cell line (8). In this study, we examined the effect
of JD on EC109/Taxol cells both in vitro and in vivo.
JD exhibited a strong activity against EC109/Taxol cells. The
JD-induced inhibition of cancer growth was confirmed in nude mouse
models. The results indicated that EC109/Taxol xenografts were
sensitive to JD.
Cell cycle perturbation plays an important role in
carcinogenesis, and cell proliferation, apoptosis, differentiation
and senescence are all cell cycle-dependent (16). In the next set of experiments, we
examined the effects of JD on the cell cycle distribution of
EC109/Taxol cells. EC109/Taxol cells were treated with JD at
various concentrations (0, 10, 20 and 40 µM) for 24 h. JD
caused an accumulation of cells in the G2/M phase and diminished
cells in the S and G0/G1 phases compared with the untreated control
cells. The results suggested that JD caused an obvious G2/M arrest
pattern in a concentration-dependent manner, with a concomitant
decrease in terms of the number of cells in other phases of the
cell cycle.
Apoptosis is a regulated type of physiological cell
death in which a controlled sequence of events leads to cell
elimination essential for the maintenance of homeostasis (17,18). As the death of tumor cells by
chemotherapy is mediated mainly by the induction of apoptosis, the
deregulation of apoptosis would confer resistance to anticancer
agents. In this study, apoptosis, as determined using Hoechst 33258
staining and flow cytometric analysis (Fig. 5), was confirmed to be responsible
for the JD-mediated anti-resistance activity in EC109/Taxol cells.
Thereby, our research provides new approaches to overcoming drug
resistance by inducing apoptosis in resistant EC109/Taxol cell
lines.
For further study, we elucidated the possible
mechanisms responsible for the apoptosis induced by JD. Although
the mechanism of drug resistance is not completely defined, the
deficiency in apoptosis and abnormally activated survival signaling
pathways have been observed in most cancers with MDR. Apoptosis
deficiency may be involved in cancer initiation, progression and
treatment failure in cancers with MDR (19). Apoptosis is regulated at multiple
levels in the extrinsic and intrinsic pathways (20). The extrinsic pathway involves
death receptors on the cell surface that can directly activate
caspase-8. The intrinsic pathway of apoptosis centers on the
mitochondria and is associated with the loss of the mitochondrial
membrane potential (MMP), the release of cytochrome c into
the cytosol and the activation of caspase-9/3 (21,22). The Bcl-2 and caspase families are
considered as the most important proteins for regulating apoptosis
in the intrinsic pathway. The Bcl-2 family can be divided into two
types: anti-apoptotic and pro-apoptotic proteins (23). The ratio of Bcl-2/Bax is a
critical determinant for cell apoptosis. The dysregulation of the
Bcl-2 family has been shown to induce the destruction of the
mitochondrial membrane, which is accompanied by the release of
intramembranous proteins into the cytosol, such as cytochrome
c and other apoptosis-inducing factors, and to subsequently
induce the activation of the caspase cascade by activating
caspase-9 and caspase-3 (24,25). Bax is constitutively located
within the cytosol, whereas the activation of Bax involves its
translocation from the cytosol to the outer mitochondrial membrane
and, subsequently, its insertion into the membrane (26). Bax can release cytochrome c
after the formation of the Bax/Bak hetero-oligomer, whereas Bcl-2
can interact with activator proteins or Bax/Bak, thereby
sequestering these proteins (27-29). The BH3-only protein PUMA is a
pro-apoptotic protein that is normally expressed at a low level.
Its upregulation leads to cytochrome c release, which
results in apoptosis (30). The
tumor suppressor and transcription factor p53 is a key modulator of
cellular stress responses, and the activation of p53 can trigger
apoptosis in many cell types (31,32). Therefore, the dysregulation of the
Bcl-2 and caspase families and of p53, as well as their respective
signaling pathways, has been considered the most important
mechanism in the development of apoptotic deficiency and the
phenotype of MDR in cancer cells. The regulation of these protein
kinases may be an effective regimen for the treatment of cancers
with apoptotic deficiency and cells with MDR (33).
Our results indicated that treatment with JD
significantly downregulated anti-apoptotic Bcl-2 protein
expression, upregulated PUMA expression and did not alter
pro-apoptotic Bax protein expression, thus shifting the Bax/Bcl-2
ratio in favor of apoptosis. Moreover, JD upregulated the
expression of p53, cleaved-caspase-9 and cleaved-caspase-3 in
EC109/Taxol cells and downregulated the expression of procaspase-3
and procas-pase-9 in EC109/Taxol cells in a concentration-dependent
manner. We therefore suggested that the induction of apoptosis in
EC109/Taxol cells by JD may be due to the activation of the
mitochondria-mediated intrinsic apoptosis pathway. Further research
on the specific mechanisms of action and the efficacy of JD in
other cancer cell lines is currently underway.
In conclusion, to the best of our knowledge, our
results demonstrate for the first time that JD exerts an
anti-proliferative and anti-MDR effect on EC109/Taxol cells. JD
treatment via intravenous injection in mice effectively prevented
the growth of EC109/Taxol xenografts without exerting any
significant toxicity. These findings suggest the possibility that
JD may be a promising drug candidate and provide new supporting
evidence for the manipulation of the dysregulated apoptotic pathway
as a strategy with which to overcome drug resistance.
Acknowledgments
This study was supported by the National Natural
Science Foundation of China (project nos. 21372206 and 81430085,
for H.L.), the Natural Science Foundation of Henan Province of
China (the foundation and frontier technology research program)
(project no. 152300410030 for C.W.), the Science and Technology
Research Key Project in the Henan province Department of Education
(project no. 14B350011 for C.W.) and the Specialized Research
Foundation of the Doctoral Program of High College for new teachers
(no. 20104101120012 for C.W.).
References
|
1
|
Sui X, Chen R, Wang Z, Huang Z, Kong N,
Zhang M, Han W, Lou F, Yang J, Zhang Q, et al: Autophagy and
chemotherapy resistance: a promising therapeutic target for cancer
treatment. Cell Death Dis. 4:e8382013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lu HP and Chao CC: Cancer cells acquire
resistance to anticancer drugs: an update. Biomed J. 35:464–472.
2012. View Article : Google Scholar
|
|
3
|
Ambudkar SV, Dey S, Hrycyna CA,
Ramachandra M, Pastan I and Gottesman MM: Biochemical, cellular,
and pharmacological aspects of the multidrug transporter. Annu Rev
Pharmacol Toxicol. 39:361–398. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Baguley BC: Multidrug resistance in
cancer. Methods Mol Biol. 596:1–14. 2010. View Article : Google Scholar
|
|
5
|
Zhou Q, Li Y, Jin J, Lang L, Zhu Z, Fang W
and Chen X: Lx2-32c, a novel taxane derivative, exerts
anti-resistance activity by initiating intrinsic apoptosis pathway
in vitro and inhibits the growth of resistant tumor in vivo. Biol
Pharm Bull. 35:2170–2179. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mor G, Montagna MK and Alvero AB:
Modulation of apoptosis to reverse chemoresistance. Methods Mol
Biol. 414:1–12. 2008.PubMed/NCBI
|
|
7
|
Hsu PC, Hung HC, Liao YF, Liu CC, Tsay GJ
and Liu GY: Ornithine decarboxylase attenuates leukemic
chemotherapy drugs-induced cell apoptosis and arrest in human
promyelocytic HL-60 cells. Leuk Res. 32:1530–1540. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang C, Guo LB, Ma JY, Li YM and Liu HM:
Establishment and characterization of a paclitaxel resistant human
esophageal carcinoma cell line. Int J Oncol. 43:1607–1617.
2013.PubMed/NCBI
|
|
9
|
Liu Z, Ouyang L, Peng H and Zhang WZ:
Oridonin: Targeting programmed cell death pathways as an
anti-tumour agent. Cell Prolif. 45:499–507. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li CY, Wang EQ, Cheng Y and Bao JK:
Oridonin: An active diterpenoid targeting cell cycle arrest,
apoptotic and autophagic pathways for cancer therapeutics. Int J
Biochem Cell Biol. 43:701–704. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang C, Jiang L, Wang S, Shi H, Wang J,
Wang R, Li Y, Dou Y, Liu Y, Hou G, et al: The antitumor activity of
the novel compound Jesridonin on human esophageal carcinoma cells.
PLoS One. 10:e01302842015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tian QE, Li HD, Yan M, Cai HL, Tan QY and
Zhang WY: Astragalus polysaccharides can regulate cytokine and
P-glycoprotein expression in H22 tumor-bearing mice. World J
Gastroenterol. 18:7079–7086. 2012. View Article : Google Scholar
|
|
13
|
Wacheck V and Zangemeister-Wittke U:
Antisense molecules for targeted cancer therapy. Crit Rev Oncol
Hematol. 59:65–73. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Uchiyama-Kokubu N and Watanabe T:
Establishment and characterization of adriamycin-resistant human
colorectal adenocarcinoma HCT-15 cell lines with multidrug
resistance. Anticancer Drugs. 12:769–779. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Fulda S and Debatin KM: Extrinsic versus
intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene.
25:4798–4811. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang J, Zhu X, Li H, Li B, Sun L, Xie T,
Zhu T, Zhou H and Ye Z: Piperine inhibits proliferation of human
osteosarcoma cells via G2/M phase arrest and metastasis by
suppressing MMP-2/-9 expression. Int Immunopharmacol. 24:50–58.
2015. View Article : Google Scholar
|
|
17
|
Hafezi F, Steinbach JP, Marti A, Munz K,
Wang ZQ, Wagner EF, Aguzzi A and Remé CE: The absence of c-fos
prevents light-induced apoptotic cell death of photoreceptors in
retinal degeneration in vivo. Nat Med. 3:346–349. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cao B, Chen H, Gao Y, Niu C, Zhang Y and
Li L: CIP-36, a novel topoisomerase II-targeting agent, induces the
apoptosis of multidrug-resistant cancer cells in vitro. Int J Mol
Med. 35:771–776. 2015.PubMed/NCBI
|
|
19
|
Uboldi S, Bernasconi S, Romano M, Marchini
S, Fuso Nerini I, Damia G, Ganzinelli M, Marangon E, Sala F, Clivio
L, et al: Characterization of a new trabectedin-resistant myxoid
liposarcoma cell line that shows collateral sensitivity to
methylating agents. Int J Cancer. 131:59–69. 2012. View Article : Google Scholar
|
|
20
|
Bai HW, Badaboina S, Park CH, Choi BY, Na
YH and Chung BY: Centipedegrass extract induces apoptosis through
the activation of caspases and the downregulation of I3K/Akt and
MAPK phosphorylation in leukemia cells. Int J Mol Med. 35:511–518.
2015.
|
|
21
|
Kroemer G and Reed JC: Mitochondrial
control of cell death. Nat Med. 6:513–519. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Fares M, Abedi-Valugerdi M, Hassan M and
Potácová Z: DNA damage, lysosomal degradation and Bcl-xL
deamidation in doxycycline- and minocycline-induced cell death in
the K562 leukemic cell line. Biochem Biophys Res Commun.
463:268–274. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Peng J, Ding J, Tan C, Baggenstoss B,
Zhang Z, Lapolla SM and Lin J: Oligomerization of membrane-bound
Bcl-2 is involved in its pore formation induced by tBid. Apoptosis.
14:1145–1153. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kluck RM, Bossy-Wetzel E, Green DR and
Newmeyer DD: The release of cytochrome c from mitochondria: A
primary site for Bcl-2 regulation of apoptosis. Science.
275:1132–1136. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Degli Esposti M and Dive C: Mitochondrial
membrane permeabilisation by Bax/Bak. Biochem Biophys Res Commun.
304:455–461. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chipuk JE, Kuwana T, Bouchier-Hayes L,
Droin NM, Newmeyer DD, Schuler M and Green DR: Direct activation of
Bax by 53 mediates mitochondrial membrane permeabilization and
apoptosis. Science. 303:1010–1014. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kang MH and Reynolds CP: Bcl-2 inhibitors:
Targeting mitochondrial apoptotic pathways in cancer therapy. Clin
Cancer Res. 15:1126–1132. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bodur C and Basaga H: Bcl-2 inhibitors:
Emerging drugs in cancer therapy. Curr Med Chem. 19:1804–1820.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Nikoletopoulou V, Markaki M, Palikaras K
and Tavernarakis N: Crosstalk between apoptosis, necrosis and
autophagy. Biochim Biophys Acta. 1833:3448–3459. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Seervi M, Sobhan PK, Joseph J, Ann Mathew
K and Santhoshkumar TR: ERO1α-dependent endoplasmic
reticulum-mitochondrial calcium flux contributes to ER stress and
mitochondrial permeabilization by procaspase-activating compound-1
(PAC-1). Cell Death Dis. 4:e9682013. View Article : Google Scholar
|
|
31
|
Culmsee C and Mattson MP: 53 in neuronal
apoptosis. Biochem Biophys Res Commun. 331:761–777. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Tacar O and Dass CR: Doxorubicin-induced
death in tumour cells and cardiomyocytes: Is autophagy the key to
improving future clinical outcomes? J Pharm Pharmacol.
65:1577–1589. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
West KA, Castillo SS and Dennis PA:
Activation of the I3K/Akt pathway and chemotherapeutic resistance.
Drug Resist Updat. 5:234–248. 2002. View Article : Google Scholar
|